From signatures to models: understanding cancer using microarrays (original) (raw)
Genomics provides powerful tools with which to probe the components and behavior of biological systems. Microarrays, high-throughput chromatin immunoprecipitation1,2 (ChIP) and tissue microarrays3 inform us on different perspectives of the molecular mechanisms underlying cellular functions. The staggering volume of molecular data resulting from the rapid adoption of such techniques has underscored the importance of computational analysis as a key link between data generation and the formulation of new hypotheses. It is widely believed that genomics will transform our understanding of the mechanisms underlying the function of cells and organisms, and revolutionize the diagnosis and management of disease by offering an unprecedented comprehensive view of the molecular underpinnings of pathology4,5. Gene-expression profiling has been applied extensively in cancer research. Gene-expression microarrays have been analyzed using clustering algorithms that group genes and samples on the basis of expression profiles, and statistical methods that score genes on the basis of their relevance to various clinical attributes (Supplementary Note online). Using these methods, investigators have identified new classes of hematological malignancies, predicted prognosis in lung cancer and breast cancer and made many mechanistic observations (Supplementary Fig. 1 online). Despite the natural caution associated with the implementation of new technologies in the clinical arena, the utility of the results of microarray analysis as an effective diagnostic tool at the point of care is already being assessed6.
Approaches such as clustering and identification of gene signatures, though successful, tend to ignore much of the signal in the data, both in genes whose activity changes but does not pass the threshold for differential expression and in genes that are differentially expressed but unfamiliar to the researcher analyzing the list. Furthermore, because these analyses are done at the gene level, they are prone to the inherent noise that exists both in the sample population and in different stages of assaying gene expression. Moreover, simply listing genes associated with a certain tumor type is far from identifying the biological processes in which these genes are involved. Finally, clustering genes with similar expression patterns does not identify the causal molecular mechanisms that regulate them. Therefore, developing analysis methods that can extract a more biologically meaningful understanding of the processes giving rise to cancer is a key challenge. Here, we focus on ongoing research that attempts to achieve this goal, discuss challenges in its application to complex multicellular tissues and conclude with some opportunities for using these methods to improve cancer diagnosis and treatment.
A module-level view
To transcend from individual genes to biological processes, several recent methods7,8,9,10 use gene modules as the basic building blocks for analysis. These methods aim to distill a higher-order and more interpretable characterization of transcriptional changes. Moreover, by considering coherent changes in expression in larger modules, we can identify patterns that are too subtle to discern when considering expression profiles of individual genes in isolation.
Mootha et al.8 (Fig. 1a) tested biologically coherent sets of genes (e.g., pathways) for association with disease phenotypes. They applied their method to a data set of human diabetic muscle, with the goal of identifying processes that were systematically altered in diabetic muscle. Their analysis showed that, by examining the joint behavior of a set of genes, they could detect significant changes even in cases where the expression of individual genes was not significantly different. It was only in the coherent signal associated with a higher-level entity that the pattern was evident.
Figure 1: Module-level analysis.
