Protective immunity and susceptibility to infectious diseases: lessons from the 1918 influenza pandemic (original) (raw)
The very young and the very old are the most susceptible to infectious diseases. One of the main reasons for this is that the young are often immunologically naive and the old are undergoing immune senescence. This pattern of susceptibility is characteristic of most infections (Fig. 1a). These data show the death rate from influenza virus during the years 1911–1915 and illustrate the typical U-shaped curve of mortality as a function of age. These historical data from 1911–1915 highlight the markedly different mortality curve that was observed during the influenza pandemic of 1918 that killed over 50 million people worldwide, making it one of the deadliest plagues ever experienced by mankind (Fig. 1b)1. The most notable difference between the mortality curves of 1918 compared with those of 1911–1915 is that the 1918 pandemic was particularly deadly for young adults between the ages of 18–30, whereas, quite surprisingly, adults in the 30–60-year-old age group fared better. As expected, the very young (<2 years) and the elderly (>70 years) had a high mortality rate. This pattern of susceptibility resulted in the unique W mortality curve of the 1918 influenza pandemic.
Figure 1: Deaths per 100,000 in the United States caused by influenza-pneumonia.
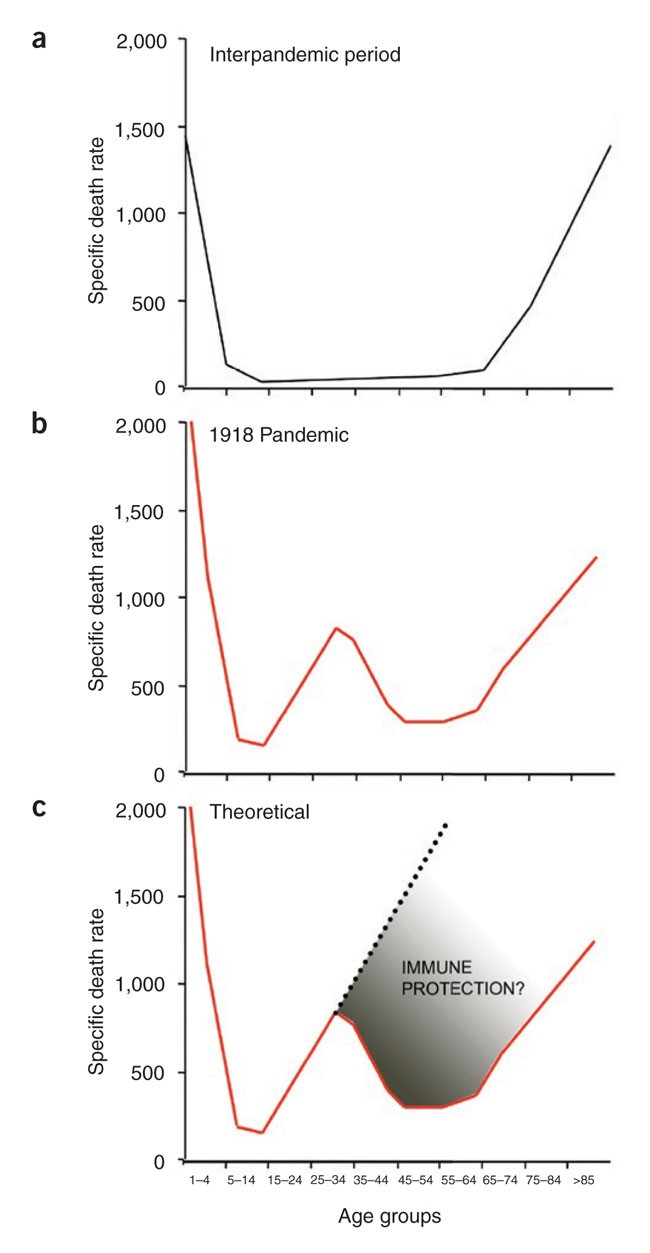
(a) A U-shaped mortality curve was observed for different age groups for the interpandemic period of 1911–1915. (b) A W-shaped mortality curve was observed for the pandemic year 1918. (c) A V-shaped mortality curve might have been observed in 1918, if the population had not been exposed previously (before 1889) to an H1-like influenza virus (the specific death rates were taken from ref. [50](/articles/ni1530#ref-CR50 "Linder, F.E. & Grove, R.D. Vital statistics rates in the United States: 1900–1940. (US Government Printing Office, Washington, D.C., 1947) http://www.cdc.gov/nchs/data/vsus/vsrates1900_40.pdf
.")).There has been much debate about the reasons for this W-shaped curve and why the young adults were more susceptible than the >30-year-old adults. Because many of these deaths were among young men fighting in World War I, it has been suggested that battle conditions (stress, fatigue, chemical exposure, etc.) may have weakened the soldiers' immune systems, thereby increasing their vulnerability to disease. However, similar mortality rates were seen in young men and women not involved in the war. Thus, one must consider the possibility that the >30 year olds may have had some degree of protective immunity against the 1918 influenza virus pandemic strain and that this immunity was lacking in the younger adults (18–30 year olds) (Fig. 1c). In this review, we will address this issue and consider how immunological memory may have shaped the W mortality curve of the 1918 influenza pandemic. We will also discuss why this 1918 pandemic flu strain was so virulent. Finally, we will end on one of the great mysteries of infectious diseases: why did children (ages 4–12) fare much better than young adults did during the 1918 influenza pandemic?
