A pathogenic picornavirus acquires an envelope by hijacking cellular membranes (original) (raw)
Main
Supernatant fluids of hepatoma cell cultures infected with low-passage, non-cytopathic HAV9 (Fig. 1A) contain two populations of virus particles that are resolved in isopycnic iodixanol gradients (Fig. 1B). One bands at a low density consistent with membrane association (1.06–1.10 g cm−3, fractions 8–12) and is not detected in a capsid antigen enzyme-linked immunosorbent assay (ELISA) (Fig. 1C, left), whereas the other bands at the density expected for picornaviruses (1.22–1.28 g cm−3, fractions 18–22) and is readily detected by ELISA. Electron microscopy of the light fractions revealed numerous virus-like particles enclosed in membranes (see a–d in Fig. 1D, and Supplementary Fig. 1a) with morphology indistinguishable from ∼27-nm HAV particles in dense fractions (see e in Fig. 1D). These membrane-bound structures ranged from 50 to 110 nm in diameter, a size similar to that of exosomes7, and contained one to four virus-like particles (Supplementary Fig. 1b). Consistent with this, viral RNA banded in gradients with capsid protein (VP2) and the exosome-associated protein flotillin-1 (Supplementary Fig. 1c).
Figure 1: Enveloped particles (eHAV) are the dominant form of virus released from infected cell cultures.
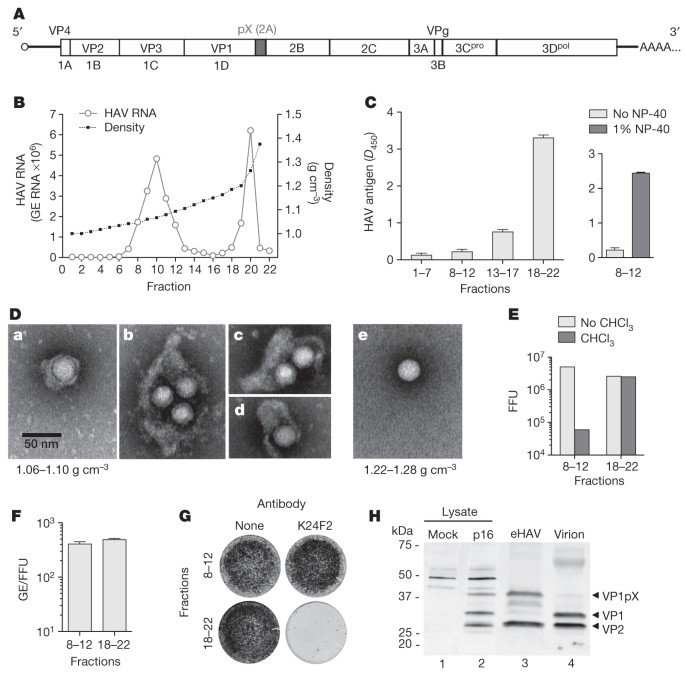
A, HAV genome organization. The polyprotein is depicted as a box with pX highlighted. B, Buoyant density of HAV particles released by Huh-7.5 cells in iodixanol gradients. eHAV bands at 1.06–1.10 g cm−3, whereas non-enveloped HAV bands at 1.22–1.28 g cm−3. GE, genome equivalents. C, Left, HAV capsid antigen was detected by ELISA only in pools of denser fractions from the gradient in B. Right, capsid antigen was detected in light fractions after treatment with 1% NP-40. Data shown are _D_450 (mean and range) in duplicate assays. D, Electron microscopic images of negatively stained eHAV (a–d from fraction 10 in B) and a non-enveloped virion (e, fraction 20 in B). E, Infectious titre of pooled fractions containing eHAV or non-enveloped virions before and after extraction with chloroform. FFU, focus-forming units. F, Specific infectivity of pooled fractions containing eHAV or non-enveloped virions, calculated by dividing the HAV RNA copy number (GE, qRT–PCR) by the infectious titre (FFU, IR-FIFA). Values shown are means and range for duplicate RT–PCR reactions. G, eHAV is resistant to neutralization by anti-capsid monoclonal antibody K24F2 (ref. 11). Antibody–virus mixtures were incubated for 1 h at 37 °C and inoculated onto cells for 1 h, followed by removal of the inoculum, washing three times with PBS, and the addition of an agarose overlay. Viral antigen was detected by IR-FIFA10. H, Immunoblots of HAV capsid proteins (VP1 and VP2) in lysates of mock-infected or HAV-infected cells (lanes 1 and 2), gradient-purified eHAV (lane 3) and chloroform-extracted non-enveloped virions (lane 4).
A modified plaque assay (infrared fluorescence immunofocus assay; IR-FIFA)10 revealed the membrane-wrapped particles to be infectious (Fig. 1E) with a specific infectivity equivalent to that of virions (Fig. 1F). Extraction with chloroform, a classic method for distinguishing enveloped from non-enveloped viruses, had no effect on standard virions but resulted in a 2log10 decrease in infectious virus in the light fraction (Fig. 1E and Supplementary Fig. 2a). We call these membrane-wrapped HAV particles ‘enveloped HAV’ (eHAV). Capsid antigen could be detected in the eHAV fraction after treatment with a nonionic detergent (1% Igepal CA-630; NP-40) (Fig. 1C, right). This shifted the viral particles to an intermediate density in iodixanol gradients (1.15–1.17 g cm−3) but did not destroy infectivity (Supplementary Fig. 2b, c). A potent, neutralizing, monoclonal antibody (mAb), K24F2 (ref. 11), failed to neutralize eHAV (Fig. 1G), providing further evidence for complete envelopment of the capsid.