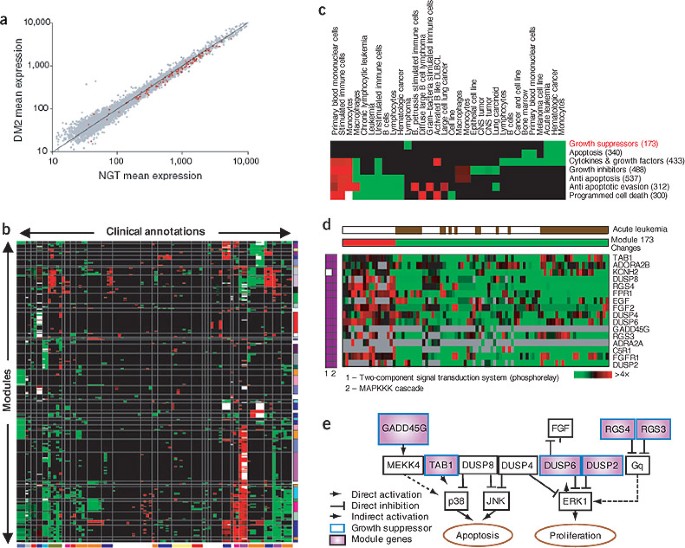
(a) Example from the gene-set enrichment analysis method of Mootha et al.8 showing that the expression of oxidative phosphorylation genes is reduced in diabetic muscle. The mean expression of all genes (gray) and of the oxidative phosphorylation genes (red) is plotted for individuals with type 2 diabetes mellitus (DM2) versus those with normal glucose tolerance (NGT). The individual genes in the set changed only modestly and could not be identified using standard analyses for differentially expressed genes. But the pattern over the set as a whole is statistically significant. (b–e) These panels illustrate how module maps7 suggest new functional roles for specific proliferation and apoptosis genes in acute leukemia. (b) The cancer module map of Segal et al.7, shown as a matrix of modules (rows) versus array clinical conditions (columns), in which a red (or green) entry for module m and condition c indicates that the arrays in which module m was significantly induced (or repressed) contained more arrays from condition c than would be expected by chance. The intensity of the entries corresponds to the fraction of arrays in the module from condition c that were significantly induced (or repressed). White entries indicate that both the induced and repressed arrays were significant for the given annotation. The rows and columns of the matrix were each clustered into distinct clusters, and the resulting clusters are indicated by vertical and horizontal lines. Related conditions (or modules) are often clustered together in the module map. But many modules are shared across conditions, indicating that tumors are characterized by combinations of a small number of shared and unique modules. (c) Submatrix of the full map in b for related growth-regulatory processes. These modules are mostly used by hematologic malignancies. In most cases, a particular condition shows either uniform induction or repression of most growth-modulating modules, both apoptotic and antiapoptotic, indicating a complex response. (d) The Growth Inhibitory Module (highlighted in red in c). Shown are all arrays in which the module's genes change significantly, and the direction of change (induction or repression) in each such array is indicated (middle; red or green, respectively). Gray pixels represent missing values. The arrays corresponding to acute leukemia are indicated by brown pixels in the top row. The membership of the module genes in the two gene sets from which the module was generated is shown (left, purple pixels). (e) Growth Inhibitory Module genes (purple) in the context of the MAPK pathways of proliferation and apoptosis (as compiled from known interactions in the literature). Most module genes are known to inhibit cell growth (bold blue border). Some are known to directly or indirectly repress ERK1, an activator of cell proliferation known to be constitutively active in acute leukemia. Others are known to activate the apoptosis repressor p38. Thus, the concerted downregulation of these growth suppressors may allow ERK1 and p38 to escape regulation, leading to uncontrolled proliferation and reduced cell death. Only DUSP2 was previously implicated in acute leukemia; other module genes are new potential targets.
Segal et al.7 applied a module-level analysis to obtain a global view of the shared and unique molecular modules underlying human cancer. They compiled a 'cancer compendium' from multiple studies and a large collection of biologically meaningful gene sets from experimental studies and human-curated annotations. They identified gene sets with similar behavior across arrays, combined them into modules and used these modules to characterize a variety of clinical conditions (e.g., tumor stage and type) by the combination of activated and deactivated modules. In the resulting 'cancer module map'7 (Fig. 1b), the activation or repression of some modules (e.g., cell cycle) was shared across multiple tumor types and could be related to general tumorigenic processes, whereas others (e.g., growth-regulatory modules; Fig. 1c) were more specific to the tissue origin or progression of particular tumors. Conversely, the module map characterized each condition by a particular combination of module activity, providing insight into the mechanisms underlying specific malignancies. For example, the Growth Inhibitory Module (Fig. 1d) consisted primarily of growth suppressors coordinately repressed in a subset of acute leukemia arrays and suggested a possible explanation for the uncontrolled proliferation and reduced cell death in these tumors (Fig. 1e). Other modules were shared across a diverse set of clinical conditions, suggestive of common tumor-progression mechanisms. For example, a bone osteoblastic module, spanning various tumor types, included both secreted growth factors and their receptors, suggesting a single mechanism for both primary tumor proliferation and metastasis to bone.
