Comparative analysis of the genome sequences of Bordetella pertussis, Bordetella parapertussis and Bordetella bronchiseptica (original) (raw)
Main
B. pertussis, B. parapertussis and B. bronchiseptica colonize the respiratory tracts of mammals. B. pertussis is strictly a human pathogen and is the etiologic agent of whooping cough, an acute respiratory disease that is particularly serious in children1. Despite extensive vaccination programs, whooping cough is still endemic in many countries, causing approximately 285,000 deaths in 2001 (ref. 2). B. pertussis strains show little genetic variation, indicating that the species derived from a common ancestor in the recent past, perhaps only a few thousand years ago3. B. parapertussis infects both humans and sheep; in human infants it causes whooping cough4,5. Phylogenetic analyses have shown that B. parapertussis strains isolated from humans (_B. parapertussis_hu) are distinct from those isolated from sheep (_B. parapertussis_ov; ref. 6). It has been suggested that _B. parapertussis_hu and _B. parapertussis_ov evolved independently from a common ancestor (B. bronchiseptica; ref. 6), and there is little or no transmission between the two reservoirs (sheep and human). Furthermore, _B. parapertussis_ov isolates are genetically diverse, whereas _B. parapertussis_hu isolates are uniform6. B. bronchiseptica has a broad host range, causing chronic and often asymptomatic respiratory infections in a wide range of animals, but only occasionally in humans7.
Many of the virulence factors characterized in the bordetellae are common to all three species. These include adhesins, such as filamentous hemagglutinin (FHA), pertactin, tracheal colonization factor and fimbriae, and toxins, such as adenylate cyclase-hemolysin, dermonecrotic toxin and tracheal cytotoxin. Other virulence factors are expressed by just one of the species, such as pertussis toxin and serum-resistance protein expressed by B. pertussis or a type-III secretion system expressed by B. bronchiseptica (reviewed in ref. 8).
Expression of many of the proteins implicated in Bordetella pathogenesis is regulated in response to environmental stimuli by a two-component regulatory system called BvgA/S (reviewed in ref. 8). Environmental stimuli cause the inner membrane sensor histidine kinase BvgS to undergo autophosphorylation before phosphorylating the BvgA protein. Phosphorylated BvgA activates the transcription of a number of virulence-activated genes (vags) by binding to sites in their promoters. One such gene encodes the regulator BvgR that represses the transcription of a number of other genes called virulence-repressed genes (vrgs). Thus, activation of Bvg markedly changes the gene expression profile of the bacterium. Many genes implicated in pathogenesis are upregulated when the Bvg system is active, suggesting that this phase (Bvg-plus) is adopted by bacteria on entering the host during the course of infection. Conversely, the Bvg-minus phase is thought to be expressed when the bacteria are in the environment. A third distinct phenotypic phase, Bvg-intermediate (Bvgi), is characterized by the lack of expression of the vrgs and by the expression of only a few of the vags. The signals to which Bvg responds in vivo are unknown, but in vitro, the system is activated by growth at 37 °C and silenced either by growth below 27 °C or by adding sulfate ions or nicotinic acid to the growth medium, regardless of growth temperature.
We sequenced and compared the complete genomes of B. pertussis Tohama I, a clinical isolate that has subsequently been used widely for studies in the laboratory; B. parapertussis strain 12822, isolated in 1993 from a baby in Germany; and B. bronchiseptica strain RB50, isolated from a rabbit. Comparison of the genomes of these bacteria elucidates the genetic background of their differences in disease severity and the factors that determine their host range.
Results
Structure of the genomes
The general properties of the three genomes are shown in Table 1 and Figure 1. Our data indicate that at the genetic level, B. pertussis and B. parapertussis each derived from a _B. bronchiseptica_-like ancestor. Because strains of B. pertussis (and human isolates of B. parapertussis) are genetically uniform, it seems possible that each evolved recently from a strain of B. bronchiseptica. We therefore estimated the time to a last common ancestor between each pair of genomes based on the pairwise synonymous substitution rates for all ∼3,000 genes that are common to the three species. The estimated times to the last common ancestors were 0.27–1.4 million years (My) for B. bronchiseptica and B. parapertussis, 0.7–3.5 My for B. bronchiseptica and B. pertussis and 0.8–4.0 My for B. parapertussis and B. pertussis, suggesting that the genetic uniformity in B. pertussis reflects a recent bottleneck rather than recent descent from B. bronchiseptica. Alternatively, B. bronchiseptica might be sufficiently genetically diverse that some isolates are more similar to the last common ancestor of B. pertussis than is strain RB50, whose genome was sequenced here. These estimates have a large range, reflecting the uncertainty over actual substitution rates in bacterial populations, and should therefore be treated with caution.
Table 1 General features of the genomes of B. pertussis, B. parapertussis and B. bronchiseptica
Figure 1: Circular representations of the genomes of B. bronchiseptica, B. parapertussis and B. pertussis.
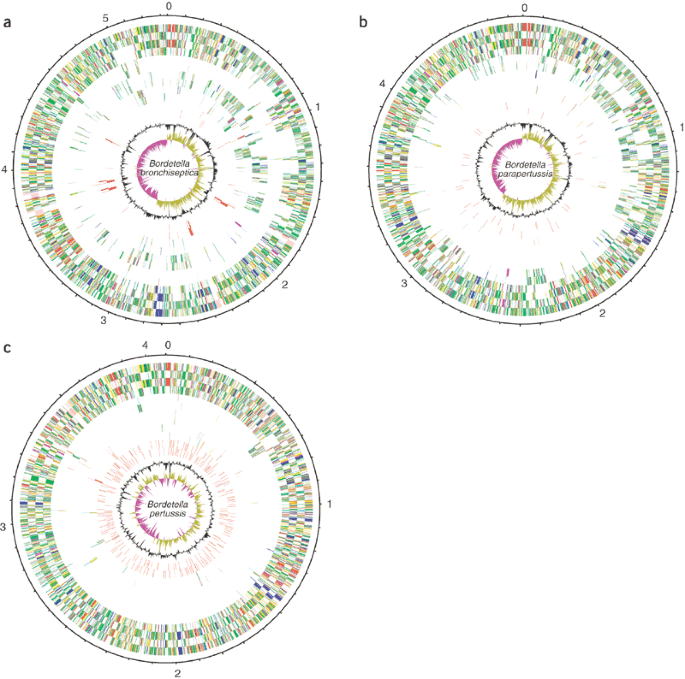
(a) B. bronchiseptica. The circles represent the following genes, numbering from the outside in: 1,2, all genes (transcribed clockwise and anti-clockwise); 3,4, core genes (shared in all three species); 5,6, genes shared only with B. pertussis; 7,8, genes shared only with B. parapertussis; 9,10, unique genes; 11,12, bacteriophage genes; 13, G+C content (plotted using a 10-kb window); 14, GC deviation ((G − C)/(G + C) plotted using a 10-kb window; khaki indicates values >1, purple <1). (b) B. parapertussis. The circles represent the same genes as in a, with the following exceptions: 5,6, genes shared only with B. bronchiseptica; 7,8, genes shared only with B. pertussis; 11,12, ISEs. (c) B. pertussis. The circles represent the same genes as in a, with the following exceptions: 5,6, genes shared only with B. bronchiseptica; 7,8, genes shared only with B. parapertussis; 11,12, ISEs. Genes are color-coding as follows: dark blue, pathogenicity/adaptation; black, energy metabolism; red, information transfer; dark green, surface-associated; cyan, degradation of large molecules; magenta, degradation of small molecules; yellow, central/intermediary metabolism; pale green, unknown; pale blue, regulators; orange, conserved hypothetical; brown, pseudogenes; pink, phage and ISEs; gray, miscellaneous.