In 12 experiments, eHAV represented 79 ± 13% (mean ± s.d.) of virus in medium from infected cell cultures. eHAV was released from multiple cell types and was also observed with high-passage cytopathic virus (Supplementary Fig. 3a–e). Gradient-purified eHAV contained mostly mature VP2 (Fig. 1H, lane 3, and Supplementary Fig. 2d), indicating that enveloped virions had undergone maturation cleavage of VP0 to VP4 + VP2. However, whereas non-enveloped virions contained fully processed VP1, eHAV contained primarily unprocessed VP1pX (Fig. 1H; compare lanes 3 and 4). pX is an unusual 8-kDa carboxy-terminal extension on VP1 that is unique to HAV among picornaviruses (Fig. 1A). It functions in virion assembly and is processed from VP1 by an unidentified host protease late in the viral lifecycle12,13. pX was protected from proteinase K in eHAV particles, but was rendered susceptible to digestion after treatment with NP-40 (Supplementary Fig. 2e). Thus, pX is fully enclosed in membranes. We conclude that most HAV is released enveloped in host membranes, a process we term ‘membrane hijacking’.
Infectious virus circulating in the blood of infected humans and chimpanzees (Pan troglodytes) possesses the buoyant density of eHAV (Fig. 2a and Supplementary Fig. 4a–e), confirming the relevance of these observations to HAV pathogenesis. However, virus in faeces is non-enveloped (Fig. 2b and Supplementary Fig. 4c). Because most faecal virus is produced in the liver14,15, the membrane may be stripped from eHAV during passage through the biliary tract to the gut. However, suspending eHAV in bile minimally altered its buoyant density (Supplementary Fig. 5a).
Figure 2: eHAV circulates in the blood of HAV-infected humans and chimpanzees.
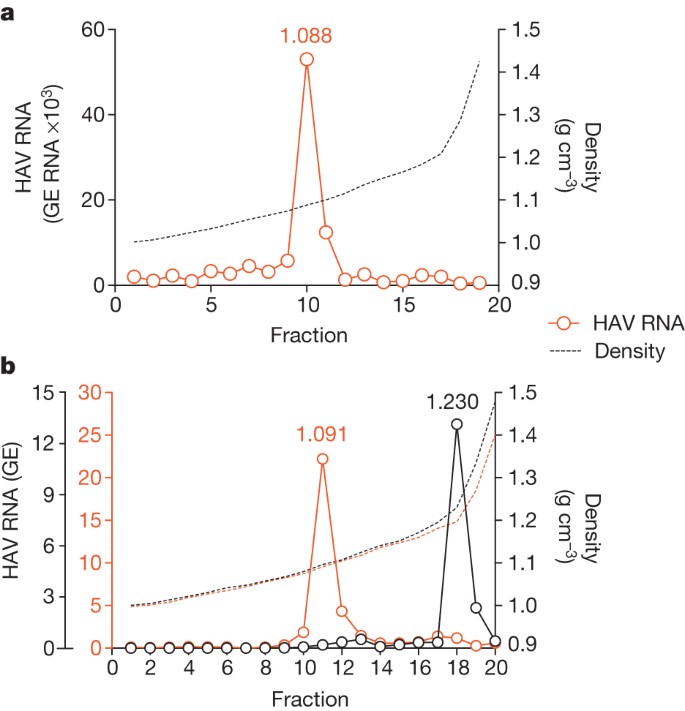
a, Distribution of HAV RNA in an iodixanol gradient loaded with early, acute-phase serum from patient BH12. b, Buoyant density of HAV particles from plasma (red open circles) and faeces (black open circles) of an experimentally infected chimpanzee, x0293 (ref. 5). Faecal HAV RNA is shown ×105; plasma RNA is shown ×102 (also see Supplementary Fig. 4). The low buoyant density of circulating virus was not due to a passive effect of serum, because the buoyant density of non-enveloped virions was not altered by suspension in 90% serum (data not shown).
It has been suggested16 that poliovirus, a distantly related picornavirus, could be released from cells wrapped in a single membrane if entrapped in an autophagosome that fuses with the plasma membrane. However, RNA-mediated interference knockdown of beclin-1, which mediates autophagosome formation, did not inhibit eHAV release (Supplementary Fig. 6a, b). In contrast, knockdown of VPS4B or ALIX, which are ESCRT-III-associated proteins that facilitate the budding of many enveloped viruses17,18,19,20, inhibited the release of eHAV from cells (Fig. 3a–c and Supplementary Figs 6c and 7a) but not viral RNA replication (Fig. 3d), intracellular assembly of capsids or encapsidation of viral RNA (Supplementary Fig. 7a–c). ESCRT complexes act sequentially to sort and load cargo into multivesicular bodies (MVBs)8. VPS4B is an ATPase that provides energy for the dissolution of ESCRT-III complexes at membrane fission, whereas ALIX contributes to exosome biogenesis and also facilitates lentivirus budding8,19,21. Knockdown of VPS4B and ALIX inhibited the release of both enveloped and non-enveloped HAV (Fig. 3c), suggesting these share a common egress pathway and that non-enveloped virus in extracellular fluids might be derived from eHAV. Knockdown of HRS and TSG101, which are ESCRT-0 and ESCRT-I proteins involved early in cargo recruitment, did not inhibit eHAV release (Fig. 3b, c). Thus, eHAV release is dependent on ESCRT-associated proteins but not on the entire ESCRT machinery, in a similar manner to the budding of some enveloped viruses8,18. Although an interaction of ALIX with the ESCRT-III protein CHMP4B facilitates the budding of human immunodeficiency virus22, CHMP4A, CHMP4B or CHMP4C knockdown minimally decreased eHAV release (Supplementary Fig. 8).
