Wt1 is required for cardiovascular progenitor cell formation through transcriptional control of Snail and E-cadherin (original) (raw)
- Letter
- Published: 20 December 2009
- Laura A Lettice1,
- Abdelkader Essafi1,
- Juan Antonio Guadix2,
- Joan Slight1,
- Víctor Velecela1,
- Emma Hall1,
- Judith Reichmann1,
- Paul S Devenney1,
- Peter Hohenstein1,
- Naoki Hosen3,
- Robert E Hill1,
- Ramón Muñoz-Chapuli2 &
- …
- Nicholas D Hastie1
Nature Genetics volume 42, pages 89–93 (2010)Cite this article
- 8578 Accesses
- 5 Altmetric
- Metrics details
Abstract
The epicardial epithelial-mesenchymal transition (EMT) is hypothesized to generate cardiovascular progenitor cells that differentiate into various cell types, including coronary smooth muscle and endothelial cells, perivascular and cardiac interstitial fibroblasts and cardiomyocytes. Here we show that an epicardial-specific knockout of the gene encoding Wilms' tumor-1 (Wt1) leads to a reduction in mesenchymal progenitor cells and their derivatives. We show that Wt1 is essential for repression of the epithelial phenotype in epicardial cells and during embryonic stem cell differentiation through direct transcriptional regulation of the genes encoding Snail (Snai1) and E-cadherin (Cdh1), two of the major mediators of EMT. Some mesodermal lineages do not form in _Wt1_-null embryoid bodies, but this effect is rescued by the expression of Snai1, underscoring the importance of EMT in generating these differentiated cells. These new insights into the molecular mechanisms regulating cardiovascular progenitor cells and EMT will shed light on the pathogenesis of heart diseases and may help the development of cell-based therapies.
Similar content being viewed by others
Main
In the developing heart, very little is known about the molecular and cellular mechanisms controlling epicardial EMT and the formation of cardiovascular progenitor cells1. Wt1 encodes a zinc-finger protein with a crucial role in the normal development of several organs, such as kidney, gonads, spleen and heart2. Coronary vascular defects in _Wt1_-mutant mice are believed to arise through defective EMT3. However, until now it has not been possible to determine whether Wt1 in the epicardium is directly involved in EMT, or whether EMT is essential for the formation of cardiovascular progenitor cells.
To investigate the role of Wt1 in the epicardium, we generated Wt1 conditional knockout mice (Supplementary Fig. 1) that were crossed with Wt1 knockin mice expressing green fluorescent protein (GFP) reporter (_Wt1_GFP/+; ref. 4) and mice transgenic for Gata5-Cre5 (see Online Methods for details of breeding). The resulting Gata5-Cre; _Wt1_loxP/GFP (referred to here as Cre+) mice died between embryonic day 16.5 (E16.5) and E18.5 as a result of cardiovascular failure. Cre+ E16.5 embryos showed edema and accumulation of blood in the systemic veins (Fig. 1a,b).
Figure 1: Heart defects in epicardial-specific _Wt1_-mutant embryos.
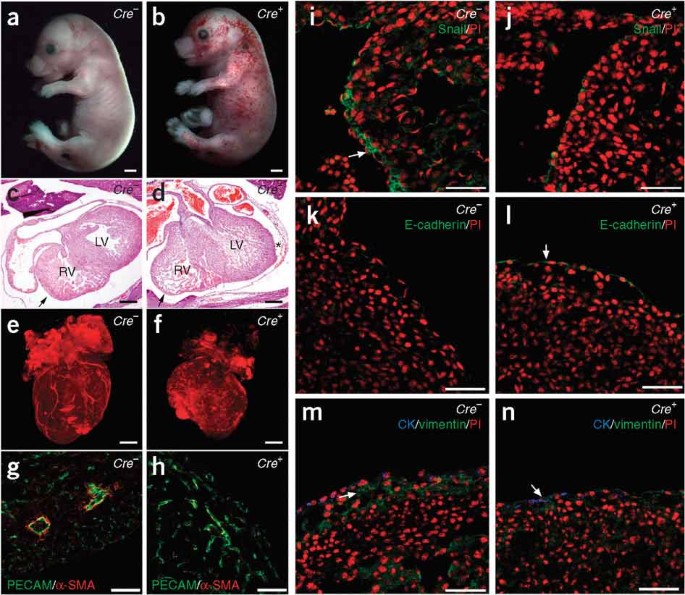
(a,b) Gata5-Cre; _Wt1_loxP/GFP (Cre+) embryos (b) showed edema and accumulation of blood in the systemic veins. A littermate control (Cre −) is shown in a. Scale bars represent 100 μm. (c,d) Hematoxylin- and eosin-stained sections of Cre − and Cre+ E16.5 embryos. The right ventricles (RV) of some of the mutant embryos (d) had thinner walls (arrows) compared to control embryos (c), whereas the left ventricles (LV) were apparently normal. Mutant embryos showed pericardial hemorrhage (d, asterisk). Scale bars represent 50 μm. (e,f) OPT images of control and mutant hearts at E16.5. Scale bars represent 50 μm. (g,h) Immunofluorescence staining for the indicated blood vessel markers. Only control embryos (g) showed arteries with a well-differentiated smooth muscle layer. (i–n) EMT markers were analyzed with antibodies to Snail (i,j), E-cadherin (k,l) and vimentin and cytokeratin (CK; m,n). Abnormal E-cadherin (l) and decreased Snail (j) and vimentin (n) expression was observed in epicardial cells from Cre+ embryos. PI, propidium iodide. Arrows point to expression of the indicated proteins in control and mutant epicardial cells. Scale bars represent 50 μm.