These results and others8,9,11,12,13 illustrate the value of analyzing complex processes such as tumorigenesis in terms of higher-level gene modules and biological processes. This type of analysis increases our ability to identify the signal in microarray data and provides results that are more interpretable than gene lists. In particular, when grouped together into a coherent module, the functional and clinical effects of pleiotropic genes might become more apparent, as would the complexity of the mechanism that has to be addressed therapeutically (Fig. 1e). Finally, a modular approach can be applied uniformly to multiple data sources from different tumor types, thereby uncovering the commonalities and differences of multiple clinical conditions.
From modules to regulatory mechanisms
The characterization of cancer processes in terms of transcriptional changes in genes or modules is only a step towards the goal of obtaining a detailed mechanistic model of the processes leading to malignancy. Recent work attempts to use gene expression and other genomic data to understand regulatory interactions between genes and how these might result in tumorigenesis.
Cellular processes are regulated by a variety of mechanisms, occurring at every step in the process of going from DNA to functional proteins. Transcriptional regulation, directly observed in gene-expression data, controls the production of mRNA transcripts. Important components in this process are _cis_-regulatory elements in a target gene's promoter region, _trans_-acting factors that bind to these DNA motifs and signaling molecules that modulate this process based on exogenous and endogenous signals. Genomic data sets offer (noisy) views of different facets of this process. Protein-DNA binding events are directly observed in ChIP-chip assays14,15. We can computationally detect cis elements in promoter sequences, on the basis of experimentally determined sites16, de novo identification or evolutionary conservation17,18. Finally, similar expression profiles allow us to identify target genes that are controlled by a shared regulatory mechanism.
Most attempts to identify regulatory relationships from genomic data have focused on the unicellular yeast Saccharomyces cerevisiae. One focus aims at reconstructing _cis_-regulatory circuits19, including identifying new cis elements, detecting their targets and identifying combinations of elements that modulate expression of a target gene. Because signal at the level of individual genes is often hard to detect, most approaches focus on regulatory modules, whose member genes are expected to be controlled by similar regulators in a similar way20,21. Early approaches identified new individual cis elements that are enriched in clusters of coexpressed genes19, or pairs of elements that act in synergy under specific conditions22. Recent extensions increase accuracy by using other sources of data, such as regulator binding (from ChIP-chip assays)21 or evolutionary conservation23,24.
More recently, several studies25,26,27,28 have attempted to identify how the set of _cis_-regulatory elements in a gene's promoter governs its behavior and explains the observed expression pattern. Segal et al.26 proposed a model of regulatory modules in which module genes shared both a similar expression profile and a similar profile of cis elements. Thus, a gene's cis element profile determined its module assignment and hence its expression profile. Beer and Tavazoie27 subsequently proposed a similar approach, which also included a finer-grained model of promoter configuration. Both groups showed that a substantial fraction of the signal in gene-expression data could be explained in terms of _cis_-element profiles, and that these profiles exhibited an interesting combinatorial organization of elements into various logic gates (OR, AND) and spatial configurations (Fig. 2a). This general framework can also accommodate transcription factor–binding data instead of (or in addition to) cis elements. For example, Bar-Joseph et al.28 identified gene modules whose expression could be explained by a shared transcription factor–binding profile (Fig. 2b), and Segal et al.25 combined expression, sequence and transcription factor–binding to identify combinations of transcription factors, their target modules and the cis elements that mediated this regulation.
Figure 2: Computational prediction of _cis_-regulatory networks.