Consistent with the relative degrees of divergence at the nucleotide level, it is apparent from the gross comparison between the genomes (Fig. 2) that B. bronchiseptica and B. parapertussis are more similar, in terms of overall organization, than B. bronchiseptica and B. pertussis. The comparison between B. bronchiseptica and B. parapertussis shows a large degree of collinearity between the genomes, primarily around the replication origin. The region around the terminus has eight rearranged blocks of DNA. Most chromosomal rearrangements between related bacteria are arranged reciprocally around the origin or terminus of replication, because, it has been suggested, most recombination occurs between, or close to, replication forks9 or because other rearrangements may occur but are selectively disadvantageous10. Most of the rearrangements in B. parapertussis (and B. pertussis) do not follow this pattern, suggesting that any such selection may be overcome by frequent recombination between large perfect repeats. Indeed, each rearrangement is bordered by identical copies of the insertion sequence elements (ISEs) IS_1001_ or IS_1002_ in B. parapertussis, suggesting that recombination between these ISEs was the primary cause of the rearrangements.
Figure 2: Linear genomic comparison of B. pertussis, B. bronchiseptica and B. parapertussis.
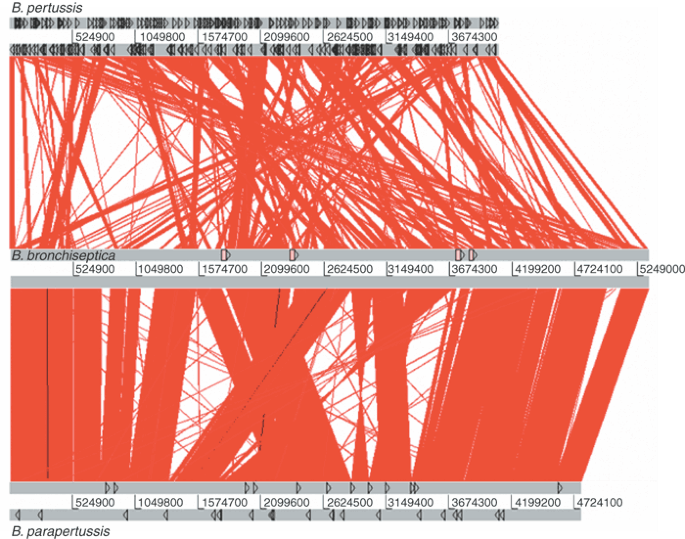
The gray bars represent the forward and reverse strands. Top, B. pertussis. Black triangles represent ISEs. Center, B. bronchiseptica. Pink boxes represent prophage. Bottom, B. parapertussis. Black triangles represent ISEs. The red lines between the genomes represent DNA:DNA similarities (BLASTN matches) between the two sequences.
Compared with B. parapertussis, B. bronchiseptica has several large discrete regions of unique DNA. Several of these are prophage insertions, but three of them (80 kb around position 750,000, 120 kb around position 1,083,000 and 65 kb around position 4,981,000) clearly are not. Two lines of evidence suggest that these large blocks of DNA are deletions in B. parapertussis rather than insertions in B. bronchiseptica. First, similar, though not identical, blocks of DNA are absent from B. pertussis, but the boundaries of the regions are different in B. parapertussis and B. pertussis, suggesting that independent deletions of similar regions occurred in the two species. Second, analysis of the B. bronchiseptica genome for signals associated with horizontally transferred DNA, such as anomalous G+C content, dinucleotide content or GC-skew11, clearly indicated that such sequences as the prophage were of recent horizontal acquisition but gave no such indication for these three large regions of difference. Similar analysis of the whole B. bronchiseptica genome suggested that little of the DNA was probably acquired recently and certainly not enough to account for the differences in size between the three genomes.
Genomic comparison of B. bronchiseptica and B. pertussis (Fig. 2) showed that rearrangements and deletions in B. pertussis were similar to those in B. parapertussis but more extreme, with short blocks of almost perfect conservation broken up by nearly 150 individual rearrangements, 88% of which are bounded by ISEs (primarily IS_481_). Again, the rearrangements do not follow the usual reciprocal pattern around the origin or terminus of replication. The large degree of rearrangement of overall genome structure is unprecedented for congeneric bacteria.
The B. pertussis genome is considerably smaller than that of B. bronchiseptica, and, although individual events are difficult to trace, much of this loss is probably due to ISE-mediated deletion events. At some point in its recent evolution, B. pertussis seems to have undergone a massive expansion of one family of ISEs (IS_481_; Table 1), and subsequent recombination between these perfect DNA repeats caused a large amount of rearrangement and deletion in the chromosome. Comparison of the genetic maps of several B. pertussis strains showed that inversions of large sections of the genome are frequent, presumably owing to such recombination12. Such ISE expansions have previously been seen in rarely recombining organisms whose effective population size was greatly reduced by an evolutionary bottleneck13. In any population, ISEs transpose to novel sites at a certain rate, but most of these novel insertions are lethal or carry a selective disadvantage, and bacteria carrying them are therefore competed out of the population. When the population size is very small, however, the degree of intraspecies competition is reduced, and bacteria carrying such non-lethal mutations are less likely to be competed out, leading to an increase in ISE accumulation in the population.
Gene complements
Comparison of the gene complements of the three species supports the inference from the gross structure that B. pertussis and B. parapertussis are independent derivatives of a _B. bronchiseptica_-like organism. Apart from ISE transposases, only 114 genes are unique to B. pertussis compared with B. bronchiseptica and B. parapertussis, and only 50 genes are unique to B. parapertussis compared with B. bronchiseptica and B. pertussis (Fig. 3). In fact, most genes that are apparently specific to B. pertussis seem to reflect diversity in B. bronchiseptica as indicated by microarray analysis (C. Cummings and D. Relman; personal communication).
Figure 3: Venn diagram showing gene complements of B. pertussis, B. parapertussis and B. bronchiseptica.