Figure 3: eHAV biogenesis requires ESCRT-associated proteins.
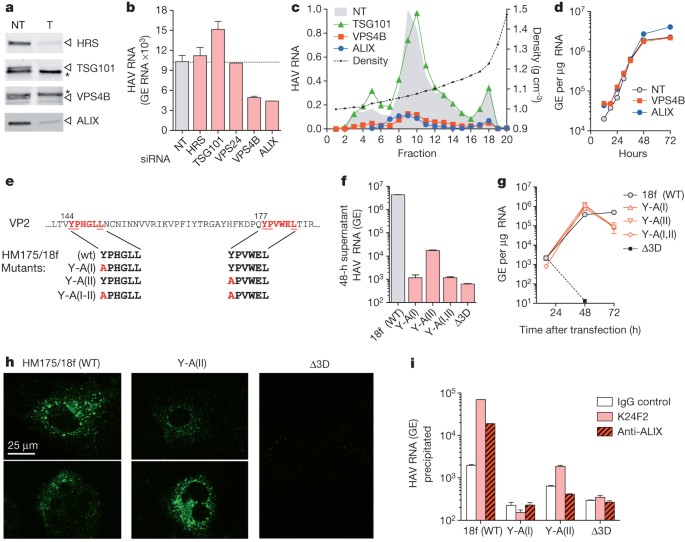
a, Immunoblots of HRS, TSG101, VPS4B and ALIX in HAV-infected cells 72 h after transfection with the indicated gene-specific targeting (T) or non-targeting (NT) control siRNAs. Asterisks, non-specific bands. qRT–PCR assays confirmed knockdown of TSG101 and VPS4B (Supplementary Fig. 6c). b, Viral RNA detected in culture fluids 48–72 h after siRNA transfection. c, Relative yield and buoyant density of virus released from siRNA transfected cells (as in a) determined in iodixanol gradients. RNA associated with eHAV particles (fractions 8–12) was decreased by 85% and 87% by depletion of VPS4B and ALIX, respectively. d, One-step growth of eHAV. Huh-7.5 cells were transfected with the indicated siRNAs for 3 days, then infected with gradient-purified eHAV at a multiplicity of infection of 20. Cell-associated HAV RNA was assayed by qRT–PCR. Knockdown efficiency was confirmed at the end of the experiment (not shown). e, Tandem YPX3L ALIX-interacting motifs (red) in VP2. Below are the motif sequences in wild-type (WT, HM175/18f) and mutant viral RNAs. f, Viral RNA released into culture fluids 24–48 h after electroporation of Huh-7.5 cells with wild-type and mutant viral RNAs. Δ3D is a replication-incompetent subgenomic replicon RNA with a frameshift mutation in 3Dpol. g, Intracellular HAV RNA after electroporation of cells with WT and VP2 mutant RNAs. h, Confocal microscopy showing K24F2 detection of capsid antigen in cells electroporated 48 h previously with WT or mutant RNAs. K24F2 recognizes a conformation-dependent assembled neutralization epitope in the viral capsid11. i, Anti-capsid (K24F2) and anti-ALIX (Bethyl) antibody-mediated immunoprecipitation of encapsidated, RNase-resistant viral RNA in detergent-treated lysates of cells electroporated with wild-type or mutant HAV RNAs. RT–PCR data are means ± s.e.m. for two or three replicate assays; all results are representative of two or three independent experiments.
Structural proteins of enveloped viruses interact with ESCRT-related proteins by means of Pro-rich late (‘L’) domain motifs: PPXY, P(S/T)AP or (L)YPX1/3L (refs 8, 17). Only two domains in the 2,227-residue-long HAV polyprotein conform to these motifs: conserved, tandem YPX3L motifs separated by 28 residues in the VP2 capsid protein (Fig. 3e). YPX1/3L motifs mediate interactions with ALIX (ref. 19), and their presence in VP2 is consistent with a requirement for ALIX. Disrupting either YPX3L motif by substituting Ala for Tyr (Fig. 3e) severely decreased virus release but did not impair viral RNA replication (Fig. 3f, g). Intracellular capsid antigen was similarly decreased, suggesting that the mutations impede capsid assembly, but the antigen was detected in cells transfected with Y-A(II) RNA in which the second YPX3L motif had been ablated (Fig. 3h). Limited capsid assembly was confirmed by K24F2 precipitation of RNase-protected viral RNA from detergent-treated cell lysates (Fig. 3i). Consistent with a role for ALIX in eHAV biogenesis, anti-ALIX antibody precipitated encapsidated (RNase-protected) viral RNA from lysates of cells transfected with wild-type, but not Y-A(II), RNA (Fig. 3i). Furthermore, ALIX banded at the same density as eHAV in iodixanol gradients (Supplementary Fig. 7d). Although details remain to be worked out, these data suggest that HAV co-opts ALIX to facilitate its envelopment in host membranes. Where this occurs is uncertain, but HAV particles have been observed within cytoplasmic vesicles in infected liver14,23.