We confirmed efficient deletion of Wt1 in epicardial cells by immunohistochemical analysis of heart sections and real-time PCR analysis of FACS-sorted GFP+ epicardial cells isolated from Cre+ mice (Supplementary Fig. 2a–c). The gross morphology of Cre+ embryos was normal, but the right ventricles of some mutant embryos had thinner free walls compared to control embryos (Fig. 1c,d, arrows), whereas the left ventricles were apparently normal. Mutant embryos also showed pericardial hemorrhaging (Fig. 1d, asterisk). We also confirmed embryonic expression of the Gata5-Cre transgene in the heart. At E10.5, we detected cells expressing Cre in the epicardium; at E12.5, _Gata5-Cre_–derived cells were abundant within the heart5 (Supplementary Fig. 3 and data not shown). Optical projection tomography (OPT) revealed that the coronary arteries did not form in Cre+ mice (Fig. 1e,f and Supplementary Movies 1 and 2). We confirmed this result by staining for the adhesion molecule PECAM-1 (CD31) and α-smooth muscle actin (α-SMA), markers for endothelial and smooth muscle cells, respectively (Fig. 1g,h).
GFP+ epicardial cells covered the surface of the myocardium in Cre+ mice, confirming the integrity of this structure in the mutant mice (Supplementary Fig. 4b). Because epicardial EMT has a very important role in the formation of coronary vascular precursor cells, we studied the expression patterns of the major markers of EMT in the Wt1 mutant epicardium. Expression of Snail, a key activator of EMT, was reduced in the epicardium of mutant hearts compared to controls (Fig. 1i,j). Conversely, the epithelial markers E-cadherin and cytokeratin were upregulated (Fig. 1k–n). Vimentin expression was also downregulated in mutant hearts (Fig. 1m,n). We also compared the levels of Snai2 (also known as Slug) in Cre+ and Cre − FACS-sorted epicardial cells by real-time PCR and confirmed that, similar to Snai1, Snai2 levels were reduced by 70% in Cre+ compared to Cre − cells.
To determine whether Wt1 has a direct and cell-autonomous role in epicardial EMT, we generated tamoxifen-inducible _Wt1_-knockout immortalized epicardial cells (Cre+ CoMEECs; Fig. 2; see Online Methods). CoMEECs had typical cobblestone morphology (Fig. 2c), highlighted by ZO-1 staining at the cellular junctions (Fig. 2e), and showed robust Wt1 and GFP expression (Fig. 2d,f). Loss of Wt1 after tamoxifen treatment led to a robust increase in E-cadherin expression in a dose-dependent manner (Fig. 2g), which was associated with downregulation of N-cadherin and α-SMA (Fig. 2g). RT-PCR analysis also revealed a marked downregulation of Snai1 after Wt1 deletion (Fig. 2h). Treatment of the Cre+ CoMEECs with tamoxifen led not only to changes in the EMT marker pattern, but also to reduced cell migration (Fig. 2i). We did not observe any difference in the markers that analyzed or in the migration properties of _Cre_− CoMEECs after tamoxifen treatment (Supplementary Fig. 5a,b).
Figure 2: Wt1 expression is necessary to maintain a mesenchymal phenotype in immortalized epicardial cells.
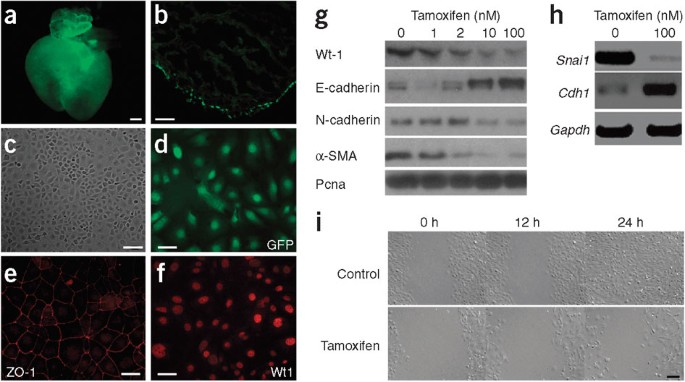
(a) Heart of _Wt1_GFP/+ knockin mouse. Scale bar represents 50 μm. (b) Direct GFP expression analysis in heart cryosection of _Wt1_GFP/+ knockin embryos showed GFP expression in epicardial cells. Scale bar represents 50 μm. (c) Phase-contrast micrograph of CoMEECs. Scale bar represents 100 μm. (d–f) GFP, ZO-1 and Wt1 expression in CoMEECs. Scale bars represent 50 μm. CoMEECs showed a cobblestone monolayer typical of epicardial cells (c,e). (g) Protein blot analysis of Cre+ tamoxifen-inducible CoMEECs cultured in the presence of various concentrations of tamoxifen. Pcna, proliferating cell nuclear antigen. (h) RT-PCR analyses of Snai1 and Cdh1 in Cre+ CoMEECs in the presence of tamoxifen. (i) The migratory behavior of Cre+ CoMEECs in the presence of tamoxifen was analyzed in an in vitro wound model. Scale bar represents 100 μm.