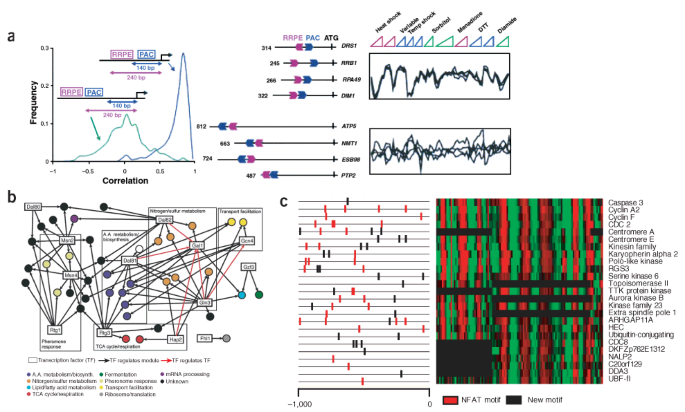
(a) One of the _cis_-regulatory modules produced by the analysis of Beer and Tavazoie27. The module is defined in terms of a coherent expression signature and is enriched for genes involved in ribosomal RNA transcription and processing. Its _cis_-regulatory profile is defined by the presence of two computationally discovered sequence elements, PAC and RRPE, in a particular positional configuration on the chromosome. Genes containing both elements in the correct configuration are tightly coregulated, whereas in genes containing only one of the two elements, or containing both elements in a different positional configuration, the distribution of correlations is close to random. The distribution of correlations (left), as well as examples of genes that do (top right) and do not (bottom right) satisfy the positional constraint, along with their expression patterns, are shown. (b) Transcriptional gene-regulation network under response to rapamycin, produced by the analysis of Bar-Joseph et al.28. The network was derived from both gene expression and ChIP data under rapamycin. The analysis resulted in 39 modules (circles) regulated by 13 transcription factors (black arrows). Red arrows between transcriptional regulators indicate that the source transcription factor binds at least one module containing the target transcription factor. The analysis resulted in new predictions regarding transcriptional regulation of the response to rapamycin, for example the regulation of nitrogen metabolism modules by Hap2. (c) One of the modules resulting from applying the method of Segal et al.26 to combined human promoters (1,000 bp upstream of predicted transcription start sites) and measured expression of human cell cycle in HeLa cells62 (E.S. & D.K., unpublished result). Shown is the expression (right) and promoter region (left) of each gene in the module. The genes assigned to this module are known to be involved in mitosis (10 of 25 genes, P < 10−9), and one of the motifs automatically identified by the method is the known binding site for the nuclear factor of activated T-cells (NFAT; red motifs), previously reported to have a role in cell-cycle progression63,64.
Despite these successes in model organisms, this approach has yet to be broadly applied in multicellular organisms. In particular, most current methods for detecting cis elements are not well suited to the large, complex genomes and long intergenic regions typical of mammals. Nevertheless, several researchers have identified regulatory circuits in expression data from synchronized HeLa cells26,29, both finding known cell cycle–regulatory elements and targets, and suggesting new ones (Fig. 2c). Some of the more successful approaches rely on additional signals, such as evolutionary conservation30, spatial clustering of cis elements in the DNA sequence30,31,32,33,34 or a global model of cis regulation and gene expression26, to improve the detection of reliable biological signals.
A complementary approach focuses on the transcription factors and signaling molecules that modulate gene expression either directly or indirectly. Although regulator activity is not observed directly, if a regulator is itself transcriptionally regulated, its expression level can serve as a proxy for its activity, allowing us to infer regulatory interactions correctly from expression profiles. Motivated by this insight, several studies35,36,37 propose algorithms that construct a Bayesian network describing the probabilistic dependencies between the expression levels of genes. These methods can detect both direct and indirect regulatory relations (e.g., between a MAP kinase and its downstream targets). Recent work38,39 extends this approach by using more realistic models of binding affinity between transcription factors and binding sites, in accordance with biochemical principles40.
A recent extension is based on the observation that many regulatory interactions are shared by all members of a gene module20,39. Segal et al.41 proposed the module-network approach for identifying modules of coregulated genes and their shared regulation program, which specified the expression profile of a module's genes as a function of the expression of the module's regulators. As with the identification of cancer modules and cis elements, this higher-level analysis improved both statistical robustness and biological interpretability. This approach was successfully applied to a yeast expression data set, identifying functionally coherent modules and known regulatory relations (Fig. 3a). It also suggested testable hypotheses regarding the role of transcription factors and signaling molecules, three of which were tested and validated experimentally.
Figure 3: Computational prediction of _trans_-regulatory networks.