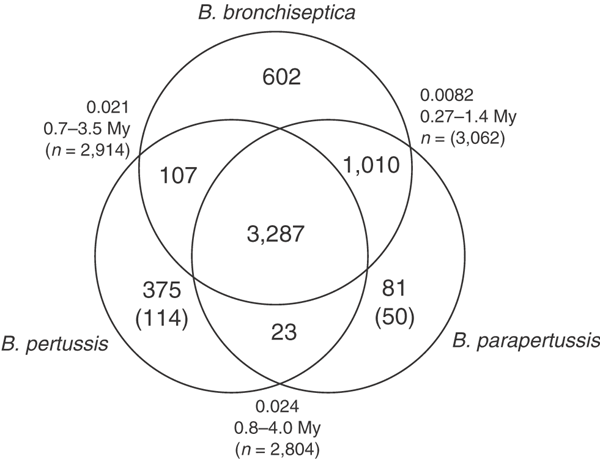
Numbers in parentheses indicate numbers of unique genes, excluding those from ISEs. Figures outside the circles indicate the average synonymous substitution rates (number of synonymous substitutions per potential substitution site) for the set of core genes between each pair of organisms and the estimated age of divergence in million years (My) calculated from these rates.
Furthermore, only 23 genes were found in both B. pertussis and B. parapertussis but not in B. bronchiseptica RB50. Thus, these derivative organisms seem to have little net gain of genes. Investigation of these genes (Supplementary Table 1 online) shows few obvious virulence factors. Conversely, B. bronchiseptica has over 600 genes not found in either of the other two species, over 1,000 shared only with B. parapertussis and just over 100 shared only with B. pertussis. Analysis of the genes that were lost in B. pertussis and B. parapertussis (Fig. 4) indicates that many are involved in membrane transport, small-molecule metabolism, regulation of gene expression and synthesis of surface structures. The three large deletions identified by comparing B. bronchiseptica and B. parapertussis contain genes that are unique to B. bronchiseptica and, therefore, that were probably lost in both B. pertussis and B. parapertussis. Some of these genes are involved in the transport and metabolism of a wide range of compounds, including amino acids, other aromatic compounds, fatty acids, nitrobenzene/benzoate, polyamines, phosphonates, phthalate, vanillate and mandelate. There is also a cluster of genes encoding a type-IV pilus (BB0776-BB0792), two siderophore receptors and several regulatory proteins.
Figure 4: Representation of genes lost in B. pertussis and B. parapertussis.
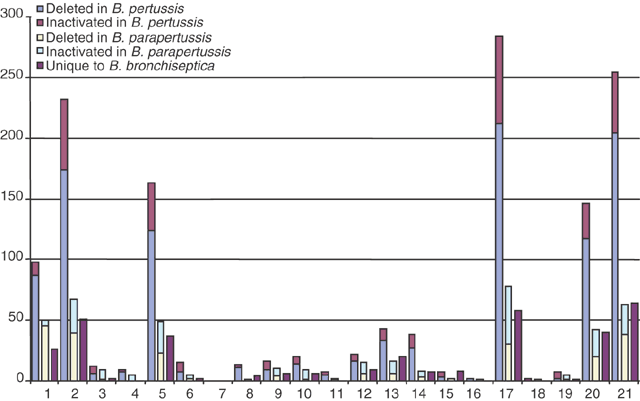
Blue, genes present in B. parapertussis and not B. pertussis (genes lost by B. pertussis); mauve, pseudogenes in B. pertussis; yellow, genes present in B. pertussis and not B. parapertussis (genes lost by B. parapertussis); pale blue, pseudogenes in B. parapertussis; purple, genes unique to B. bronchiseptica (genes lost by both B. pertussis and B. parapertussis). Functional classifications are as follows: 1, unknown; 2, conserved hypothetical; 3, cell processes; 4, protection responses; 5, transport/binding proteins; 6, adaptation; 7, cell division; 8, macromolecule degradation; 9, macromolecule synthesis/modification; 10, amino acid biosynthesis; 11, cofactor biosynthesis; 12, central/intermediary metabolism; 13, small-molecule degradation; 14, energy metabolism; 15, fatty acid biosynthesis; 16, ribonucleotide biosynthesis; 17, cell surface; 18, ribosome constituents; 19, pathogenicity; 20, regulation; 21, miscellaneous. Genes from mobile elements (ISEs and bacteriophage) were omitted from the figure.
In addition to this substantial gene deletion in B. pertussis and B. parapertussis, both seem to have lost the function of many other genes through pseudogene formation. In B. pertussis, 358 genes have been inactivated by insertion of ISEs, in-frame stop codons or frameshifts; similarly, 200 genes in B. parapertussis have been inactivated by these mechanisms. Conversely, B. bronchiseptica has only 19 pseudogenes (Supplementary Table 2 online). Again, analysis of the inactivated genes in B. pertussis and B. parapertussis indicates that they are predominantly involved in transport, small-molecule metabolism, regulation and surface structures (Fig. 4).
Metabolism
B. bronchiseptica can survive in the environment, but B. pertussis and B. parapertussis are apparently incapable of long-term survival outside the host. Despite this, much of the central and intermediary metabolism is conserved between the three genomes. Consistent with their ability to grow on defined media in culture, the genomes of all three Bordetella species seem to encode complete pathways for the biosynthesis of nucleotides, cofactors and amino acids (except cysteine, see below). Generally, Bordetella do not use sugars as carbon sources. This is probably because three genes encoding glycolytic functions (glucokinase, phosphofructokinase and fructose-1,6-bisphosphatase) are absent from the Bordetella genomes. Genes encoding the gluconeogenesis pathway are present, suggesting that glucose can be synthesized by Bordetella spp.
The metabolism of B. pertussis has been studied in some detail; it can grow in culture on glutamate and cysteine14. In laboratory media, cysteine is present at low levels and probably serves as a source of sulfur rather than carbon, suggesting that glutamate is the main carbon source for cultured B. pertussis. Given this cysteine auxotrophy, it is not surprising that the genes encoding several of the proteins of the sulfate transport (cysU and cysW) and cysteine biosynthesis pathway (cysH and cysN) are pseudogenes in B. pertussis and genes related to other key steps (cysC, cysI, cysJ and cysP) are absent. Not all the defects found in B. pertussis are present in B. parapertussis and B. bronchiseptica, but the pathway is mutated in both. This suggests that an ancestral Bordetella could use external sulfate or thiosulfate for cysteine biosynthesis but this ability was since lost. The genes related to synthesis of cysteine from acetyl-coenzyme A, serine and sulfide are present and intact, but the cysteine auxotrophy of B. pertussis suggests that this pathway does not function, and an IS_481_ insertion immediately upstream of the start codon of cysM may explain this.
It has been reported that the tricarboxylic acid cycle in the bordetellae is non-functional, based on the observation that in these bacteria, acetyl-coenzyme A and oxaloacetate did not give rise to α-ketoglutarate14. Genes encoding all the main tricarboxylic acid cycle enzymes are present in all three species, however. Thus, there is no obvious genetic defect that explains these observations. Overall, it seems that the inability of B. pertussis and B. parapertussis to survive in the environment is probably due to the loss of numerous accessory pathways for the use of alternative nutrient sources, described above, rather than a lack of central metabolic pathways.
