Global patterns of human mitochondrial DNA and Y-chromosome structure are not influenced by higher migration rates of females versus males (original) (raw)
Differences in migration rates between males and females can be inferred from analyses of within- and between-group variation (e.g., FST, a measure of genetic differentiation among populations) for the maternally inherited mtDNA and paternally inherited Y chromosome. Using this approach, one study pieced together data sets from published mtDNA, Y-chromosome and autosomal studies and found that the Y chromosome had larger between-group differences than did other portions of the genome1. But these data sets varied considerably with regard to sample sizes, populations represented and method used to assay genetic variation4, making any comparison of the extent of genetic differentiation between populations difficult. This problem is exacerbated for Y chromosomes because they lack sequence diversity5. To accommodate this difficulty, Y-chromosome researchers have adopted a strategy6,7,8,9 to estimate levels of between-group variation using single-nucleotide polymorphisms (SNPs) discovered in small panels of globally diverse males and then genotyped in much larger population samples (e.g., the global data set10 analyzed in ref. 1). This nonrandom sampling of SNPs can result in an ascertainment bias that has an unknown effect on estimates of FST values. Several researchers have suggested that an extensive study of mtDNA and Y-chromosome diversity in the same samples, using the same method to assay variation, is necessary before one can make firm conclusions regarding the relative degree of mtDNA and Y-chromosome differentiation4,11.
Here, we directly compare 6.7 kb of Y-chromosome sequence and 770 bp of the gene mitochondrial cytochrome c oxidase 3 (MTCO3) in a sample of 389 individuals representing ten globally distributed human populations. We assayed Y-chromosome variation using DNA sequences that encompass recently inserted Alu retrotransposons (e.g., the Y family of Alu elements and its subfamilies12; Table 1), because these elements may have a higher mutation rate than other noncoding DNA as a result of their high density of CpG dinucleotides13. Focusing on these regions, we uncovered a much higher density of SNPs than reported in previous surveys. SNP density was 3.15 times higher in our data set than in a study of a pseudogene and other noncoding regions on the Y chromosome5 (comparing data from 73 individuals that overlap between the two studies). Given the value of Watterson's θ (an estimator of the quantity 2Neμ, where Ne is the effective population size and μ is the mutation rate) in our present Alu data set, we estimated the probability of observing a SNP density equal to or lower than that observed in the non-Alu data set to be P = 0.007 using coalescent simulations. Thus, there is a significant increase in DNA sequence variation in regions encompassing Alu elements over the background level of noncoding diversity on the Y chromosome. Using this rich source of polymorphism, we can directly compare sequence variation between the Y chromosome and mtDNA in a manner that is free of ascertainment bias for both loci. We observed 43 Y-chromosome SNPs and 68 mtDNA SNPs (Supplementary Tables 1 and 2 online).
Table 1 Characteristics of individual Alu elements sequenced on the Y chromosome
We found no evidence that the Y chromosome has a higher level of differentiation between populations than does mtDNA. Using an analysis of molecular variance (AMOVA), we calculated the overall value of ΦST (which approximates the quantity 1/(1 + Ne_m_) assuming an equilibrium island model of population structure14, where m is the migration rate between populations) to be 0.334 for the Y chromosome and 0.382 for mtDNA. We examined distinct geographic regions individually and observed the same pattern of slightly higher ΦST values for mtDNA than for the Y chromosome (Table 2). The between-groups component of genetic variation was higher for mtDNA than for the Y chromosome in every region except Asia, where the Y-chromosome ΦST value slightly exceeded that of mtDNA (by 0.002). An AMOVA that incorporated a hierarchical grouping of populations within continents yielded similar results (Table 3). Values of ΦSC (between-group, within-continent variation) and ΦCT (between-continent variation) were similar for mtDNA and the Y chromosome, though slightly higher for mtDNA.