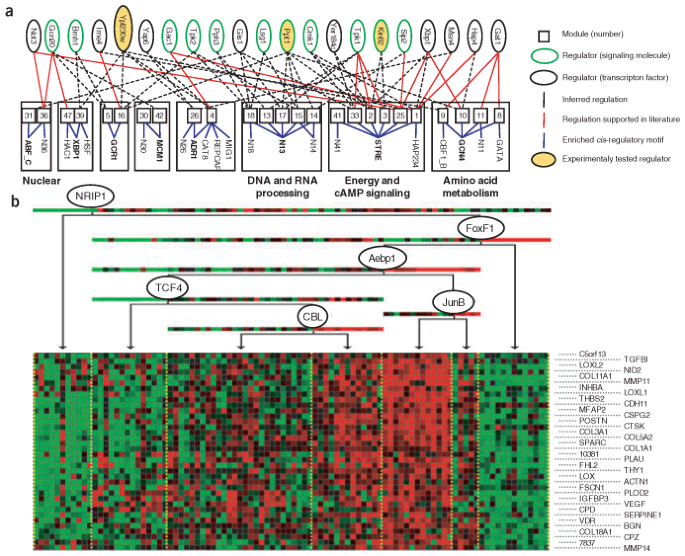
(a) Global module network for yeast stress data, derived by Segal et al.41. The graph depicts inferred modules (middle, numbered squares), their significantly enriched _cis_-regulatory motifs (bottom) and their associated regulators (top, black-bordered ovals for transcription factors, green-bordered ovals for signal-transduction molecules). Modules are connected to their significantly enriched motifs by solid blue lines. Module groups, consisting of sets of modules that share a common motif, and their associated motifs are enclosed in bold boxes; module groups whose modules are functionally related are labeled (right). Red edges between a module and a predicted regulator are supported in the literature. Three regulators, marked in yellow, correspond to previously uncharacterized regulators whose predicted role was validated experimentally41. Modules belonging to the same module group seem to share regulators and motifs, with individual modules having different combinations of these regulatory elements. (b) One of the modules identified by the module network analysis of the lung cancer data43 (N.K., E.S., N.F., A.R., & D.K., unpublished result). According to known databases, 17 of 36 of the genes in this module belong to extracellular matrix–related annotations (P < 9 × 10−10); when we manually curated the genes in this module, we found at least nine additional genes associated with fibrosis and TGFβ signaling. The genes in this module are characterized by having the lowest expression levels in normal lung, with higher expression in individuals who died with the disease. The module network procedure predicts that this module is regulated by Jun-B, a TGFβ-regulated transcription factor, and TCF4, the WNT–β-catenin target transcription factor65. The genes are overexpressed when Jun-B is underexpressed, consistent with reports suggesting that loss of Jun-B activity enabled epithelial mesenchymal transition in tumors66.
A key limitation of such approaches is that many regulators are regulated post-transcriptionally, and their activity is undetectable in gene-expression data. Nevertheless, in the context of tumorigenic processes, there is reason for optimism. Tumorigenesis often arises from some change to a cell's DNA, which in turn results in a perturbation in expression of certain key regulators. For example, the Myc oncogene is amplified in many tumors, resulting in a concomitant change in the expression of its targets42. Thus, even regulators that, under normal conditions, are regulated post-transcriptionally may undergo transcriptional regulation in tumor cells, making the regulatory processes more apparent in expression data.
Encouraged by this observation, we applied the module network procedure to a data set of lung cancer arrays43, focusing on regulation by transcription factors. In addition to the usual cancer-related functional categories (cell cycle, DNA and RNA repair, and metabolism), we found multiple modules enriched for genes associated with extracellular inflammation, immunity and extracellular matrix, processes that are increasingly recognized to be important in tumor generation and progression44,45. An in-depth analysis of one of the modules (Fig. 3b) suggested that extracellular matrix–related genes, whose expression is often increased in tumors, were not mere representatives of stromal activity but were related to tumor clinical biology and were tightly regulated by cancer-relevant transcription factors. This example illustrates the potential of this approach for identifying transcriptional regulation in complex tissues; it also shows how an unbiased discovery approach can lead the observer to unexpected conclusions (such as the possible role of fibrotic and inflammatory modules in cancer).
Comparative analysis
Taking a more global view, we can extend our analysis to encompass multiple studies across diverse organisms and conditions. In such comparative analysis, conserved patterns can help to identify true biological signals and key mechanisms, and highlight commonalities and differences. This approach is particularly compelling when applied to the available data from an increasing number of mammalian species and of animal models of cancer.