The cloaking of its capsid by membranes allows eHAV to evade neutralizing antibodies (Fig. 1G and Supplementary Fig. 9a) and explains how infectious virus co-exists with anti-HAV in serum15. However, antibodies protect against hepatitis A when passively transferred after HAV replication has been well established in the liver5,6. We therefore reasoned that eHAV might be neutralized after binding to or entering hepatocytes. Consistent with this, eHAV was neutralized when cells were exposed to antibody either immediately before inoculation with virus or up to 6 h afterwards (Fig. 4a, b). IgG and IgA anti-capsid mAbs neutralized eHAV when added to cultures after removal of the inoculum, but an IgM mAb, H14C42 (ref. 11), failed to do so despite potent neutralizing activity against non-enveloped HAV (Supplementary Fig. 9b). Taken together, these results show capsid is the target for neutralization and that eHAV membranes must rupture before neutralization, but not before eHAV is internalized and inaccessible to H14C42 in the medium. This suggests that eHAV may be neutralized after endocytosis. Chloroquine efficiently blocked infection by eHAV (but not by non-enveloped HAV) (Fig. 4c), indicating that endosomal acidification is important for eHAV entry, whereas an antibody against the HAV receptor, TIM-1 (HAVCR-1)24, inhibited both (Fig. 4d). We found eHAV membranes to be stable at pH 5.0 (Supplementary Fig. 5b), but they could nonetheless be degraded in late endosomes and lysosomes25. If so, this would render the capsid accessible to TIM-1, which undergoes constitutive endocytosis and trafficking to late endosomes and lysosomes26, as well as to anti-HAV. Many details of eHAV entry and neutralization remain to be explained. However, TRIM-21 (tripartite motif-containing 21), which is implicated in intracellular neutralization of other viruses27, seems to have no function in neutralization of eHAV (Supplementary Fig. 9c).
Figure 4: Extracellular eHAV is resistant to antibody-mediated neutralization but is neutralized by antibody after infection.
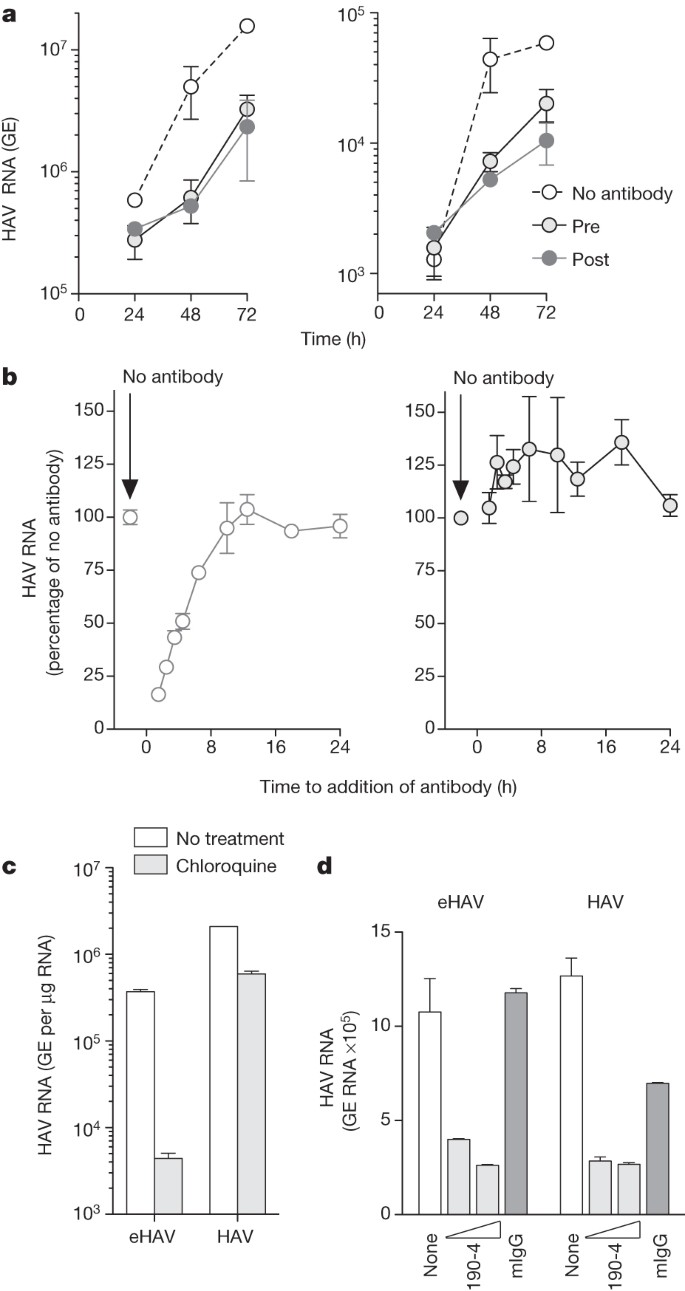
a, Neutralization of eHAV in cells treated with anti-HAV before (pre) or after (post) adsorption of virus. Left, intracellular; right, extracellular. Pretreated cells were incubated with JC plasma (1:50–1:100) for 1 h, washed extensively with PBS, then incubated with gradient-purified eHAV (multiplicity of infection about 6) for 1 h in the absence of antibody. For post-treatment, JC was added to the medium on refeeding after removal of the inoculum. Cells were subsequently re-fed at 24 h intervals with fresh medium containing no antibody. Data are means ± s.e.m. from duplicate cultures and are representative of six independent experiments. b, Adding anti-HAV to medium as late as 6 h after removal of the inoculum restricts the replication of eHAV (left) but not that of non-enveloped HAV (right). JC antibody was added at intervals after removal of the inoculum, and intracellular viral RNA was quantified at 48 h. Results are means ± s.e.m. of replicate RT–PCR assays and are representative of three independent experiments. c, Chloroquine (50 μM) blocks eHAV entry but not HAV entry. Cells were treated for 30 min before inoculation with virus, and were harvested at 48 h to assay intracellular HAV RNA. d, Anti-TIM-1 mAb 190-4 blocks entry of both eHAV and HAV. GL37 cells were incubated with 190-4 (10–100 μg ml−1) or control IgG (100 μg ml−1) for 1 h at 37 °C, then inoculated with virus for 1 h. Intracellular HAV RNA was assayed at 72 h. RT–PCR data are means ± s.e.m. for two or three replicate assays; all results are representative of two or three independent experiments.