Host range and pathogenicity
All bordetellae carry a wide range of determinants involved in host interaction and virulence, many of which have been extensively studied. Examples include the filamentous hemagglutinin FhaB; fimbriae; a large number of autotransporters including the adhesins pertactin, tracheal colonization factor and the serum-resistance protein; numerous toxins including the pertussis toxin, adenylate cyclase-hemolysin, dermonecrotic toxin15 and tracheal cytotoxin; a type-III secretion system; and lipopolysaccharide and flagella biosynthesis systems (reviewed in refs. 8 and 16). Comparisons of many of these systems in the three species identify differences that might bear on the different host ranges and virulence profiles of these organisms.
FhaB (BP1879) is a large secreted protein involved in attachment to host cells17 and is a member of a growing family of hemagglutinin/hemolysin proteins present in a number of Gram-negative plant and animal pathogens. An ortholog of fhaB is present in each of the three species (BP1879, BB2993 and BPP3027); although each seems to be intact, there are internal variations in sequence and length of the repetitive internal sequences18. Two other genes encoding FhaB-like proteins are present in B. pertussis, BP2667 and BP2907. BP2667 (fhaS) is orthologous to BB2312 and BPP1243, and BP2907 (fhaL) to BB1936 and BPP2489. In the former case, there are again differences in the internal sequences between the three Bordetella spp., although the fhaS orthologs seem to be more similar to each other than are the fhaL orthologs. FHA is important in adherence of the bacteria to host cells. It binds directly to ciliary membrane glycosphingolipids that are thought to be host receptors for Bordetella adherence19. Differential binding of the three Bordetella species to ciliated cells from different hosts suggests that host specificity may be partly dependent on this receptor-ligand interaction20. The identification of additional FHA-like genes that vary between Bordetella species is of interest to the study of the host-pathogen interaction and host specificity.
As described above, the genome of B. bronchiseptica contains a type-IV pilus biosynthesis operon that is absent in the other two species. Other fimbrial systems are also encoded by the genomes. The previously described chaperone-usher fimbrial system (fimA-fimD) is present in all three, although the fimbrial gene fimA, which has been inactivated by a deletion in B. pertussis (BP1880; ref. 21) and by a frameshift in B. parapertussis (BPP3026), is apparently intact in B. bronchiseptica (BB2992). Other isolated fimbrial genes exist: the serotype 3 subunit (fim3; ref. 22) is intact in all three species; fimN23 is present in B. bronchiseptica and B. parapertussis (although with a variable C terminus) but deleted from B. pertussis; fimX24 is intact in B. pertussis and B. bronchiseptica but frameshifted in B. parapertussis; the serotype 2 fimbrial subunit (fim2; ref. 25) is present in all three species but with a variable C terminus in B. pertussis; and there is a novel subunit upstream of fimN present in B. bronchiseptica and B. parapertussis but deleted from B. pertussis.
Variation is also seen in the complement of autotransporters. These are members of a large family of exported proteins that encode an integral outer-membrane pore, enabling them to cross the outer membrane26. The bordetellae encode 21 of these proteins, a few (serum-resistance protein BrkA, pertactin, SphB1, TcfA and Vag8) with documented functions in host interaction and virulence26,27. Each of the three species contains a different complement of autotransporters (Table 2). B. bronchiseptica includes what might be considered the complete set of genes, albeit with one represented by a pseudogene. B. parapertussis and B. pertussis each have fewer autotransporter genes and more autotransporter pseudogenes than B. bronchiseptica. As with the variation in sequence of the FHA and fimbrial proteins, this variable complement of autotransporter proteins may have a direct effect on the ability of each species to interact with, and cause disease in, different hosts.
Table 2 Autotransporters encoded in the genomes of B. pertussis, B. parapertussis and B. bronchiseptica
Iron acquisition is of great importance to mammalian pathogens, as iron is present in limiting conditions in the host, and any iron that is present is sequestered by heme and other iron-chelating compounds. Many pathogens produce iron-chelating compounds called siderophores to acquire iron, and the three bordetellae each contain an operon encoding the production, export and uptake of the siderophore alcaligin28 (BP2456-BP2463, BB3893-BB3900 and BPP3443-BPP3450). Iron from siderophores and host iron-binding complexes is internalized through TonB-dependent outer-membrane ferric complex receptors; the three Bordetella species encode up to 16 of these proteins (Table 3), suggesting that they scavenge host iron-binding complexes and xenosiderophores in addition to using their own. As with the autotransporters, B. bronchiseptica encodes a complete set of these receptors, with B. parapertussis and B_. pertussis_ having fewer genes and more pseudogenes. The genes encoding several of these receptors have adjacent cognate regulators (AraC, AsnC family and MarR family transcriptional regulators, as well as a two-component system that consists of a sensor protein and an extracytoplasmic function sigma factor). Like the TonB-dependent receptors, all these regulators are intact in B. bronchiseptica. In contrast, a few of them are pseudogenes in B. pertussis and B. parapertussis even when their cognate TonB-dependent receptor genes are intact. Again, it is possible that these different iron-uptake system gene complements affect the range of environments in which the bacteria can survive.
Table 3 TonB-dependent ferric complex receptors encoded in the genomes of B. pertussis, B. parapertussis and B. bronchiseptica
Three structures of great importance for virulence and host interaction in Gram-negative pathogens are type-III secretion systems, O-antigens and flagella; each of these show species specific variations among the three Bordetella species. Type-III secretion systems are specific mechanisms for the export of virulence factors into host cells29. The three bordetellae encode a novel type-III secretion system for which the secreted effectors have not yet been identified30,31. B. bronchiseptica encodes the full, intact operon (BB1609-BB1638), which is expressed. The operon in B. parapertussis (BPP2212-BPP2241) contains two identifiable pseudogenes in regulatory and structural proteins (BPP2241 and BP2262) and is not expressed. Curiously, the operon in B. pertussis does not seem to be expressed30,31 but contains no clearly identifiable pseudogenes. It is possible that its inactivity is dependant on _cis_- or _trans_-acting regulatory mutations.
O-antigens are the membrane-distal domains of some lipopolysaccharides and are involved in many bacterial species in evasion of host innate and acquired immunity mechanisms. B. bronchiseptica and B. parapertussis have been reported to express an identical O-antigen32, but variable modifications to the terminal sugar residue of the O-antigen polymer were recently discovered33. These modifications involve, for example, formylation of the amino groups of the terminal sugar residue. The precise pattern of modification differs between different strains of B. bronchiseptica and correlates with different reactivities of the O-antigens to monoclonal antibodies. Within the O-antigen biosynthesis locus of B. bronchiseptica RB50 reported here is a region that differs from the partial locus sequenced from strain CN7635E and reported previously34. This alternative region of the locus contains genes that are predicted to code for formyl transferases, and these are probably responsible for the variable O-antigen modifications between strains. The 'modification region' from B. parapertussis sequenced here is identical to the partial locus of B. bronchiseptica CN7635E previously reported, and these two strains share an identical reactivity profile to monoclonal antibodies against O-antigen, which is different from that of B. bronchiseptica RB50 (A.P., unpublished observations). Thus, this genetic region of the O-antigen biosynthetic locus probably directs differential modification of the lipopolysaccharide molecule, possibly forming the genetic basis for O-antigenic variation. The O-antigen biosynthesis locus in B. pertussis has been deleted by IS_1002_ insertion and consequently, B. pertussis does not express O-antigen34.