Several works have explored the conservation of coexpression relationships and gene modules across a diverse range of organisms46,47,48. These works showed that conserved coexpression relationships were more likely to correspond to true functional interactions (Fig. 4a) and allowed us to study the change in the role of functional modules over evolution (Fig. 4b). This analysis can highlight functional modules that have a key role in a process of interest. For example, McCarroll et al.48 identified a common expression signature in aging between flies and worms, which included genes involved in mitochondrial metabolism, DNA repair and cellular transport.
Figure 4: Multispecies analysis of gene expression data.
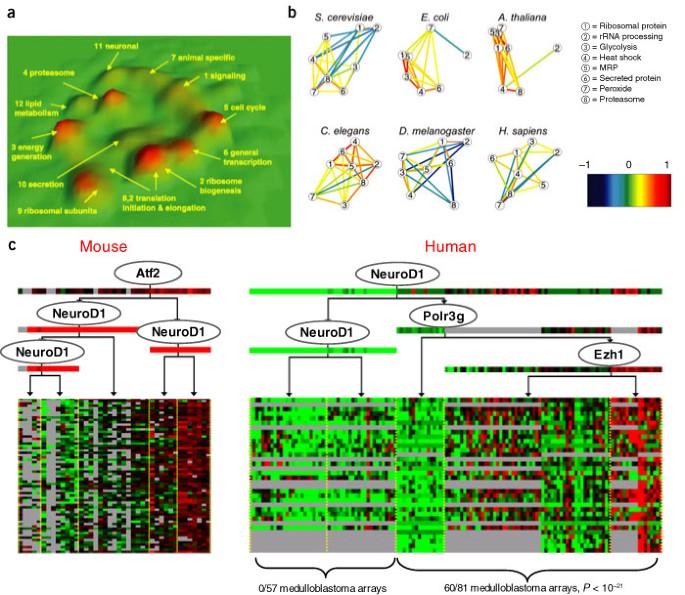
(a) Three-dimensional representation of a multispecies coexpression network produced by Stuart et al.46. The network links metagenes (sets of orthologous genes) across four diverse organisms (human, fly, worm and yeast) that are coexpressed in at least two organisms. The network is visualized as a terrain map, in which highly correlated metagenes are placed in proximity in the _x_-y plane, and the density of genes in a region is shown by the altitude in the z direction. The visualization uncovers 12 components of highly interconnected metagenes, which were enriched for metagenes involved in similar biological processes. Links in the network suggest potential interactions between genes that have been conserved across evolution and are therefore likely to correspond to functional relationships. Stuart et al.46 showed that the network can be used to predict a gene's function, by labeling it with the annotations of its neighboring genes in the network. They show that, for most functional categories, the multispecies network performed significantly better in making such predictions than any single-species coexpression network. (b) Analysis of interaction between eight transcriptional modules across six organisms, as derived by Bergmann et al.47. Eight modules, whose function is known in yeast, were used to generate corresponding homolog modules in five other organisms. Correlations between the module-expression profiles were computed for all pairs of modules. Modules are shown as circles; significant correlations between them, by colored lines and by distances between the modules. Most of the relations between modules differ among organisms. For example, heat shock and protein synthesis are anticorrelated in yeast and fly and correlated in the other four organisms. (c) One of the modules learned in a joint model over expression profiles from normal mouse samples and human brain tumors67 using a multispecies extension to the module network framework of Segal et al.41 (E.S. & D.K., unpublished result). The module was found in both human and mouse and included 34 orthologous genes from the two organisms. Most of the module genes were previously shown to be expressed in brain68 (18 of 34, P < 10−12), supporting their combination into a single module. Furthermore, a conserved regulatory role was predicted for the neurogenic differentiation factor NeuroD in both human and mouse (ovals in regulation programs). The expression of NeuroD splits the human arrays into two groups; all 60 medulloblastoma arrays in the data set are in the group in which NeuroD is overexpressed, a finding supported by the suggested role of NeuroD in medulloblastoma tumors69.