Virulence of the influenza pandemic strain
Influenza viruses belong to the orthomyxovirus family and come in three types: A, B and C. Only influenza A and B viruses are important for causing disease in humans. These viruses have a negative-sense, segmented RNA genome and can code for up to 11 proteins2. By virtue of possessing a segmented genome, influenza viruses can easily reassort (exchange RNA segments between human and animal viruses), and thereby acquire new antigenic properties (antigenic shift). The fact that influenza viruses have an error-prone RNA-dependent RNA polymerase explains the fact that mutations occur frequently and that, through selection, new antigenic variants emerge (antigenic drift). The 1918 virus was responsible for one of the most devastating pandemics in recorded history, and a question of great interest has been why this particular influenza virus strain was so virulent.
A major breakthrough toward addressing this question was made when available pathology materials from patients who had died during the 1918 pandemic were used to obtain the entire sequence of the 1918 virus3 and to subsequently reconstruct the extinct strain in the laboratory using reverse genetics4,5. The virus turned out to be highly virulent in intranasally inoculated mice, with a lethal dose 50 (LD50) that was more than 1,000-fold lower than that of other human (non–mouse adapted) influenza virus strains. In embryonated eggs, the 1918 virus was 106-fold more virulent than the human control strain, as measured by the dose required to kill an 8-d-old embryo, and the 1918 influenza strain grew to titers that were at least one log unit higher than those of control influenza viruses in tissue culture of human bronchial epithelial cells4. Further studies in mice showed excessive immune-cell infiltration in the lung following infection with viruses containing genes from the 1918 strain and higher lung virus titers than in the controls. Specifically, an increased influx of neutrophils and alveolar macrophages and an increase in the production of cytokines and chemokines were observed in lung tissues with a virus expressing only two proteins, hemagglutinin (HA) and neuraminidase (NA), from the 1918 strain6. In mice infected with a virus expressing all eight genes from the 1918 virus, a marked activation of pro-inflammatory and cell death pathways was observed, which was less pronounced in reassortant viruses that only contained a subset of genes from the 1918 virus7. A question of substantial interest is whether the enhanced production of inflammatory cytokines and associated pathology that was seen after infection with the 1918 pandemic influenza virus strain is due to some specific interactions of the viral genes of the 1918 virus with the immune system or if this is primarily a reflection of the rapid growth and spread of this virus. The two possibilities are not mutually exclusive, and it is conceivable that both contribute to the complex pathogenesis that is seen in vivo.
Pathogenicity of the pandemic strain was also studied in the cynomolgus macaque (Macaca fascicularis) model. Macaques infected with the 1918 virus became symptomatic within 24 h of infection and had to be euthanized by day 8 as a result of the severity of the symptoms. The animals showed severe respiratory signs, with an increase in respiration rate and a decrease in lung function, as measured by a decrease in blood oxygen saturation. Also, interleukin-6 (IL-6), IL-8 and the chemokines CCL2 (monocyte chemotactic protein 1) and CCL5 (RANTES) were elevated in infected animals. Notably, compared with a control non-1918 influenza virus, the 1918 virus demonstrated reduced activation of the RNA helicase sensor proteins RIG-I and MDA-5 in infected macaques. These data suggest that the NS1 protein of the 1918 virus, which is an interferon antagonist, has an important immunomodulatory role. By effectively downregulating the innate immune response of the host, the NS1 protein may very well have contributed to the extraordinary virulence of the 1918 virus in humans. Although one can measure the contribution to virulence of individual genes of the 1918 virus (as, for example, in the case of the 1918 virus NS1 gene and the 1918 HA and NA genes), it appears that the interplay—or combination of the natural biological functions—of all eight 1918 genes results in a virus with the highest virulence. Thus, the 1918 virus is a unique influenza virus strain by virtue of its 'matching' genes or because of genes that express viral proteins that affect hundreds of cellular proteins during replication. By the same token, any reassortment of genes in the 1918 virus with RNAs from other influenza viruses has, in most cases, led to a decrease in virulence, highlighting the extraordinary gene constellation of the 1918 virus4,5,6,7.