We found that the bordetellae contain a locus that seems to encode a type II polysaccharide capsule. Capsules are often key contributors to the ability of pathogens to withstand host defense mechanisms35. Several references to a Bordetella capsule have been made in the literature36,37, but it is not clear what structure is being referred to, and a capsule has not been isolated and characterized from the bordetellae. The locus arrangement for the Bordetella capsule is typical of that of type II capsules38, comprising three regions involved in export/modification, biosynthesis and transport. The genes involved in biosynthesis of the Bordetella capsule encode products that are homologous to the Salmonella typhi Vi antigen biosynthesis enzymes, suggesting that the product of the Bordetella locus may be similar to the N-acetyl galactosaminuronic acid Vi antigen polymer. Of the three Bordetella spp. that we sequenced, only B. bronchiseptica contains an intact capsule locus (BB2918-2934). In B. pertussis, the central part of the locus (BP1619-1631) is intact, but the 3′ region of BP1618 underwent an inversion event and at the other end, an ISE-mediated rearrangement deleted part of region 1 (which contains genes involved in export/modification) and moved two of these export genes to a different part of the chromosome (BP1654 and BP1655, which is interrupted by the ISE). In B. parapertussis, the ortholog of BP1618 is also mutated but in this case by a point mutation that introduces an internal stop codon (BPP2967). In addition, an export gene from region 1 (BPP2951), one of those deleted from B. pertussis, suffered a frameshift mutation that altered the 3′ end of the coding sequence. The fact that the capsule locus is intact in B. bronchiseptica but mutated in B. pertussis and B. parapertussis might suggest that the putative capsule is not involved in pathogenesis in the mammalian host, but perhaps contributes to the survival of B. bronchiseptica in the environment. Given that all but the ends of the loci are intact in B. pertussis and B. parapertussis, however, it is possible that some capsule expression may occur in these species.
Flagella are virulence factors for a number of organisms. B. bronchiseptica is motile and encodes a full flagellar operon, whereas the flagellar operons of both B. pertussis and B. parapertussis are inactivated by multiple pseudogenes and ISE insertions, leading to a lack of motility. This is reminiscent of the situation in Yersinia pestis, which also seems to have recently lost the ability to make flagella, coincident with a change in niche from a gut to a systemic pathogen13. Like O-antigen, flagella are preferentially expressed in the Bvg-minus phase, suggesting that they may be primarily involved with survival of the bacteria in the environment. Thus, the lack of expression of these factors in B. pertussis may reflect adaptation to its host-restricted niche and supports the hypothesis that B. pertussis does not have an environmental phase to its life cycle. Loss of surface structures, such as flagella, fimbriae and polysaccharides, probably enhances the virulence of B. pertussis (and, to a lesser extent, B. parapertussis) by reducing the number of targets that are available for recognition by the human immune system.
The bordetellae produce a number of different toxins, of which the pertussis toxin produced exclusively by B. pertussis is best characterized. The genes encoding the toxin (BP3783-BP3787) are immediately followed by genes encoding a type-IV secretion system (BP3788-BP3796) involved in its export. In confirmation of previous studies, the pertussis toxin structural and export operons are present in B. parapertussis (BPP4304-BPP4316) and B. bronchiseptica (BB4890-BB4902). The B. parapertussis toxin subunit gene ptxB is represented by a pseudogene, but there are no identifiable pseudogenes in the B. bronchiseptica operon. The lack of expression of pertussis toxin in B. parapertussis and B. bronchiseptica is due to differences in the promoter regions of the operons, which have been interpreted as promoter inactivation mutations in these organisms39. A comparison of the individual changes in the ptxA promoter region (Fig. 5), however, shows that 62% of the base changes are due to changes in B. pertussis, whereas the sequence is conserved in the other two species (Fig. 5). Several of these changes fall in the promoter or activator (BvgA) binding sites, and the effect of these is to increase the similarity to the σ70 or putative BvgA-binding consensus sequence (TTTCGTA; ref. 40) in B. pertussis. Thus, it is at least possible that recent mutations in B. pertussis increased the regulated expression of pertussis toxin in this organism. This apparent increase in virulence due to mutation is similar in some ways to the increase in virulence caused by certain chromosomal deletions in Escherichia coli41 and by specific mutations in Salmonella42.
Figure 5: The ptxA promoter regions of B. pertussis, B. parapertussis and B. bronchiseptica.
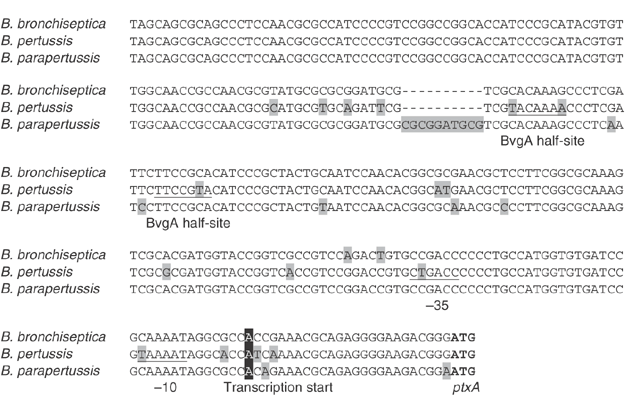
The upstream sequences of ptxA from each species are shown aligned. Changes in one sequence are highlighted in light gray, and the initiation codon of ptxA is shown in bold. The characterized transcription start site39 is highlighted in black, and the predicted −10 and −35 promoter sequences39 are underlined. The BvgA binding half-sites40 are also underlined.
Discussion
The genomic comparison between B. bronchiseptica, B. parapertussis and B. pertussis helps to explain the difference in host range and pathogenesis between these three closely related organisms. Contrary to what might be expected, the broader host range of B. bronchiseptica and the increased virulence of B. pertussis over B. parapertussis do not seem to be due to recent acquisition of host interaction factors or virulence determinants. There is little evidence overall of recent large-scale acquisition of genes by any of the species. Conversely, it seems that many of the individual traits of the organisms could be due to independent mechanisms of large-scale gene inactivation and loss in the two derivative species, B. parapertussis and B. pertussis. The limited host range of these species compared to that of a _B. bronchiseptica_-like putative ancestor can be easily explained by the documented loss of host interaction mechanisms. In addition, the increased virulence for humans of B. pertussis could also be due to an overexpression or constitutive expression of virulence traits that are subject to tighter temporal or environmental control in the other species or to the removal of structures or systems that might hinder virulence or survival in the single host. It may be that B. pertussis followed this evolutionary path owing to the opportunities for increased transmission rate provided by the increase in size and density of the populations of its specific host, Homo sapiens. This would support the hypothesis that, when transmission rates are high, the ability to survive in the environment and keep damage to the host to a minimum are no longer selectively advantageous42.