We next examined whether Snai1, one of the master regulators of EMT6, is directly regulated by Wt1. We identified three conserved potential Wt1 binding sites in the Snai1 genomic sequence (Fig. 3a). The −KTS Wt1 isoform (Lys-Thr-Ser tripeptide deleted from exon 9) functions as a transcription factor2 and was able to activate the Snai1 fragment containing the promoter binding site in a dose-dependent manner (Fig. 3b). The other fragments were insensitive to Wt1 activation (data not shown). The transcriptional activation was abolished when we mutated the functional binding site (Fig. 3c). Chromatin immunoprecipitation (ChIP) assays showed in vivo binding of Wt1 to the endogenous Snai1 promoter but not to the intronic and 3′ UTR regions in epicardial cells (Fig. 3d). The endogenous Snai1 promoter of epicardial cells was enriched in acetylated Lys9 and trimethylated Lys4 of histone H3 (H3K9ac and H3K4me3, respectively), but depleted in trimethylated Lys27 of histone H3 (H3K27me3), which is compatible with the activated state of Snai1 in these cells (Fig. 3d).
Figure 3: Wt1 is an activator of Snai1 and a repressor of Cdh1 in epicardial cells.
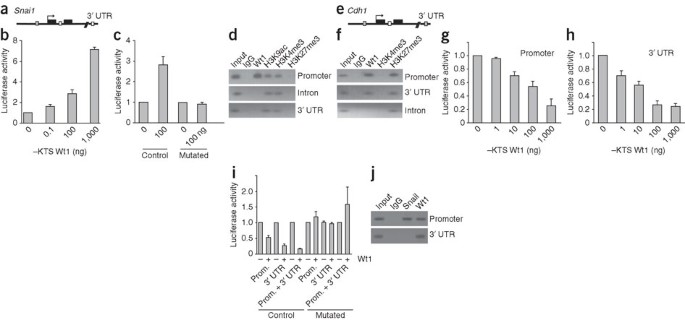
(a) Putative conserved Wt1 binding sites in the Snai1 locus. ▪, functional binding site; □, putative but nonfunctional binding site; ▪, exons. (b) Luciferase activity of reporter construct carrying mouse Snai1 promoter in epicardial cells in the presence of indicated amounts of −KTS Wt1 expression vector. (c) Luciferase activity of wild-type (control) or mutated Snai1 promoters in the presence of −KTS Wt1 isoform. (d) Cell extracts from epicardial cells were subjected to ChIP using antibodies to Wt1, H3K9ac, H3K4me3 and H3K27me3, or an irrelevant antibody (IgG). The input was used as a positive control for PCR of the Snai1 (d) promoter, intronic regions and 3′ UTR. (e) Putative conserved Wt1 binding sites in the Cdh1 locus. (f) ChIP and PCR analysis of Cdh1, as in d. (g,h) Luciferase activity of constructs carrying DNA fragments identified by the Cdh1 ChIP assay in epicardial cells, together with various concentrations of −KTS Wt1 isoform. (i) Luciferase activity of control or mutated Cdh1 constructs in the presence of 100 ng of −KTS Wt1 isoform. (j) ChIP assays for Snail and Wt1 at the endogenous Cdh1 promoter in epicardial cells. All graphs show mean values ± s.e.m. from three independent experiments.
Wt1 has been shown to transcriptionally activate the Cdh1 promoter in NIH3T3 cells7. We examined whether Wt1 directly represses Cdh1 in epicardial cells (Fig. 3). ChIP with primers flanking the previously identified Wt1 binding sequence in the Cdh1 promoter confirmed that Wt1 binds directly to the endogenous Cdh1 promoter in epicardial cells (Fig. 3f). Consistent with a repressed state, the Cdh1 promoter was depleted in H3K4me3 and enriched for H3K27me3 (Fig. 3f). An extended analysis of the Cdh1 genomic sequence revealed two new conserved potential Wt1 binding sites in the intron and 3′ UTR (Fig. 3e). ChIP with primers flanking these regions showed in vivo binding of Wt1 in epicardial cells to the 3′ UTR but not to the intronic region (Fig. 3f). In epicardial cells, the −KTS Wt1 isoform had a repressive effect on the Cdh1 promoter fragment containing the Wt1 binding site shown previously to be activated in NIH3T3 cells (Fig. 3g and data not shown) and on the 3′ UTR construct (Fig. 3h). The transcriptional repression was abolished when the functional binding sites were mutated (Fig. 3i). The +KTS isoform was able to regulate the Snai1 and Cdh1 constructs, but to a much lesser degree (data not shown).
Our finding that Wt1 can directly repress Cdh1 raised the question of whether Snail is also a repressor of Cdh1 in this system. ChIP analysis showed that Snail does interact with the Cdh1 promoter in epicardial cells (Fig. 3j). Furthermore, knockdown of Snai1 in epicardial cells with a short hairpin RNA targeting Snai1 led to an increase in Cdh1 promoter activity (Supplementary Fig. 6). These results led us to propose a model in which Wt1 promotes downregulation of Cdh1 directly and indirectly by increasing levels of Snail.
To determine whether Wt1 regulates EMT and cardiovascular differentiation in another cellular system, we investigated Wt1 in embryonic stem cells8. Fluorescence analysis (_Wt1_GFP/+ knockin embryonic stem cells) and protein blotting showed that Wt1 was almost undetectable in undifferentiated embryonic stem cells (day 0); Wt1 expression was first detectable on day 3 and peaked between days 9 and 11 during embryoid body differentiation (a proliferative period for undifferentiated mesenchymal cells; Fig. 4a,b). Wt1 expression declined thereafter, as various terminally differentiated cell types started appearing (data not shown).
Figure 4: Wt1 is required for EMT in embryonic stem cells.