Genetic variation in influenza virus and immune memory
Prior to addressing the important issue of immunological memory and the 1918 pandemic influenza virus strain, it is essential to first consider the degree of genetic variation in influenza viruses and the epidemiology of the various influenza virus strains that have been in circulation among the human population.
A hallmark of influenza viruses is their ability to undergo genetic shift and drift. Specifically, reassortment can lead to influenza viruses acquiring RNA segments, most likely from avian influenza viruses, that can lead to new pandemic (globally epidemic) strains. The pandemic 1957 strain sported a new HA (subtype 2) and a new NA (subtype 2) and caused worldwide morbidity and mortality. In 1968, a new pandemic strain had only the HA (subtype 3) exchanged, and in 1977 an H1N1 virus appeared that had circulated around 1950 in the human population (Fig. 2). In 1977, it was mostly young people born after the end of the H1 period (1957 and later) that came down with the disease when infected with this new (recycled) virus. Individuals older than 20–25 years of age had ostensibly been exposed to similar H1 strains and were thus partially protected. It is likely that an antigenic shift also occurred in 1918, when an H1N1 virus caused the major pandemic of the 20th century (Fig. 2)4. As for the subtype strain circulating before 1918, only indirect evidence from serologic patient data are available that suggest an H3-like virus circulated in humans starting in 1889 (ref. 8). Viruses circulating before 1889 were postulated to be of the H1 subtype9,10,11,12. In each case in 1889, 1918, 1957, 1968 and 1977, a large segment of the population lacked protective antibodies against these previously unknown (reassortant) viruses, and it is thought that this single antigenic shift is the single most important factor responsible for the outbreaks of pandemics.
Figure 2: Influenza A and B viruses circulating in the human population.
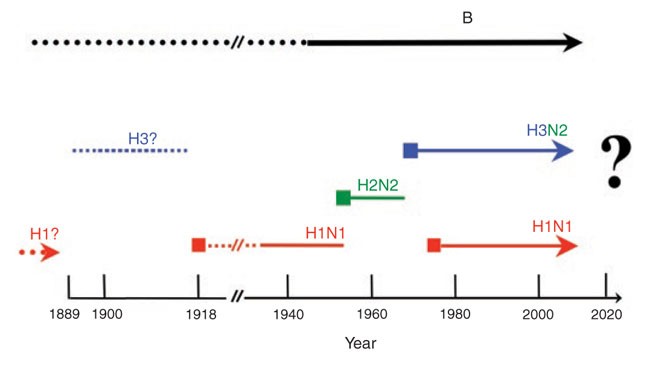
Influenza A viruses with three different hemagglutinin subtypes (H1, H2 and H3) and two different neuraminidase subtypes (N1 and N2) have been identified, and the introductions of these (antigenic shift as a result of reassortment) strains were associated with pandemics. All influenza viruses also undergo continuing antigenic change (antigenic drift as a result of mutation) during interpandemic years. Broken lines indicate that no virus isolates are available from that time period.
However, influenza viruses also undergo antigenic drift and can change their surface glycoproteins by accumulation of nucleotide mutations in the glycoprotein gene. Such drift variants can re-infect and cause disease in individuals who were infected just 2–4 years earlier with a virus belonging to the same subtype. Why influenza viruses undergo antigenic drift remains unclear. Measles and mumps viruses are also negative-sense RNA viruses and their RNA-dependent RNA polymerases are probably as error-prone as that of influenza virus. However, these viruses stay more or less the same antigenically, as evidenced by our present day use of measles and mumps vaccine strains that were first introduced in humans in the 1960s. Although we have no satisfactory explanation for the molecular basis of the continuing antigenic change in influenza viruses, we nevertheless recognize this by changing the vaccine formulation of the three influenza virus components on an annual or biannual schedule. Thus, the trivalent influenza virus vaccine for the 2007–2008 season contains A/Wisconsin/67/2005(H3N2), A/Solomon Islands/3/2006(H1N1) and B/Malaysia/2506/2006) components. As a direct demonstration of the consequences of antigenic drift in influenza, the A/Solomon Islands/3/2006 (H1N1) virus isolated in 2006 replaced the A/New Caledonia/20/1999 (H1N1) virus in the vaccine preparations from the previous seasons; the latter virus was first isolated in 1999, and thus does not adequately protect against the new antigenic drift variants circulating in the human population at the present time.
Immunological memory to the 1918 influenza virus
Why were adults in the 30–60-year-old group more resistant to the 1918 pandemic influenza virus strain than the 18–30-year-old young adults? Did people older than 30 years in 1918 have some level of protective immunity to the influenza virus pandemic strain, and could this immune memory explain the W-shaped 1918 mortality curve?