Methods
DNA preparation and sequencing.
B. pertussis Tohama I was a gift from S. Stibitz (Food and Drug Administration, Bethesda, Maryland). We grew bacteria on Bordet Gengou agar (Difco) supplemented with 15% defibrinated horse blood (TCS Biologicals) at 37 °C for 3 d. We isolated DNA by the agarose plug method43. We obtained the initial genome assembly from 87,500 paired-end sequences (giving 8.9-fold coverage) derived from three pUC18 genomic shotgun libraries (with insert sizes of 1.0–4.0 kb) using dye terminator chemistry on ABI377 automated sequencers. We used 2,560 paired-end sequences from a pBACe3.6 library with insert sizes of 10–20 kb (a clone coverage of 4.7-fold) as a scaffold. We generated another 41,700 sequencing reads during finishing and verified the final assembly by comparison with the published restriction map44.
B. bronchiseptica strain RB50 was isolated from a rabbit and was a gift from J. F. Miller (University of California Los Angeles). We obtained the initial genome assembly from 99,000 paired-end sequences (giving 7.95-fold coverage) derived from two pUC18 genomic shotgun libraries (with insert sizes of 1.0–2.0 kb) using dye terminator chemistry on ABI377 and ABI 3700 automated sequencers. We used 4,480 paired-end sequences from two pBACe3.6 libraries with insert sizes of 15–23 kb and 23–48 kb (a total clone coverage of 12.7-fold) as a scaffold. We generated another 22,660 sequencing reads during finishing.
B. parapertussis strain 12822 was a gift from U. Heininger (der Friedrich-Alexander-Universitat Erlangen-Nurnberg, Germany). We obtained the initial genome assembly from 83,850 paired-end sequences (giving 8.6-fold coverage) derived from a pUC18 genomic shotgun library (with insert sizes of 2.0–4.0 kb) using dye terminator chemistry on ABI 3700 automated sequencers. We used 3,020 paired-end sequences from a pBACe3.6 library with insert sizes of 23–48 kb (a clone coverage of 11.0-fold) as a scaffold. We generated another 5,450 sequencing reads during finishing.
Annotation and analysis.
We assembled, finished and annotated the sequences as described previously45, using Artemis46 to collate data and facilitate annotation. We compared the DNA and encoded protein sequences of the three species using the Artemis Comparison Tool (K. Rutherford, unpublished data). We identified orthologous gene sets by reciprocal best-match FASTA comparisons. Pseudogenes had one or more mutations that would prevent complete translation and were identified by direct comparison between the three genomes. We then checked each of the inactivating mutations against the original sequencing data. We calculated synonymous substitution frequency (Ds) values from orthologous gene pairs from the core gene set according to the method of Nei and Gojobori47 and estimated ages of divergence (Age = Ds/mutation rate) from the mutation rates calculated by Whittam48 or Guttman and Dykhuizen49.
URLs.
The Artemis Comparison Tool is available from http://www.sanger.ac.uk/Software/ACT/. The full annotation of each of the sequences and further information are available from http://www.sanger.ac.uk/Projects/B_pertussis/.
EMBL accession numbers.
B. pertussis, BX470248; B. parapertussis, BX470249; B. bronchiseptica, BX470250.
Note: Supplementary information is available on the Nature Genetics website.
Accession codes
Accessions
GenBank/EMBL/DDBJ
References
- Cherry, J.D. Historical review of pertussis and the classical vaccine. J. Infect. Dis. 174, S259–S263 (1996).
Article Google Scholar - World Health Organization. The World Health Report 2002: Reducing risks, promoting healthy life. (World Health Organization, Geneva, 2002).
- van Loo, I.H., Heuvelman, K.J., King, A.J. & Mooi, F.R. Multilocus sequence typing of Bordetella pertussis based on surface protein genes. J. Clin. Microbiol. 40, 1994–2001 (2002).
Article CAS Google Scholar - Porter, J.F., Connor, K. & Donachie, W. Isolation and characterization of _Bordetella parapertussis_-like bacteria from ovine lungs. Microbiology 140, 255–261 (1994).
Article Google Scholar - Heininger, U. et al. Clinical characteristics of illness caused by Bordetella parapertussis compared with illness caused by Bordetella pertussis. Pediatr. Infect. Dis. J. 13, 306–309 (1994).
Article CAS Google Scholar - van der Zee, A., Mooi, F., Van Embden, J. & Musser, J. Molecular evolution and host adaptation of Bordetella spp.: phylogenetic analysis using multilocus enzyme electrophoresis and typing with three insertion sequences. J. Bacteriol. 179, 6609–6617 (1997).
Article CAS Google Scholar - Goodnow, R.A. Biology of Bordetella bronchiseptica. Microbiol. Rev. 44, 722–738 (1980).
CAS PubMed PubMed Central Google Scholar - Cotter, P.A. & Miller, J.F. Bordetella. in Principles of Bacterial Pathogenesis (ed. Groisman, E.) 619–674 (Academic, San Diego, 2001).
Book Google Scholar - Tillier, E.R. & Collins, R.A. Genome rearrangement by replication-directed translocation. Nat. Genet. 26, 195–197 (2000).
Article CAS Google Scholar - Mackiewicz, P., Mackiewicz, D., Kowalczuk, M. & Cebrat, S. Flip-flop around the origin and terminus of replication in prokaryotic genomes. Genome Biol. 2, INTERACTIONS1004 (2001).
- Karlin, S. Detecting anomalous gene clusters and pathogenicity islands in diverse bacterial genomes. Trends Microbiol. 9, 335–343 (2001).
Article CAS Google Scholar - Stibitz, S. & Yang, M.S. Genomic plasticity in natural populations of Bordetella pertussis. J. Bacteriol. 181, 5512–5515 (1999).
CAS PubMed PubMed Central Google Scholar - Parkhill, J. et al. Genome sequence of Yersinia pestis, the causative agent of plague. Nature 413, 523–527 (2001).
Article CAS Google Scholar - Thalen, M. et al. Rational medium design for Bordetella pertussis: basic metabolism. J. Biotechnol. 75, 147–159 (1999).
Article CAS Google Scholar - Brockmeier, S.L. et al. Role of the dermonecrotic toxin of Bordetella bronchiseptica in the pathogenesis of respiratory disease in swine. Infect. Immun. 70, 481–490 (2002).
Article CAS Google Scholar - Mattoo, S., Foreman-Wykert, A.K., Cotter, P.A. & Miller, J.F. Mechanisms of Bordetella pathogenesis. Front. Biosci. 6, E168–E186 (2001).
Article CAS Google Scholar - Coutte, L. et al. Role of adhesin release for mucosal colonization by a bacterial pathogen. J. Exp. Med. 197, 735–742 (2003).