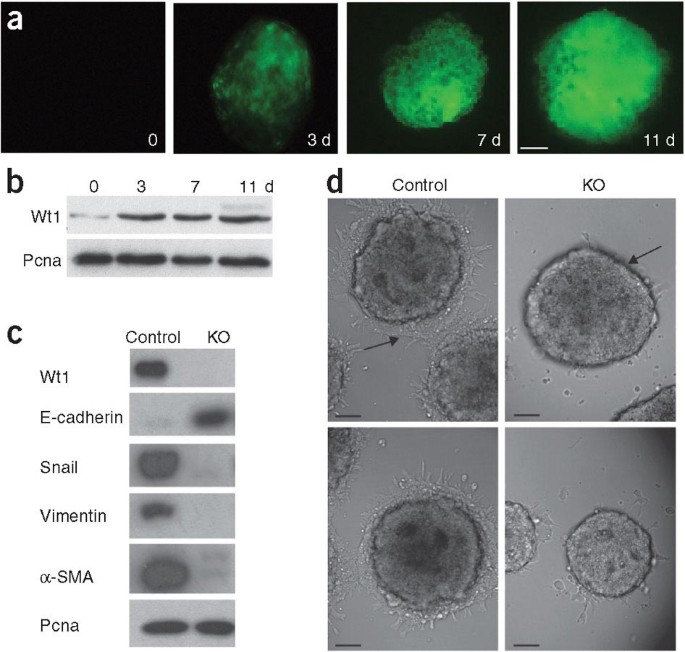
(a) _Wt1_GFP/+ expression during embryoid body differentiation of _Wt1_GFP/+ knockin embryonic stem cells. Scale bar represents 50 μm. (b) Protein blot analysis of endogenous Wt1 protein in embryoid bodies. (c) Protein blot analysis of epithelial and mesenchymal protein markers in wild-type (control) and _Wt1_-knockout (KO) embryoid bodies. (d) Phase-contrast microscopy of control and _Wt1_-knockout embryoid body migrating cells. Arrows in d indicate presence of migratory cells in control embryoid bodies and absence from knockout embryoid bodies. Scale bars represent 50 μm.
To determine whether Wt1 is required for EMT in embryonic stem cells, we examined the expression of the principal epithelial and mesenchymal markers in wild type (control) and _Wt1_-knockout embryoid bodies (Fig. 4c). Control embryoid bodies expressed high levels of vimentin, α-SMA and Snail, but not E-cadherin, indicating that these cells had undergone EMT. By contrast, _Wt1_-knockout embryoid bodies maintained high levels of E-cadherin and did not induce the mesenchymal markers, reflecting their inability to undergo EMT (Fig. 4c). Additionally, cells derived from control embryoid bodies had the capacity to migrate, whereas cells derived from _Wt1_-knockout embryoid bodies showed a clear impairment in their migration properties (Fig. 4d).
Because _Wt1_-knockout embryoid bodies were unable to undergo EMT, we examined whether Wt1 is required for the generation of mesodermal lineages from embryonic stem cells. We analyzed the expression patterns of the primitive streak genes encoding Brachyury (T) and Goosecoid (Gsc). Both of these genes were normally expressed in _Wt1_-knockout embryoid bodies (Fig. 5a). The more mature mesodermal markers, including hematopoietic (Kdr and Hbb-b1), endothelial (Kdr, Tek and Cdh5) and cardiac (Kdr, Nkx2-5, Hand1 and Isl1) markers clearly showed reduced expression in _Wt1_-knockout embryoid bodies compared to control embryoid bodies (Fig. 5a). The ectodermal (Sox2 and Nes) and endodermal (Fgf5, Afp and Hnf4a) markers showed no differences (Fig. 5b).
Figure 5: Wt1 is required for the formation of some mesodermal lineages in embryoid body differentiation.

(a,b) RT-PCR analysis of expression of mesodermal (a) and endodermal and ectodermal (b) markers in wild-type (control) and _Wt1_-knockout (KO) embryoid bodies. (c) RT-PCR analysis of expression of Snai1 and Cdh1 in _Wt1_-knockout _Snai1_-expressing clones (Snai1 c1 and Snai2 c2) and _Wt1_-knockout GFP-expressing clones (c1 and c2). (d) RT-PCR analysis of expression of mesodermal markers in Snai1 and GFP clones. (e) Numbers of beating cardiomyocytes in control and _Wt1_-knockout clones. Graph shows mean values ± s.e.m. from three independent experiments.
To examine whether the EMT defect in _Wt1_-knockout embryoid bodies is the principal cause of impairment in the formation of mesoderm precursors, we expressed Snai1 in _Wt1_-knockout embryonic stem cells. _Snai1_-positive clones expressed lower levels of Cdh1 and higher levels of Snai2 and Twist1 than did control GFP-expressing clones (Fig. 5c,d), suggesting a rescue of the EMT process. Expression of the endothelial and cardiac mesodermal markers was also rescued after Snai1 expression (Fig. 5d). To confirm the presence of mature functional mesoderm, we analyzed the formation of beating cardiomyocytes. These were observed in control embryoid bodies and _Snai1_-rescued clones but were absent from the control GFP clones (Fig. 5e).
Our findings support a crucial role for Wt1 in the generation of mesenchymal cardiovascular progenitor cells in the epicardium and during embryonic stem cell differentiation, through direct regulation of the Snail transcription factor and E-cadherin. A model built on our principal findings is shown in Supplementary Figure 7. Wt1 expression is reactivated in hearts after ischemia9, so it would be interesting to determine whether Wt1 is required for the regeneration and repair of damaged hearts through the pathways described here.