Applying a similar approach to cancer data from mouse and human can shed light on the mechanisms underlying tumorigenesis. For example, Sweet-Cordero et al.49 used three different mouse models of lung cancer to identify signatures of specific genetic alterations that lead to tumorigenesis. They projected the genes in each signature to their human orthologs and used a gene-set–based method8 to test for activity of these signatures in different human lung tumors. This design used changes observed in controlled manipulations in mouse disease models to draw insights about disease manifestations in humans.
This approach transfers results of an analysis done in mouse to inform a subsequent analysis in human; we can also carry out a joint analysis that explicitly searches for patterns conserved across multiple species. Along these lines, we analyzed a human-mouse data set of normal and tumor brain tissue using an extension50 of the module network approach41. This analysis suggested regulatory modules that were conserved across human and mouse, and proposed new hypotheses regarding regulation in medulloblastoma (Fig. 4c).
Challenges and opportunities
The reconstruction of the molecular mechanisms that underlie a complex process, such as tumorigenesis, is a formidable challenge. This challenge arises in part from difficulties associated with microarray assays, including noise in the data and limited reproducibility across platforms and researchers51,52. Moreover, most analyses implicitly treat mRNA expression as a surrogate for protein activity level, an assumption that does not account for processes such as mRNA stability, protein degradation and post-translational modifications. In addition, when we attempt to find complex patterns in data, we invariably encounter multiple alternative explanations of the data (e.g., clusters, regulatory modules, etc.). Therefore, the results of such analysis are sensitive to the choice of model and parameters and the specific data used, and must be interpreted with care.
Nevertheless, the successes obtained by combining genomic techniques and computational algorithms to reconstruct networks (albeit primarily in model organisms) are encouraging. Three recurring themes form the basis for this success. The first is the analysis of data at the level of biological modules, rather than individual genes, an approach that produces results that are biologically interpretable and statistically robust. The second is the use of biological knowledge in developing analytic techniques, either directly (e.g., to define functionally coherent gene sets) or indirectly (e.g., to construct biologically realistic models). As we create more realistic biological models, we can hope for better biological understanding and more focused predictions to inform further experiments. The third theme is the integration of multiple sources of data in the analysis. By putting together different partial (and noisy) views of a single complex process (gene expression, promoter sequences, protein-DNA binding, protein-protein interactions and more), we can often obtain a much more accurate and complete picture. In addition, by considering data from different conditions or cell types, we can obtain a more global understanding of the function of the same set of building blocks in different contexts. Finally, the integration of data across organisms allows us to identify functional components based on their conservation and, conversely, to recognize the mechanisms that are the basis for biological diversity.
Although genomic approaches are prevalent in cancer research, we are still far from reconstructing molecular mechanisms in human cancer. In fact, the methods we describe do not always scale easily to mammalian systems. Unlike yeast genomes, mammalian genomes are less compact, and enhancers are more dispersed and remote. Both regulatory and signaling networks are larger and more elaborate, and the control of many genes and processes involves undefined epigenetic mechanisms, a higher degree of combinatorial regulation and multiple signaling pathways. Furthermore, many interactions are context-specific, as different components of the molecular network are active in different cellular states and phenotypes.
Much of the added complexity in applying genomic analysis to cancer is related to multicellularity, which can confound the analysis of data from tissue samples that contain heterogeneous population of cells. Most genomic techniques measure an average signal in a sample from a cell population. This is a concern even when studying unicellular organisms or cell cultures as the averaging process tends to obscure variations between cells53,54,55. When analyzing a heterogeneous tissue, this problem is more pronounced because the signal for different cell types is obfuscated; differential regulation of genes associated to changes in cell state can be hard to distinguish or can even disappear entirely. Moreover, the averaging effect introduces an additional source of noise as the proportions of the different cell types are typically different across samples. This variability may swamp the variability resulting from other, perhaps more relevant, differences between the samples. Another, more challenging issue is raised by intercellular signaling in tissues. Interactions between cells often lead to complex behaviors, which are hard to distinguish from the regulatory processes in the cell itself and cannot be emulated in in vitro cell-culture assays. Finally, this epigenetic variability is further confounded in tumor samples, where considerable genetic variability occurs between and within samples.