Our findings show how a ‘non-enveloped’ picornavirus promotes its egress in the absence of cell lysis, and reveal a previously unrecognized strategy by which a virus cloaks itself in host membranes to evade neutralizing antibodies. The host membrane enveloping eHAV is likely to facilitate its spread within the liver. Antibodies restrict the replication of eHAV when added to cultures several hours after infection, but further studies will be needed to determine how this happens and whether this can explain why immune globulin and vaccines protect against hepatitis when administered long after exposure, when virus is already circulating in blood5. Our results suggest that categorizing viruses into those that are enveloped and those that are not, long a tradition in virology1, is overly simplistic, and that efforts should be made to identify other examples of membrane hijacking by ‘non-enveloped’ viruses.
Methods Summary
Non-cytopathic, cell-culture-adapted HM175/p16 HAV9 was propagated in Huh-7.5 cells. Reverse molecular genetics studies were performed with pHM175/18f, a molecular clone of a related cytopathic variant28. Buoyant density was assessed in 8–40% iodixanol (Opti-Prep) gradients centrifuged at 141,000_g_ for 48 h at 4 °C. Viral RNA was measured by quantitative PCR with reverse transcription (qRT–PCR) with primers targeting the 5′ untranslated region. Infectivity was quantified by IR-FIFA10. For RNA-mediated interference studies, cells were transfected with SmartPool short interfering RNAs (siRNAs) (Dharmacon) and samples were collected 48–72 h later for viral RNA quantification. To analyse VP2–ALIX interactions, cell lysates were prepared 48 h after electroporation of mutant and wild-type viral RNAs, treated with RNase and immunoprecipitated. RNA extracted from immunoprecipitates was assayed by HAV-specific qRT–PCR. For intracellular neutralization, cells were incubated with eHAV for 1 h at 37 °C, then washed extensively. Antibodies were added at intervals, and intracellular and extracellular HAV RNA was quantified at 48–72 h. For standard neutralization assays, virus was incubated with antibodies for 1 h at 37 °C, then inoculated onto cells for IR-FIFA10.
Online Methods
Reagents and antibodies
Chemical reagents were purchased from Sigma unless otherwise noted. A human convalescent plasma (JC plasma) sample with high-titre anti-HAV antibody, collected 90 days after the onset of acute hepatitis A, was described previously11,29. Other antibodies were obtained as follows: anti-TSG101 (Abcam), anti-HRS (Bethyl), anti-ALIX (Pierce and Bethyl), anti-VPS4B (Sigma), anti-beclin-1 (Cell Signaling), anti-flotillin-1 (BD Biosciences) and anti-HAV mAbs K24F2 and K32F2 (Commonwealth Serum Laboratories, Victoria, Australia). The sources of other anti-HAV mAbs have been described previously11. Anti-TIM-1 mAb 190-4 was a gift from G. Kaplan. Guinea-pig anti-peptide antibodies against HAV VP1 (residues 211–228) and VP2 (residues 240–259) were a gift from D. Sangar.
Cells and viruses
Huh-7.5 and HepG2 (both human hepatoma cells), MRC-5 (human lung fibroblasts), FRhK-4 (fetal rhesus monkey kidney cells), BS-C-1 and GL37 (ref. 30) (both African green monkey kidney) cells were cultured in DMEM medium supplemented with 10% FBS. Cell-culture adapted variants of the HM175 strain of HAV, HM175/p16 (refs 9, 31) (non-cytopathic) and HM175/18f (refs 28, 32) (rapidly replicating, cytopathic), have been described previously9,32. Infectious virus titres were determined by a modification of the IR-FIFA described previously10 performed in FRhK-4 cells.
Human samples and clinical information
All human samples and related clinical information were obtained with the approval of the Institutional Review Boards of the Seoul National University Bundang Hospital and the University of North Carolina at Chapel Hill.
Chimpanzee samples
All chimpanzee materials were archived samples collected during previous studies5 conducted prior to 15 December 2011.
HAV capsid antigen ELISA assay
HAV capsid antigen was detected with an ELISA modification of a previously described radioimmunoassay33. Immobilized antigen captured by JC antibody was detected by sequential incubation of ELISA plates with K24F2 (1:1,000 dilution) and horseradish peroxidase-conjugated goat-anti-mouse antibody (1:10,000) at room temperature (20–23 °C) for 1 h each. After washing with PBS-T and substrate (3,3′,5,5′-tetramethylbenzidine) addition, _D_450 was determined with a Synergy 2 (BioTek) microplate reader.
qRT–PCR assay for HAV RNA
Total cellular RNA was extracted from cell lysates with the RNeasy Kit (Qiagen); RNA from culture supernatants and gradient fractions was extracted with the QiaAmp viral RNA Isolation Kit (Qiagen). HAV RNA was quantified by real-time qRT–PCR against a synthetic RNA standard as described previously5. Intracellular viral RNA was normalized to total RNA abundance or glyceraldehyde-3-phosphate dehydrogenase mRNA level determined in a SYBR green one-step qRT–PCR assay (Bio-Rad). For experiments involving electroporation of synthetic viral RNAs, a two-step qRT–PCR procedure was conducted in which complementary DNA was synthesized with (dT)20 as primer, followed by RNaseH digestion and qPCR assay with primers targeting the HAV 5′ untranslated RNA segment.