The embryonic stem cell–embryoid body model has been particularly useful in elucidating the molecular events involved in the specification of cell lineage differentiation10,11. Our data suggest that in _Wt1_-null embryonic stem cells, disruptions during EMT cause defects in the differentiation of some mesodermal lineages. This conclusion is based on the finding that Snail, the master regulator of EMT, rescued the _Wt1_-null phenotype. The role of Wt1 in coronary blood vessel cell formation has been shown previously3,12 and in the present study. However, our data provide the first evidence that Wt1 is also required for the formation of cardiomyocytes, through the regulation of EMT.
It is unclear whether the role of Wt1 in embryoid bodies recapitulates the transitions in the epicardium that are required for the formation of cardiovascular progenitor cells, or whether it reflects an earlier function of the Wt1 gene in mesoderm formation. Notably, a recently developed data set that samples the mouse transcriptome from gastrulation through organogenesis13 revealed a massive increase in Wt1 expression during gastrulation, implicating this gene in the extensive morphological changes that occur within the embryo throughout this stage. Despite early embryonic expression of Wt1, the deficient embryo shows no overt phenotype just after gastrulation, although the formation of several mesodermal tissues is impaired. More extensive analysis will be required to evaluate whether Wt1 has an earlier role in mesoderm formation.
We have compiled evidence that Wt1 regulates the reverse process—mesenchymal-epithelial transition—in the kidney, and we have begun to dissect the molecular mechanisms involved (A.E., P.H., O.M.M.-E. and N.D.H., unpublished data). Together, all of our findings suggest that Wt1 has a major role regulating the balance between two fundamental cell states in several mesodermal tissues. In this context, it is interesting to consider the pattern of expression of Wt1 during development. The highest sites of expression are in the mesothelium, podocytes of the kidneys and Sertoli cells of the testis. All of these cells have dual epithelial and mesenchymal properties. The other major sites of expression are mesenchymal cell populations whose fates have not been determined. Further studies will be necessary to elucidate whether these cells are progenitors for particular cell types in the developing fetus.
Our findings may have relevance far beyond the cardiovascular field. For example, Wt1 and Snail are both expressed at high levels in a range of adult cancers. Expression of both genes is correlated with poor prognosis in breast cancer14,15. A recent study showed that introduction of Snai1 into mammary epithelial cells converts them into cells with properties of mammary cancer stem cells16. We speculate that Wt1 regulates Snai1 and EMT in various contexts and is involved in establishing cancer stem cells and tumor cells with invasive properties.
Methods
Generation of _loxP_-flanked Wt1 targeting construct and _Wt1_loxP/loxP mice.
A genomic fragment 5.8 kb in length surrounding exon 1 of the mouse Wt1 locus (representing nucleotides 104,963,917 to 104,969,768 of chromosome 2 in Ensembl release 50) was subcloned into PolyIII. An oligonucleotide containing a loxP site marked with a PstI restriction enzyme recognition site was cloned in the unique AatII restriction site at position 2270 within this fragment. A neomycin cassette, flanked by FLP recognition target sites and carrying a single loxP site (from vector p451), was targeted by bacterial recombination17 to position 3955. This produced a targeting vector with two external homology arms of 2.2 kb and 1.9 kb and an internal homology arm of 1.7 kb (the completed targeting vector is shown in Supplementary Fig. 1a–e). The insert was removed from the vector backbone by digestion with NotI and purified using an Elutrap (Schleicher & Schuell). After homologous recombination in E14Tg2AIV embryonic stem cells (a gift from A. Smith) using standard techniques18, 307 clones were screened by Southern blotting of PstI-digested genomic DNA using a 3′ probe external to the targeting vector. Of the nine clones correctly targeted at the 3′ end, subsequent screening using an internal probe showed that three correctly carried the lone 5′ loxP site (Supplementary Fig. 1c). Two of these lines were electroporated with a vector expressing FLPe recombinase to remove the neomycin cassette (Supplementary Fig. 1d) and subsequently with a vector expressing Cre recombinase to verify accurate excision of exon 1 (Supplementary Fig. 1e). Clones were reanalyzed by Southern blotting (Supplementary Fig. 1f), karyotyped and injected into mouse blastocysts. Germline transmission of the mutant allele was initially shown by Southern blotting, but subsequent genotyping was done by PCR using the primers _Wt1_loxP/loxP (Supplementary Table 1).
Generation of epicardial-specific _Wt1_-mutant mice.
Epicardial _Wt1_-mutant mice (Gata5-Cre; _Wt1_loxP/GFP) were generated by cross-breeding Wt1 _l_oxP/loxP mice with mice heterozygous for _Wt1_GFP/+ knockin4 and Gata5-Cre5. Transgenic mice were genotyped by PCR using the primers Gata5cre, Wt1-GFP and Wt1loxP/loxP (Supplementary Table 1). Mouse studies were conducted under guidance issued by the UK Medical Research Council and the UK Home Office.
Histology and immunohistochemistry.
For histological analysis, 6-μm-thick wax sections were stained with hematoxylin and eosin. For immunostaining, antigens were retrieved by boiling samples in a pressure cooker for 4 min in TEG buffer (10 mM Tris and 0.5 mM EGTA, pH 9.0). Slides were then incubated in 50 mM NH4Cl in PBS (pH 7.4) for 30 min and blocked in 1% BSA, 0.2% gelatin and 0.05% saponin three times for 10 min each. Samples were incubated overnight at 4 °C with antibodies to Wt1 (1:800; C19, Santa Cruz Biotechnology) or GFP (1:800; Abcam) diluted with 0.1% BSA and 0.3% Triton X-100 in PBS (pH 7.4), followed by washing with 0.1% BSA, 0.2% gelatin and 0.05% saponin.