In light of these challenges, is there hope for systematic mechanistic insights from genomic and computational studies? We believe that a positive answer lies in the combination of computational and experimental insights. Computational methods should be developed to tackle cell and tissue heterogeneity56,57. For example, Stuart et al.56 used histological evaluation of tissue heterogeneity to deconvolve expression profiles and identify cell type–specific expression responses. Experimentally, most cancer genomic studies have focused on tumor samples from the human population and have therefore suffered from inevitable confounding genetic and environmental factors, tissue heterogeneity, lack of time courses for disease progression and unavailability of perturbations instrumental in identifying regulatory events. Recent studies9,49,58 suggest that careful design can greatly improve the utility of such studies, by combining the study of human tissue samples with tissue culture and animal models, to obtain a more controlled and comprehensive view. For example, Lamb et al.9 used expression-profiling in a cell-culture model with genetic perturbations to identify a 'cyclin D signature', followed by computational analysis of a compendium of human tumor expression profiles to find the transcription factor that mediated this response in tumors. Similarly, Kang et al.59 found a set of genes involved in osteoclastic metastasis by combining expression profiling on human cell cultures with phenotypic effects in animal models. Finally, new biological assays, such as in situ gene-expression signatures using laser capture microdissection60,61 or fluorescence microscopy data, can provide more refined observations about gene activation in individual cancer cells54,55.
Successful identification of mechanisms from genomic data will also require more sophisticated computational methods. Much progress can be made along the themes of modularity, incorporation of biological knowledge and data integration across techniques, conditions or organisms. It is important to develop methods that combine data across experimental systems that address the same phenomenon (e.g., different cancers in humans or the same cancer type in human and mouse) and isolate key mechanisms and root causes of the disease. The development of such computational methods should go hand in hand with that of multipronged experiments combining cell culture, animal models and human tumor samples.
A major challenge for analysis is the identification of the correct context and functional importance of different events and mechanisms. This issue is particularly pronounced in cancer, in which aberrant and normal processes are intertwined. A cancer cell has a mixture of different processes: processes that are the source for tumorigenesis (e.g., a constitutively active Ras mutant); processes that are normal on their own but are suborned by tumors and support their proliferation (e.g., cell division or angiogenesis); processes that may represent the normal host response to the tumor and may even be protective (e.g., immune response and inflammatory-cell infiltration); and perhaps processes that are simply a by-product of cancer and have no functional role. Although some of the modular approaches outlined above enhance our ability to analyze disease process–relevant signatures, we are still far from understanding the role that these signatures have in cancer. We may be able to derive a more comprehensive perspective on cancer processes by integrating existing assays with histopathologic, clinical and environmental information on the one hand, and with measurements of genetic variation, such as SNPs or DNA copy-number changes, on the other.
Finally, when considering the analysis of cancer data, we must keep in mind that our ultimate goal is to improve diagnosis and treatment of the disease. How can the methods we described above help in achieving this goal? Understanding cancer processes and identifying new drug targets is one contribution, but many of the key regulators and basic pathways of carcinogenesis were identified long before the introduction of high-throughput methods, through the careful hypothesis-based work of molecular and cell biologists. Modular analysis can place the complex interactions of these pathways in the biological context of the tumor microenvironment. Previous analyses may tell us that abnormal WNT–β-catenin pathway activation is important in certain solid tumors and increased activation of EGF receptors is important in others. The results of modular analyses can uncover a certain tumor's use of bone-survival machinery (that promotes bone metastasis) or information about its ability to create a proangiogenic microenvironment or evade immune surveillance; any one of these characteristics is potentially crucial to the disease mechanism and the final outcome for an affected individual.
An understanding of the complexity of the pathways that create and sustain tumors can enable a better use of available therapies by using rational combinations in accordance with the pathways that characterize a certain cancer. Furthermore, a detailed view of the tumor's microenvironment could lead to better design of therapeutic interventions that would help to reverse or contain the carcinogenic process. The availability of multiple secreted and membrane proteins that characterize tumors should allow the identification of combinatorial markers for early detection and noninvasive disease classification, whereas the functional and regulatory characterization of tumors should allow personalized treatment of cancer that is based not on histological appearance but on a global and detailed mechanistic understanding of an individual's disease.
Note: Supplementary information is available on the Nature Genetics website.