Virus neutralization
To assess eHAV neutralization following binding or entry of eHAV into cells, Huh-7 cells (FT3-7 subline) were incubated with gradient-purified eHAV for 1 h at 37 °C, then washed extensively and refed with fresh medium. Antibodies were added at intervals, and intracellular and extracellular HAV RNA was quantified 48–72 h after virus inoculation. For standard neutralization assays, virus was incubated with antibodies for 1 h at 37 °C, then inoculated onto FRhK-4 cells which were processed subsequently for IR-FIFA10.
Immunoblotting
Cells were lysed with CelLytic Lysis Buffer (Invitrogen) in the presence of protease inhibitor cocktail (Roche). Proteins were extracted from gradient fractions by precipitation with trichloroacetic acid. Immunoblots were performed with standard procedures and the indicated antibodies. Protein bands were detected with an Odyssey Infrared Imaging System (LI-COR Biosciences).
Immunofluorescence microscopy
Huh-7.5 cells seeded on four-well chamber slides were fixed with 4% paraformaldehyde, permeabilized with 0.2% Triton X-100 in PBS, and blocked with 10% goat serum. Cells were then incubated with mAb K24F2 (1:300 dilution in 1% BSA) for 1 h at room temperature, washed, and incubated with fluorescein isothiocyanate-conjugated goat anti-mouse antibody (1:300; Southern Biotech) for 1 h. Images were collected with a Zeiss 510 Meta laser-scanning confocal microscope.
Isopycnic gradient centrifugation of virus
Cell-culture supernatant fluids were centrifuged at 1,000_g_ for 10 min at 4 °C to remove cells and debris, further clarified by centrifugation twice at 10,000_g_ for 30 min, and concentrated by ultracentrifugation at 100,000_g_ for 1 h at 4 °C. The resulting pellet was resuspended in PBS, loaded onto a 8–40% iodixanol (Opti-Prep) step gradient, and centrifuged at 141,000_g_ in a Superspin 630 rotor for 48 h at 4 °C in a Sorvall Ultra-80 ultracentrifuge. Approximately 20 fractions were collected from the top of the gradient; density was determined with a refractometer.
Rate-zonal centrifugation of HAV in sucrose gradients
Lysates were prepared from HAV (HM175/p16)-infected Huh-7.5 cells, loaded onto a 7.5–45% (w/v) sucrose gradient in Hank’s balanced buffer, and centrifuged at 4 °C in a Beckman SW55i rotor for 3 h at 42,000 r.p.m. (167,000_g_). Fractions were assayed by HAV antigen ELISA and HAV RNA-specific qRT–PCR to identify pentamers, procapsids and mature HAV virions.
Immunoprecipitation of HAV
Cells (2 × 106) were lysed in the presence of detergent with CelLytic lysis buffer (Invitrogen), treated with 50 μg ml−1 RNase A (Qiagen) and 25 U ml−1 RNase T1 (Ambion) at 37 °C for 15 min, and incubated overnight with antigen-specific and isotype-matched antibodies at 4 °C. Protein G beads were added and incubated for a further 2 h. Beads were extensively washed with lysis buffer, and captured immunoprecipitates were subjected to RNA extraction for qRT–PCR, or immunoblotting, as above.
Virus isolation from iodixanol gradient fractions
Huh-7.5 cells in 12-well culture plates were inoculated with 10–100 μl of peak iodixanol gradient fractions containing eHAV recovered from human or chimpanzee serum or plasma. Cells were re-fed weekly, and split 1:3 at 2 weeks. RNA was extracted from supernatant culture fluids 8, 23 and 30 days after inoculation and assayed for HAV RNA by qRT–PCR as described above. To confirm viral isolates, RNA was extracted from supernatant culture fluids with the QIAamp Viral RNA Mini Kit (Qiagen), and cDNA was transcribed with random primers (Superscript III First-Strand Synthesis System; Invitrogen). HAV cDNA was amplified by nested RT–PCR with primer sets bracketing the VP1pX-2B junction (nucleotides 2908–3306 in wild-type HM-175) (PrimeSTAR; TaKaRa). Second-round PCR products were isolated from agarose gels (Mini-Elute Gel Extraction Kit; Qiagen) and directly sequenced.
Generation and characterization of HAV VP2 mutants
Mutagenesis of the infectious cDNA clone, pT7-HM175/18f, containing the complete sequence of HM175/18f virus28, was accomplished with the Quick Change II Site-directed Mutagenesis Kit (Agilent). PCR-derived fragments were sequenced to ensure sequence fidelity. Plasmids were linearized and viral RNA was synthesized with the T7 Megascript Kit (Ambion). Synthetic viral RNA (10 μg) was mixed with 5 × 106 Huh-7.5 cells in a 4-mm cuvette and pulsed once at 250 V, 950 μF and 100 Ω in a Gene Pulser Xcell Total electroporation system (Bio-Rad). Cells and supernatants were harvested at subsequent intervals for RNA extraction and qRT–PCR assay.