Antibodies to the following molecules were used for standard immunohistochemistry procedures: α-SMA (1:100; clone 1A4, Sigma), E-cadherin (1:800; BD Bioscience), cytokeratin (1:200; Dako), vimentin (1:200; Dako), PECAM-1 (1:50; clone Mec13.3, BD PharMingen) and Snail19 (1:25; gift from A. García de Herreros). Samples were incubated with the secondary antibodies and counterstained with Vectashield with DAPI or propidium iodide (Vector Laboratories).
Real-time PCR of FACS-sorted GFP+ epicardial cells.
The heart ventricles of Cre+; _Wt1_loxP/GFP and Cre −; _Wt1_loxP/GFP mice were trypsinized for 15 min at 37 °C. RNA isolated from FACS-sorted GFP+ epicardial cells was reverse-transcribed using a First Strand cDNA kit for RT-PCR (Roche). Analysis of gene expression was carried out by TaqMan real-time PCR. The levels of Wt1, Snai1 and Snai2 were normalized to that of the housekeeping gene Gapdh (Roche). Relative values are shown as ratios of gene to Gapdh.
Generation of CoMEECs.
To generate _Wt1_loxP/GFP immortalized epicardial cell lines, _Wt1_GFP/+ mice were crossed with 'Immorto' mice carrying the Tg(H2-K1-tsA58) transgene (Jackson Laboratory) carrying a temperature-sensitive simian virus 40 large T antigen20. Wt1 GFP/+; Tg(H2-K1-tsA58)+/− mice were mated with _Wt1_loxP/loxP mice. Hearts from E11.5 _Wt1_loxP/GFP; Tg(H2-K1-tsA58)+/− mice were placed in 24-well gelatinized dishes. After 24 h, the hearts were removed, and the epicardial monolayer of cells attached to the gelatin-coated surface was grown until confluent. Cells were propagated at 33 °C in DMEM with 10% heat-inactivated FCS and 20 ng/ml mouse gamma-interferon (PeproTech). We generated tamoxifen-inducible _Wt1_-mutant cells by transfecting _Wt1_loxP/GFP immortalized epicardial cells with a CAGGs-CreERT2-IRES-puroR expression construct (gift from L. Grotewold). Stable clones were generated after selection with 5 μg/ml puromycin.
Migration assays.
Immortalized epicardial cells that had been cultured for 6 d in the presence or absence of 100 nM of tamoxifen were seeded in six-well culture dishes at a density of 0.5 × 106 cells per well. A wound was incised 24 h later in the central area of the confluent culture and carefully washed to remove detached cells, and fresh medium was added before a further 24 h of incubation. The images were captured using a live-cell imaging system (Zeiss Axiovert 200 fluorescence microscope).
Embryonic stem cell culture and embryoid body differentiation.
_Wt1_-knockout21, _Snai1_-rescued, _Wt1_GFP/+ knockin and wild-type E14tg2aIV embryonic stem cells were used in this study. Briefly, undifferentiated embryonic stem cells were cultured on gelatin-coated dishes in BHK-21 medium (Glasgow minimum essential medium, GIBCO) supplemented with 10% FCS, 2 mM L-glutamine, 1 mM sodium pyruvate (Sigma), 0.1 mM nonessential amino acids (Sigma), 0.1 mM mercaptoethanol (Sigma) and leukemia inhibitory factor. Embryoid bodies were formed by culturing embryonic stem cells (1 × 106 per 9-cm bacterial dish) for the indicated number of days on nonadherent bacterial plates in medium without leukemia inhibitory factor.
For migration analysis, day 11 embryoid bodies were transferred to gelatinized plates in DMEM medium. After 6 h of migration, representative images were recorded from each type of embryoid body.
To analyze the differentiation of cardiomyocytes, embryoid bodies were transferred into gelatinized plates on day 7 of differentiation. Embryoid bodies were monitored for beating from days 1 to 7. Spontaneously contracting embryoid bodies were counted by visual inspection under a light microscope.
Generation of Snai1 transfectant cells.
The constructs expressing Myc-tagged Snail and GFP were made by cloning the coding regions of Snai1 and GFP into pcDNA6.2-v5-DEST (Invitrogen). Snai1 and GFP vectors (100 μg each) were electroporated into 1 × 107 _Wt1_-knockout embryonic stem cells. Stable clones were generated after selection in 10 μg/ml blasticidin. Two independent clones (c1 and c2) from each construct were analyzed during the rescue experiments.
Protein blot and RT-PCR analysis.
For protein blotting, lysates from day 7–11 embryoid bodies or epicardial cells treated with tamoxifen for 6 d were analyzed using antibodies to E-cadherin (1:2,000; Transduction Laboratories), Wt1 (1:2,000; C-19, Santa Cruz Biotechnology), vimentin (1:1,000; Abcam), α-SMA (1:5,000; Sigma), proliferating cell nuclear antigen (1:10,000; Santa Cruz Biotechnology) and Snail (1:100)19.
For gene expression analysis, total RNA was isolated from immortalized epicardial cells treated with tamoxifen for 6 d, and from day 3, 7 and 11 embryoid bodies, using an RNA purification kit (Invitrogen). Total RNA was reverse-transcribed as described above. Primers used are listed in Supplementary Table 1.
Luciferase assays and ChIP.