Article CAS Google Scholar - Jacob-Dubuisson, F. et al. Molecular characterization of Bordetella bronchiseptica filamentous haemagglutinin and its secretion machinery. Microbiology 146, 1211–1221 (2000).
Article CAS Google Scholar - Tuomanen, E. et al. Receptor analogs and monoclonal antibodies that inhibit adherence of Bordetella pertussis to human ciliated respiratory epithelial cells. J. Exp. Med. 168, 267–277 (1988).
Article CAS Google Scholar - Tuomanen, E.I., Nedelman, J., Hendley, J.O. & Hewlett, E.L. Species specificity of Bordetella adherence to human and animal ciliated respiratory epithelial cells. Infect. Immun. 42, 692–695 (1983).
CAS PubMed PubMed Central Google Scholar - Willems, R.J. van der Heide, H.G. & Mooi, F.R. Characterization of a Bordetella pertussis fimbrial gene cluster which is located directly downstream of the filamentous haemagglutinin gene. Mol. Microbiol. 6, 2661–2671 (1992).
Article CAS Google Scholar - Mooi, F.R., ter Avest, A. & van der Heide, H.G. Structure of the Bordetella pertussis gene coding for the serotype 3 fimbrial subunit. FEMS Microbiol. Lett. 54, 327–331 (1990).
Article CAS Google Scholar - Kania, S.A. et al. Characterization of fimN, a new Bordetella bronchiseptica major fimbrial subunit gene. Gene 256, 149–155 (2000).
Article CAS Google Scholar - Pedroni, P. et al. Cloning of a novel pilin-like gene from Bordetella pertussis: homology to the fim2 gene. Mol. Microbiol. 2, 539–543 (1988).
Article CAS Google Scholar - Livey, I., Duggleby, C.J. & Robinson, A. Cloning and nucleotide sequence analysis of the serotype 2 fimbrial subunit gene of Bordetella pertussis. Mol. Microbiol. 1, 203–209 (1987).
Article CAS Google Scholar - Henderson, I.R. & Nataro, J.P. Virulence functions of autotransporter proteins. Infect. Immun. 69, 1231–1243 (2001).
Article CAS Google Scholar - Coutte, L., Antoine, R., Drobecq, H., Locht, C. & Jacob-Dubuisson, F. Subtilisin-like autotransporter serves as maturation protease in a bacterial secretion pathway. EMBO J. 20, 5040–5048 (2001).
Article CAS Google Scholar - Moore, C.H., Foster, L.A., Gerbig, D.G. Jr., Dyer, D.W. & Gibson, B.W. Identification of alcaligin as the siderophore produced by Bordetella pertussis and B. bronchiseptica. J. Bacteriol. 177, 1116–1118 (1995).
Article CAS Google Scholar - Galan, J.E. & Collmer, A. Type III secretion machines: bacterial devices for protein delivery into host cells. Science 284, 1322–1328 (1999).
Article CAS Google Scholar - Yuk, M.H., Harvill, E.T. & Miller, J.F. The BvgAS virulence control system regulates type III secretion in Bordetella bronchiseptica. Mol. Microbiol. 28, 945–959 (1998).
Article CAS Google Scholar - Stockbauer, K.E., Foreman-Wykert, A.K. & Miller, J.F. Bordetella type III secretion induces caspase 1-independent necrosis. Cell. Microbiol. 5, 123–132 (2003).
Article CAS Google Scholar - Di Fabio, J.L., Caroff, M., Karibian, D., Richards, J.C. & Perry, M.B. Characterization of the common antigenic lipopolysaccharide O-chains produced by Bordetella bronchiseptica and Bordetella parapertussis. FEMS Microbiol. Lett. 76, 275–281 (1992).
Article CAS Google Scholar - Vinogradov, E., Peppler, M.S. & Perry, M.B. The structure of the nonreducing terminal groups in the O-specific polysaccharides from two strains of Bordetella bronchiseptica. Eur. J. Biochem. 267, 7230–7237 (2000).
Article CAS Google Scholar - Preston, A. et al. Genetic basis for lipopolysaccharide O-antigen biosynthesis in bordetellae. Infect. Immun. 67, 3763–3767 (1999).
CAS PubMed PubMed Central Google Scholar - Moxon, E.R. & Kroll, J.S. The role of bacterial polysaccharide capsules as virulence factors. Curr. Top. Microbiol. Immunol. 150, 65–85 (1990).
CAS PubMed Google Scholar - Weiss, A.A., Melton, A.R., Walker, K.E., Andraos-Selim, C. & Meidl, J.J. Use of the promoter fusion transposon Tn_5 lac_ to identify mutations in Bordetella pertussis vir-regulated genes. Infect. Immun. 57, 2674–2682 (1989).
CAS PubMed PubMed Central Google Scholar - Kuwano, A., Ito, T., Tachi, H. & Hiramune, T. Comparison of the inhibitory effect of sulfamonomethoxine and other sulfonamides on capsule formation of Bordetella bronchiseptica. J. Vet. Med. Sci. 54, 1057–1059 (1992).
Article CAS Google Scholar - Frosch, M., Edwards, U., Bousset, K., Krausse, B. & Weisgerber, C. Evidence for a common molecular origin of the capsule gene loci in gram-negative bacteria expressing group II capsular polysaccharides. Mol. Microbiol. 5, 1251–1263 (1991).
Article CAS Google Scholar - Arico, B. & Rappuoli, R. Bordetella parapertussis and Bordetella bronchiseptica contain transcriptionally silent pertussis toxin genes. J. Bacteriol. 169, 2847–2853 (1987).
Article CAS Google Scholar - Boucher, P.E. & Stibitz, S. Synergistic binding of RNA polymerase and BvgA phosphate to the pertussis toxin promoter of Bordetella pertussis. J. Bacteriol. 177, 6486–6491 (1995).
Article CAS Google Scholar - Maurelli, A.T., Fernandez, R.E., Bloch, C.A., Rode, C.K. & Fasano, A. “Black holes” and bacterial pathogenicity: a large genomic deletion that enhances the virulence of Shigella spp. and enteroinvasive Escherichia coli. Proc. Natl. Acad. Sci. USA 95, 3943–3948 (1998).
Article CAS Google Scholar - Foreman-Wykert, A.K. & Miller, J.F. Hypervirulence and pathogen fitness. Trends. Microbiol. 11, 105–108 (2003).
Article CAS Google Scholar - Sambrook, J. & Russell, D.W. Molecular cloning: a laboratory manual vol. 3 (Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York, 2001).
Google Scholar - Stibitz, S. & Garletts, T.L. Derivation of a physical map of the chromosome of Bordetella pertussis Tohama I. J. Bacteriol. 174, 7770–7777 (1992).
Article CAS Google Scholar - Parkhill, J. et al. Complete DNA sequence of a serogroup A strain of Neisseria meningitidis Z2491. Nature 404, 502–506 (2000).