Electron microscopy
Gradient fractions (2.5 μl each) were spread onto the surface of a glow-discharged 400 mesh Formvar/carbon-coated copper grid and allowed to adsorb for 5 min. The grid was inverted onto a droplet of 1% glutaraldehyde in 0.15 M sodium phosphate pH 7.4 for 1 min, followed by rinsing with deionized water and staining with 3% ammonium molybdate pH 7.0. The grid was air-dried before examination in a LEO EM910 transmission electron microscope (Carl Zeiss SMT, Inc.) operating at 80 kV. Digital images were acquired with a Gatan Orius SC1000 CCD Digital Camera and Digital Micrograph 3.11.0 software (Gatan, Inc.).
siRNA knockdown of protein expression
Huh-7.5 cells were transfected with gene-specific SmartPool siRNAs (Dharmacon; see Supplementary Table 1) with SilentFect (Bio-Rad) transfection reagent. Knockdown efficiency was determined at 48–72 h by qRT–PCR (SYBR Green One-step qRT–PCR Kit; BioRad) measurement of mRNAs, or by immunoblotting as described.
References
- Harrison, S. C. in Fields Virology (eds Knipe, D. M. et al.) Ch. 3 59–98 (Lippincott Williams & Wilkins, 2007)
Google Scholar - Feng, Z. & Lemon, S. M. in The Picornaviruses (eds Ehrenfeld, E., Domingo, E. & Roos, R. P. ) Ch. 25 383–396 (ASM Press, 2010)
Google Scholar - Lemon, S. M. Type A viral hepatitis: new developments in an old disease. N. Engl. J. Med. 313, 1059–1067 (1985)
Article CAS Google Scholar - Martin, A. & Lemon, S. M. in Hepatitis Viruses (ed. Ou, J. ) 23–50 (Kluwer Academic, 2002)
Book Google Scholar - Lanford, R. E. et al. Acute hepatitis A virus infection is associated with a limited type I interferon response and persistence of intrahepatic viral RNA. Proc. Natl Acad. Sci. USA 108, 11223–11228 (2011)
Article ADS CAS Google Scholar - Victor, J. C. et al. Hepatitis A vaccine versus immune globulin for postexposure prophylaxis. N. Engl. J. Med. 357, 1685–1694 (2007)
Article CAS Google Scholar - Bobrie, A., Colombo, M., Raposo, G. & Thery, C. Exosome secretion: molecular mechanisms and roles in immune responses. Traffic 12, 1659–1668 (2011)
Article CAS Google Scholar - Hurley, J. H. The ESCRT complexes. Crit. Rev. Biochem. Mol. Biol. 45, 463–487 (2010)
Article CAS Google Scholar - Jansen, R. W., Newbold, J. E. & Lemon, S. M. Complete nucleotide sequence of a cell culture-adapted variant of hepatitis A virus: comparison with wild-type virus with restricted capacity for in vitro replication. Virology 163, 299–307 (1988)
Article CAS Google Scholar - Counihan, N. A., Daniel, L. M., Chojnacki, J. & Anderson, D. A. Infrared fluorescent immunofocus assay (IR-FIFA) for the quantitation of non-cytopathic and minimally cytopathic viruses. J. Virol. Methods 133, 62–69 (2006)
Article CAS Google Scholar - Ping, L. H. & Lemon, S. M. Antigenic structure of human hepatitis A virus defined by analysis of escape mutants selected against murine monoclonal antibodies. J. Virol. 66, 2208–2216 (1992)
CAS PubMed PubMed Central Google Scholar - Cohen, L., Benichou, D. & Martin, A. Analysis of deletion mutants indicates that the 2A polypeptide of hepatitis A virus participates in virion morphogenesis. J. Virol. 76, 7495–7505 (2002)
Article CAS Google Scholar - Graff, J. et al. Hepatitis A virus capsid protein VP1 has a heterogeneous C terminus. J. Virol. 73, 6015–6023 (1999)
CAS PubMed PubMed Central Google Scholar - Schulman, A. N. et al. Hepatitis A antigen particles in liver, bile, and stool of chimpanzees. J. Infect. Dis. 134, 80–84 (1976)
Article CAS Google Scholar - Asher, L. V. S. et al. Pathogenesis of hepatitis A in orally inoculated owl monkeys (Aotus trivergatus). J. Med. Virol. 47, 260–268 (1995)
Article CAS Google Scholar - Taylor, M. P., Burgon, T. B., Kirkegaard, K. & Jackson, W. T. Role of microtubules in extracellular release of poliovirus. J. Virol. 83, 6599–6609 (2009)
Article CAS Google Scholar - Ren, X. & Hurley, J. H. Proline-rich regions and motifs in trafficking: from ESCRT interaction to viral exploitation. Traffic 12, 1282–1290 (2011)
Article CAS Google Scholar - Chen, B. J. & Lamb, R. A. Mechanisms for enveloped virus budding: can some viruses do without an ESCRT? Virology 372, 221–232 (2008)
Article CAS Google Scholar - Sette, P. et al. The Phe105 loop of Alix Bro1 domain plays a key role in HIV-1 release. Structure 19, 1485–1495 (2011)
Article CAS Google Scholar - Fujii, K., Hurley, J. H. & Freed, E. O. Beyond Tsg101: the role of Alix in ‘ESCRTing’ HIV-1. Nature Rev. Microbiol. 5, 912–916 (2007)
Article CAS Google Scholar - Baietti, M. F. et al. Syndecan-syntenin-ALIX regulates the biogenesis of exosomes. Nature Cell Biol. 14, 677–685 (2012)
Article CAS Google Scholar - Morita, E. et al. ESCRT-III protein requirements for HIV-1 budding. Cell Host Microbe 9, 235–242 (2011)
Article CAS Google Scholar - Shimizu, Y. K. et al. Detection of hepatitis A antigen in human liver. Infect. Immun. 36, 320–324 (1982)
CAS PubMed PubMed Central Google Scholar - Feigelstock, D. et al. The human homolog of HAVcr-1 codes for a hepatitis A virus cellular receptor. J. Virol. 72, 6621–6628 (1998)
CAS PubMed PubMed Central Google Scholar - Kolter, T. & Sandhoff, K. Lysosomal degradation of membrane lipids. FEBS Lett. 584, 1700–1712 (2010)