ChIP-positive fragments were cloned into a pGL3 plasmid (Supplementary Table 1). These reporter constructs (0.1 μg) were transfected into immortalized epicardial cells in the presence of the indicated amounts of expression construct encoding the −KTS Wt1 isoform22. The total amount of transfected DNA was normalized to that of LacZ–expressing plasmid. Renilla luciferase–expressing plasmid was also cotransfected as a control for efficiency. Twenty-four hours after transfection, firefly and Renilla luciferase activities were measured using the Dual Luciferase Reporter Assay System (Promega). To mutate the putative Wt1 binding sequences, a QuikChange Site-Directed Mutagenesis Kit (Stratagene) was used.
ChIP assays were conducted on immortalized epicardial cells. Immunoprecipitation of the cross-linked chromatin was carried out with rabbit polyclonal antibodies to Wt1 (C-19; Santa Cruz Biotechnology) and Snail (Abcam). Rabbit serum served as a negative control, and a 1:10 dilution of the input sample served as a positive control. The modification status of histones at the Snai1 and Cdh1 promoters was checked. Immunoprecipitation of the cross-linked chromatin was carried out with antibodies to H3K9ac (Upstate Biotechnology), H3K4me3 (Abcam) and H3K27me3 (Upstate Biotechnology). The amplified DNA was separated on 2% agarose gel and visualized with ethidium bromide. Primers used are listed in Supplementary Table 1.
Snai1 knockdown experiments.
Immortalized epicardial cells were cultured in the presence of _Snai1_-targeting and control short hairpin RNA lentiviral particles (sc-38399-V and sc108080, respectively; Santa Cruz Biotechnology). After 3 d of culture, cells were transfected with the Cdh1 promoter. Renilla luciferase plasmid was cotransfected as a control for efficiency. Firefly and Renilla luciferase activities were measured as described above.
OPT scanning.
Embryos were fixed in 4% paraformaldehyde. The samples were then embedded and imaged as previously described23.
References
- Wessels, A. & Perez-Pomares, J.M. The epicardium and epicardially derived cells (EPDCs) as cardiac stem cells. Anat. Rec. A Discov. Mol. Cell. Evol. Biol. 276, 43–57 (2004).
Article CAS Google Scholar - Hohenstein, P. & Hastie, N.D. The many facets of the Wilms' tumour gene, WT1. Hum. Mol. Genet. 15, R196–R201 (2006).
Article CAS Google Scholar - Moore, A.W., McInnes, L., Kreidberg, J., Hastie, N.D. & Schedl, A. YAC complementation shows a requirement for Wt1 in the development of epicardium, adrenal gland and throughout nephrogenesis. Development 126, 1845–1857 (1999).
CAS Google Scholar - Hosen, N. et al. The Wilms' tumor gene WT1-GFP knock-in mouse reveals the dynamic regulation of WT1 expression in normal and leukemic hematopoiesis. Leukemia 21, 1783–1791 (2007).
Article CAS Google Scholar - Merki, E. et al. Epicardial retinoid X receptor alpha is required for myocardial growth and coronary artery formation. Proc. Natl. Acad. Sci. USA 102, 18455–18460 (2005).
Article CAS Google Scholar - Nieto, M.A. The snail superfamily of zinc-finger transcription factors. Nat. Rev. Mol. Cell Biol. 3, 155–166 (2002).
Article CAS Google Scholar - Hosono, S. et al. E-cadherin is a WT1 target gene. J. Biol. Chem. 275, 10943–10953 (2000).
Article CAS Google Scholar - Spencer, H.L. et al. E-cadherin inhibits cell surface localization of the pro-migratory 5T4 oncofetal antigen in mouse embryonic stem cells. Mol. Biol. Cell 18, 2838–2851 (2007).
Article CAS Google Scholar - Wagner, K.D. et al. The Wilms' tumor suppressor Wt1 is expressed in the coronary vasculature after myocardial infarction. FASEB J. 16, 1117–1119 (2002).
Article CAS Google Scholar - Keller, G. Embryonic stem cell differentiation: emergence of a new era in biology and medicine. Genes Dev. 19, 1129–1155 (2005).
Article CAS Google Scholar - Nishikawa, S., Jakt, L.M. & Era, T. Embryonic stem-cell culture as a tool for developmental cell biology. Nat. Rev. Mol. Cell Biol. 8, 502–507 (2007).
Article CAS Google Scholar - Wagner, N. et al. Coronary vessel development requires activation of the TrkB neurotrophin receptor by the Wilms' tumor transcription factor Wt1. Genes Dev. 19, 2631–2642 (2005).
Article CAS Google Scholar - Mitiku, N. & Baker, J.C. Genomic analysis of gastrulation and organogenesis in the mouse. Dev. Cell 13, 897–907 (2007).
Article CAS Google Scholar - Miyoshi, Y. et al. High expression of Wilms' tumor suppressor gene predicts poor prognosis in breast cancer patients. Clin. Cancer Res. 8, 1167–1171 (2002).
CAS PubMed Google Scholar - Blanco, M.J. et al. Correlation of Snail expression with histological grade and lymph node status in breast carcinomas. Oncogene 21, 3241–3246 (2002).
CAS Google Scholar - Mani, S.A. et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 133, 704–715 (2008).
Article CAS Google Scholar - Liu, P., Jenkins, N.A. & Copeland, N.G. A highly efficient recombineering-based method for generating conditional knockout mutations. Genome Res. 13, 476–484 (2003).