Article CAS Google Scholar - Rutherford, K. et al. Artemis: sequence visualization and annotation. Bioinformatics 16, 944–945 (2000).
Article CAS Google Scholar - Nei, M. & Gojobori, T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol. Biol. Evol. 3, 418–426 (1986).
CAS PubMed Google Scholar - Whittam, T.S. Genetic variation and evolutionary processes in natural populations of Escherichia coli. in Escherichia coli and Salmonella: Cellular and Molecular Biology (eds. Neidhardt, F.C. & Curtiss, R.) 2708–2720 (ASM Press, Washington, D.C., 1996).
Google Scholar - Guttman, D.S. & Dykhuizen, D.E. Clonal divergence in Escherichia coli as a result of recombination, not mutation. Science 266, 1380–1383 (1994).
Article CAS Google Scholar
Acknowledgements
We are grateful to S. Stibitz for help with reconciling our assembly with his map and to D. Relman and C. Cummings for access to unpublished data. We acknowledge support from the Wellcome Trust Sanger Institute core sequencing and informatics groups. This work was supported by the Wellcome Trust through its Beowulf Genomics initiative.
Author information
Authors and Affiliations
- The Sanger Institute, Wellcome Trust Genome Campus, Hinxton, CB10 1SA, Cambridge, UK
Julian Parkhill, Mohammed Sebaihia, Lee D Murphy, Nicholas Thomson, David E Harris, Matthew T G Holden, Carol M Churcher, Stephen D Bentley, Karen L Mungall, Ana M Cerdeño-Tárraga, Keith James, Barbara Harris, Michael A Quail, Rebecca Atkin, Steven Baker, David Basham, Nathalie Bason, Inna Cherevach, Tracey Chillingworth, Matthew Collins, Anne Cronin, Paul Davis, Jonathan Doggett, Theresa Feltwell, Arlette Goble, Nancy Hamlin, Heidi Hauser, Simon Holroyd, Kay Jagels, Sampsa Leather, Sharon Moule, Halina Norberczak, Susan O'Neil, Doug Ormond, Claire Price, Ester Rabbinowitsch, Simon Rutter, Mandy Sanders, David Saunders, Katherine Seeger, Sarah Sharp, Mark Simmonds, Jason Skelton, Robert Squares, Steven Squares, Kim Stevens, Louise Unwin, Sally Whitehead & Bart G Barrell - Department of Clinical Veterinary Medicine, The Centre for Veterinary Science, University of Cambridge, Madingley Road, Cambridge, CB3 0ES, UK
Andrew Preston & Duncan J Maskell - Drew University, 36 Madison Avenue, Madison, 07940, New Jersey, USA
Louise Temple - Department of Molecular Biology, Max-Planck Institut für Infektionsbiologie, Schumannstr. 21/22, Berlin, 10117, Germany
Mark Achtman
Authors
- Julian Parkhill
You can also search for this author inPubMed Google Scholar - Mohammed Sebaihia
You can also search for this author inPubMed Google Scholar - Andrew Preston
You can also search for this author inPubMed Google Scholar - Lee D Murphy
You can also search for this author inPubMed Google Scholar - Nicholas Thomson
You can also search for this author inPubMed Google Scholar - David E Harris
You can also search for this author inPubMed Google Scholar - Matthew T G Holden
You can also search for this author inPubMed Google Scholar - Carol M Churcher
You can also search for this author inPubMed Google Scholar - Stephen D Bentley
You can also search for this author inPubMed Google Scholar - Karen L Mungall
You can also search for this author inPubMed Google Scholar - Ana M Cerdeño-Tárraga
You can also search for this author inPubMed Google Scholar - Louise Temple
You can also search for this author inPubMed Google Scholar - Keith James
You can also search for this author inPubMed Google Scholar - Barbara Harris
You can also search for this author inPubMed Google Scholar - Michael A Quail
You can also search for this author inPubMed Google Scholar - Mark Achtman
You can also search for this author inPubMed Google Scholar - Rebecca Atkin
You can also search for this author inPubMed Google Scholar - Steven Baker
You can also search for this author inPubMed Google Scholar - David Basham
You can also search for this author inPubMed Google Scholar - Nathalie Bason
You can also search for this author inPubMed Google Scholar - Inna Cherevach
You can also search for this author inPubMed Google Scholar - Tracey Chillingworth
You can also search for this author inPubMed Google Scholar - Matthew Collins
You can also search for this author inPubMed Google Scholar - Anne Cronin
You can also search for this author inPubMed Google Scholar - Paul Davis
You can also search for this author inPubMed Google Scholar - Jonathan Doggett
You can also search for this author inPubMed Google Scholar - Theresa Feltwell
You can also search for this author inPubMed Google Scholar - Arlette Goble
You can also search for this author inPubMed Google Scholar - Nancy Hamlin
You can also search for this author inPubMed Google Scholar - Heidi Hauser
You can also search for this author inPubMed Google Scholar - Simon Holroyd
You can also search for this author inPubMed Google Scholar - Kay Jagels
You can also search for this author inPubMed Google Scholar - Sampsa Leather
You can also search for this author inPubMed Google Scholar - Sharon Moule
You can also search for this author inPubMed Google Scholar - Halina Norberczak
You can also search for this author inPubMed Google Scholar - Susan O'Neil
You can also search for this author inPubMed Google Scholar - Doug Ormond
You can also search for this author inPubMed Google Scholar - Claire Price
You can also search for this author inPubMed Google Scholar - Ester Rabbinowitsch
You can also search for this author inPubMed Google Scholar - Simon Rutter
You can also search for this author inPubMed Google Scholar - Mandy Sanders
You can also search for this author inPubMed Google Scholar - David Saunders
You can also search for this author inPubMed Google Scholar - Katherine Seeger
You can also search for this author inPubMed Google Scholar - Sarah Sharp
You can also search for this author inPubMed Google Scholar - Mark Simmonds
You can also search for this author inPubMed Google Scholar - Jason Skelton
You can also search for this author inPubMed Google Scholar - Robert Squares
You can also search for this author inPubMed Google Scholar - Steven Squares
You can also search for this author inPubMed Google Scholar - Kim Stevens
You can also search for this author inPubMed Google Scholar - Louise Unwin
You can also search for this author inPubMed Google Scholar - Sally Whitehead
You can also search for this author inPubMed Google Scholar - Bart G Barrell
You can also search for this author inPubMed Google Scholar - Duncan J Maskell
You can also search for this author inPubMed Google Scholar
Corresponding authors
Correspondence toJulian Parkhill or Duncan J Maskell.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
About this article
Cite this article
Parkhill, J., Sebaihia, M., Preston, A. et al. Comparative analysis of the genome sequences of Bordetella pertussis, Bordetella parapertussis and Bordetella bronchiseptica.Nat Genet 35, 32–40 (2003). https://doi.org/10.1038/ng1227
- Received: 06 June 2003
- Accepted: 23 July 2003
- Published: 10 August 2003
- Issue Date: 01 September 2003
- DOI: https://doi.org/10.1038/ng1227