Article CAS Google Scholar - Balasubramanian, S. et al. TIM family proteins promote the lysosomal degradation of the nuclear receptor NUR77. Sci. Signal. 5, ra90 (2012)
Article Google Scholar - Mallery, D. L. et al. Antibodies mediate intracellular immunity through tripartite motif-containing 21 (TRIM21). Proc. Natl Acad. Sci. USA 107, 19985–19990 (2010)
Article ADS CAS Google Scholar - Zhang, H. C. et al. An infectious cDNA clone of a cytopathic hepatitis A virus: genomic regions associated with rapid replication and cytopathic effect. Virology 212, 686–697 (1995)
Article CAS Google Scholar - Lemon, S. M. et al. Specific immunoglobulin M response to hepatitis A virus determined by solid-phase radioimmunoassay. Infect. Immun. 28, 927–936 (1980)
CAS PubMed PubMed Central Google Scholar - Feigelstock, D., Thompson, P., Mattoo, P. & Kaplan, G. G. Polymorphisms of the hepatitis A virus cellular receptor 1 in African green monkey kidney cells result in antigenic variants that do not react with protective monoclonal antibody 190/4. J. Virol. 72, 6218–6222 (1998)
CAS PubMed PubMed Central Google Scholar - Taylor, K. L. et al. Attenuation phenotype of a cell culture-adapted variant of hepatitis A virus (HM175/p16) in susceptible new world owl monkeys. J. Infect. Dis. 168, 592–601 (1993)
Article CAS Google Scholar - Lemon, S. M. et al. Antigenic and genetic variation in cytopathic hepatitis A virus variants arising during persistent infection: evidence for genetic recombination. J. Virol. 65, 2056–2065 (1991)
CAS PubMed PubMed Central Google Scholar - Lemon, S. M. et al. Transmission of hepatitis A virus among recently captured Panamanian owl monkeys. J. Med. Virol. 10, 25–36 (1982)
Article CAS Google Scholar
Acknowledgements
We thank M. Duncan for helpful discussions; staff of the Michael Hooker Microscopy Facility for assistance with confocal microscopy; G. Kaplan for the gift of 190-4 antibody and GL37 cells; M. Fried for anti-HAV-negative human sera; A. Blikslager for porcine bile; and C. Rice for Huh-7.5 cells. This work was supported in part by a grant from the National Institutes of Health (R01-AI103083) and the University Cancer Research Fund.
Author information
Authors and Affiliations
- Lineberger Comprehensive Cancer Center, The University of North Carolina at Chapel Hill, Chapel Hill, 27599-7292, North Carolina, USA
Zongdi Feng, Lucinda Hensley, Kevin L. McKnight, Fengyu Hu, LiFang Ping & Stanley M. Lemon - Department of Pathology and Laboratory Medicine, The University of North Carolina at Chapel Hill, Chapel Hill, 27599-7292, North Carolina, USA
Victoria Madden - Department of Internal Medicine, Seoul National University Bundang Hospital, Seongnam-si, Gyeonggi-do, 463-707, South Korea,
Sook-Hyang Jeong - and Department of Pediatrics, Center for Vaccines and Immunity, The Research Institute at The Nationwide Children’s Hospital, College of Medicine, The Ohio State University, Columbus, 43205, Ohio, USA
Christopher Walker - Department of Virology and Immunology, Texas Biomedical Research Institute, San Antonio, 78227, Texas, USA
Robert E. Lanford - Division of Infectious Disease, Department of Medicine, The University of North Carolina at Chapel Hill, Chapel Hill, 27599-7292, North Carolina, USA
Stanley M. Lemon - Department of Microbiology and Immunology, The University of North Carolina at Chapel Hill, Chapel Hill, 27599-7292, North Carolina, USA
Stanley M. Lemon
Authors
- Zongdi Feng
You can also search for this author inPubMed Google Scholar - Lucinda Hensley
You can also search for this author inPubMed Google Scholar - Kevin L. McKnight
You can also search for this author inPubMed Google Scholar - Fengyu Hu
You can also search for this author inPubMed Google Scholar - Victoria Madden
You can also search for this author inPubMed Google Scholar - LiFang Ping
You can also search for this author inPubMed Google Scholar - Sook-Hyang Jeong
You can also search for this author inPubMed Google Scholar - Christopher Walker
You can also search for this author inPubMed Google Scholar - Robert E. Lanford
You can also search for this author inPubMed Google Scholar - Stanley M. Lemon
You can also search for this author inPubMed Google Scholar
Contributions
Z.F., C.H., K.L.M., F.H., V.M. and L.-F.P. designed experiments, collected and analysed data. S.-H.J., C.W. and R.L. provided unique human and primate samples for analysis. Z.F. and S.M.L. conceived the study, analysed data and wrote the paper. All authors discussed the results and commented on the manuscript.
Corresponding author
Correspondence toStanley M. Lemon.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
PowerPoint slides
Rights and permissions
About this article
Cite this article
Feng, Z., Hensley, L., McKnight, K. et al. A pathogenic picornavirus acquires an envelope by hijacking cellular membranes.Nature 496, 367–371 (2013). https://doi.org/10.1038/nature12029
- Received: 09 August 2012
- Accepted: 20 February 2013
- Published: 31 March 2013
- Issue Date: 18 April 2013
- DOI: https://doi.org/10.1038/nature12029