Article CAS Google Scholar - Lettice, L.A. et al. The mouse bagpipe gene controls development of axial skeleton, skull, and spleen. Proc. Natl. Acad. Sci. USA 96, 9695–9700 (1999).
Article CAS Google Scholar - Francí, C. et al. Expression of Snail protein in tumor-stroma interface. Oncogene 25, 5134–5144 (2006).
Article Google Scholar - Jat, P.S. et al. Direct derivation of conditionally immortal cell lines from an H-2Kb-tsA58 transgenic mouse. Proc. Natl. Acad. Sci. USA 88, 5096–5100 (1991).
Article CAS Google Scholar - Spraggon, L. et al. hnRNP-U directly interacts with WT1 and modulates WT1 transcriptional activation. Oncogene 26, 1484–1491 (2007).
Article CAS Google Scholar - Niksic, M., Slight, J., Sanford, J.R., Caceres, J.F. & Hastie, N.D. The Wilms' tumour protein (WT1) shuttles between nucleus and cytoplasm and is present in functional polysomes. Hum. Mol. Genet. 13, 463–471 (2004).
Article CAS Google Scholar - Sharpe, J. et al. Optical projection tomography as a tool for 3D microscopy and gene expression studies. Science 296, 541–545 (2002).
Article CAS Google Scholar
Acknowledgements
We thank A. Cano for the Snai1 promoter (Universidad Autónoma de Madrid, Spain), A. García de Herreros for the antibody to Snail (IMIM-Hospital del Mar, Barcelona, Spain), P. Ruiz-Lozano for the Gata5-Cre transgenic mice (Burnham Institute for Medical Research, La Jolla, California), A. Smith for E14Tg2AIV embryonic stem cells (Wellcome Trust Centre for Stem Cell Research, Cambridge, UK), L. Grotewold for CreERT2-puroR expression construct (University of Edinburgh, UK), H. Morrison for the OPT analysis, F. Kilanowski for assistance with gene targeting, A. Thornburn for help maintaining mouse colonies, C. Nicol for assistance with graphics (Supplementary Fig. 7) and all members of N.D.H.'s laboratory for helpful discussions and comments. This work was supported by the UK Medical Research Council (core grant to N.D.H.) and the Spanish Ministry of Science (grant BFU08-02384 to R.M.-C.). A.E. was supported by EuReGene, an FP6 grant from the European Union (05085). O.M.M.-E. was supported by an EU Marie Curie (FP6) personal fellowship.
Author information
Authors and Affiliations
- MRC Human Genetics Unit and the Institute for Genetics and Molecular Medicine, Edinburgh, UK
Ofelia M Martínez-Estrada, Laura A Lettice, Abdelkader Essafi, Joan Slight, Víctor Velecela, Emma Hall, Judith Reichmann, Paul S Devenney, Peter Hohenstein, Robert E Hill & Nicholas D Hastie - Department of Animal Biology, Faculty of Science, University of Málaga, Málaga, Spain
Juan Antonio Guadix & Ramón Muñoz-Chapuli - Department of Cancer Stem Cell Biology, Osaka University Graduate School of Medicine, Osaka, Japan
Naoki Hosen
Authors
- Ofelia M Martínez-Estrada
You can also search for this author inPubMed Google Scholar - Laura A Lettice
You can also search for this author inPubMed Google Scholar - Abdelkader Essafi
You can also search for this author inPubMed Google Scholar - Juan Antonio Guadix
You can also search for this author inPubMed Google Scholar - Joan Slight
You can also search for this author inPubMed Google Scholar - Víctor Velecela
You can also search for this author inPubMed Google Scholar - Emma Hall
You can also search for this author inPubMed Google Scholar - Judith Reichmann
You can also search for this author inPubMed Google Scholar - Paul S Devenney
You can also search for this author inPubMed Google Scholar - Peter Hohenstein
You can also search for this author inPubMed Google Scholar - Naoki Hosen
You can also search for this author inPubMed Google Scholar - Robert E Hill
You can also search for this author inPubMed Google Scholar - Ramón Muñoz-Chapuli
You can also search for this author inPubMed Google Scholar - Nicholas D Hastie
You can also search for this author inPubMed Google Scholar
Contributions
O.M.M.-E. designed, conducted and analyzed experiments and wrote the manuscript. L.A.L. generated _Wt1_loxP/loxP mice. A.E. conducted part of the promoter and ChIP experiments. J.A.G. conducted immunohistochemistry experiments. J.S. assisted with the embryonic stem experiments. V.V. conducted immunohistochemistry and flow cytometry experiments. E.H. and J.R. conducted initial experiments with immortalized epicardial cells. P.S.D. conducted blastocyst microinjection and helped with mouse maintenance. P.H. helped set up genetic crosses and is responsible for the mouse database. N.H. provided the _Wt1_GFP/+ knockin mice. R.E.H. contributed to discussion. R.M.-C. designed experiments and cowrote the manuscript. N.D.H. obtained funding, helped with the design and analysis of experiments and cowrote the manuscript.
Corresponding author
Correspondence toNicholas D Hastie.
Supplementary information
Rights and permissions
About this article
Cite this article
Martínez-Estrada, O., Lettice, L., Essafi, A. et al. Wt1 is required for cardiovascular progenitor cell formation through transcriptional control of Snail and E-cadherin.Nat Genet 42, 89–93 (2010). https://doi.org/10.1038/ng.494
- Received: 07 July 2009
- Accepted: 02 November 2009
- Published: 20 December 2009
- Issue Date: January 2010
- DOI: https://doi.org/10.1038/ng.494