Table 2 AMOVA results for the Y chromosome and mtDNA at global and continental scales
Table 3 AMOVA grouping populations by continent of origin
In addition to the overall similarity of between-group components of variation for the Y chromosome and mtDNA, there was a strong and statistically significant correlation of ΦST values between pairs of populations for these loci (Fig. 1; Mantel correlation coefficient = 0.688; P < 0.001). This result has several important implications. First, it indicates that putative gene flow among populations is relatively symmetrical for females and males. The fact that ΦST values for these loci covary so strongly suggests that population-specific processes, such as variation in rates of migration for females versus males, do not influence our divergence data. Second, it indicates that there is no obvious trend towards different rates of divergence for mtDNA versus the Y chromosome. Although the nonindependence of the data points in Figure 1 precludes conventional statistical analyses, we noted that the slope of the regression line suggests a faster increase in divergence between populations for mtDNA than for the Y chromosome, contrary to the pattern that would be expected if females had a higher rate of migration. Finally, the strong correlation between ΦST values for these two compartments of the genome indicates that demographic, rather than locus-specific, evolutionary forces are the primary determinants of genetic distance between the populations we surveyed. Positive directional selection, for instance, operating in a subset of populations on either the Y chromosome or mtDNA would tend to uncouple ΦST values between loci. We see no evidence for such a process in our data.
Figure 1: Pairwise mtDNA and Y-chromosome ΦST values.
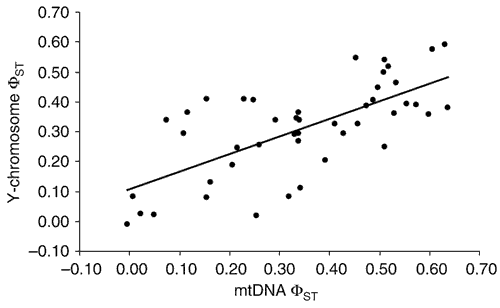
A Mantel test indicates that there is a significant correlation between pairwise mtDNA and Y-chromosome ΦST values (Mantel correlation coefficient = 0.688; P < 0.001). Although nonindependence of data points precludes statistical analysis of the regression line, mtDNA ΦST values seem to increase more quickly between populations than do those for the Y chromosome (regression equation, ΦST(Y) = 0.589 × ΦST(mtDNA) + 0.107).
Our survey of the Y chromosome and mtDNA found markedly different between-group components of variation than have been reported in previous global studies. Some Y-chromosome studies rely on predetermined SNPs and find that between-population components of genetic variation are much higher than we estimated1,11. Our data suggest that ascertainment biases associated with the use of particular SNPs may result in overestimates of genetic distance between populations. In contrast, previous studies of mtDNA often focus on hypervariable portions of the control region (where high mutation rates may cause a downward bias in estimates of between-group variation15) rather than coding DNA11. We obtained much higher estimates of between-group variation when we compared mtDNA coding sequences with hypervariable regions in the same panel of individuals (Supplementary Note online).
Our interpretation of the between-group components of genetic variation for the Y chromosome and mtDNA in terms of rates of migration relies on the assumption that the effective population sizes of the sexes (and thus of the Y chromosome and mtDNA) are equal. Among human populations, forces that skew the breeding sex ratio probably do so by increasing the number of females relative to males (e.g., owing to the widespread practice of polygyny and rarity of polyandry among cultures16,17, a higher variance in male lifetime reproductive success18 or higher rates of male mortality19); the magnitude of this skew is not known. If the effective size of the human female population is indeed somewhat larger than that of males, then our observation of roughly equal between-group components of variation for the Y chromosome and mtDNA implies a lower rate of migration for females than for males among the widely spaced populations we surveyed.
We did not detect the signature of a higher migration rate among populations for females than for males in our global survey, but this does not contradict the evidence for patrilocality effects at local scales. For instance, in a comparison of genetic variation in Northern Thailand, patrilocal villages were characterized by lower levels of variation for the Y chromosome than for mtDNA and higher Y-chromosome genetic distances between villages, whereas the opposite was true among matrilocal groups. Similar patterns were observed among patrilocal Bedouin tribes from the Sinai Peninsula3. One of the outstanding questions raised by studies such as these is the extent to which local cultural practices influence genetic patterns at the regional and global scale4. At present, there are too few studies that specifically examine these issues of scale with respect to Y-chromosome versus mtDNA differentiation to draw firm conclusions. But our results, taken together with several regional-scale studies that did not detect a genetic signal of increased migration among females versus males20,21,22, suggest that broader-scale genetic patterns may not always reflect the sum of local cultural processes. This may be because other demographic events (e.g., long-distance migrations) become proportionately more important at larger geographic scales, or because behavioral customs of individual populations do not have the temporal or geographic stability necessary to influence global patterning. Although we are unable to distinguish among these hypotheses, our results suggest that the role of female migration is no more important than that of male migration at the continental and global scales.
Note: Supplementary information is available on the Nature Genetics website.