If, as postulated (Fig. 2), an H3 influenza strain was in circulation from 1889–1918 and H1-type viruses were present before 1889, then people born in or after 1889 would have been immunologically naive to the 1918 H1 pandemic strain (that is, at least to the HA of the 1918 H1 strain). In contrast, individuals born before 1889 (>30 year olds in 1918) would have had prior exposure to H1-type influenza viruses. How would this encounter have resulted in protective immunity 30 years later? The viral proteins that are immunologically relevant for protective antibody responses are HA and, to a much lesser extent, NA13,14,15, both of which are viral surface glycoproteins, and are thus targets for protective antibodies.
Pre-existing antibody is the first level of defense against pathogens, and if there were individuals in 1918 with circulating HA-specific antibody that was reactive against the H1 pandemic strain, then those individuals would have fared better during the pandemic. It is now well-established that circulating antibody can be detected in the serum for decades after acute viral infections and even after some subunit protein vaccines, such as tetanus and diphtheria16,17,18. Thus, it is plausible that some of the individuals in the 30–60-year-old group still had some circulating antibody against the pandemic strain. Several studies have now shown that one of the major mechanisms for maintaining antibody levels in the serum for extended periods of time is the long-lived plasma cell that resides in the bone marrow16,17,19,20,21. Plasma cells are end-stage differentiated cells that constitutively produce antibody in the absence of antigen. Antigen is, of course, needed for the differentiation of naive or memory B cells into antibody-secreting cells, but it is not required for maintaining antibody production by fully differentiated plasma cells. Not all plasma cells are long-lived, but a proportion of these cells can live for extended periods of time in the bone marrow and constitute the major source of long-term antibody production after infection or vaccination. These long-lived plasma cells are not only the major source of antibody in the serum, but can also contribute to antibody in the mucosa by the process of transudation.
In addition to plasma cells, memory B cells can also be involved in protective immunity by making rapid recall responses and producing high-affinity antibody17,22. Memory B cells cannot prevent infection, but can control the spread of virus infection by rapidly differentiating into antibody-secreting cells and producing antibody that neutralizes the virus. A notable feature of the memory B cell response is its longevity. Several studies have demonstrated that memory B cells induced by our commonly used childhood vaccines (tetanus, measles, polio, etc.) persist for years in humans16,17,18,22. In one of the most striking examples, it was shown that memory B cells generated after smallpox vaccination were still detectable 40–50 years after immunization23,24. This is particularly noteworthy, as smallpox was eradicated in the 1970s and smallpox–specific memory B cells were maintained for >30 years in the absence of re-exposure to the pathogen. In light of these extensive studies demonstrating the longevity of human memory B cells, it is very likely that individuals who were exposed to the H1 virus in 1889 or earlier would have still have had some memory B cells that were specific for H1 influenza virus in 1918, and it is possible that these memory B cells also contributed toward protective immunity against the pandemic flu strain.
Immune memory and protective immunity against infectious diseases consist of three key components: pre-existing antibodies in the blood and at mucosal sites, memory B cells and memory T cells. Both CD4+ and CD8+ memory T cells provide a critical second line of defense against pathogens as a result of their higher numbers (compared with their naive counterparts), faster responses (can elaborate effector functions much faster than naive T cells) and better location (present in both lymphoid and nonlymphoid tissues)22,25. Could memory T cells have had any role in protection against the 1918 pandemic strain? Infection with influenza virus generates a broad range of CD4+ and CD8+ T cells that are reactive against most of the viral proteins13, and many of these T cell epitopes are conserved across the various influenza virus strains. Substantial progress has been made in understanding the mechanisms by which influenza virus–specific T cells control infection in the lung (reviewed in refs. 13,26). Also, several studies in animal models have shown that memory T cells do contribute to protective immunity against influenza virus, and there are also human clinical data that are consistent with this notion13,27,28. But given the extreme virulence of the pandemic flu strain and the rapid appearance of clinical disease after infection with this virus, it is unlikely that memory T cells alone would have been of much benefit during the pandemic. However, in individuals that still had residual humoral immunity against the H1 virus, memory T cells could have acted in concert with the H1-specific plasma cells and memory B cells to confer some degree of protection against the pandemic flu strain.
Infectious diseases and the honeymoon period
The influenza epidemic reached Alaska by the end of 1918 and took a terrible toll on the local population. Because of the geographic isolation of Alaska, it is likely that most of the natives had not been exposed to the 1889 H1 influenza virus and, consequently, a large percentage of the local population was immunologically naive. As a result of this, the Alaskan natives showed almost no resistance to the H1N1 pandemic strain, and there were many instances where villages lost their entire adult population (the W mortality curve was not observed among these isolated populations). Notably, the only survivors were the children in some of these villages. A photograph of the 'Flu Orphans' is shown in Figure 3. Why did the children survive and the parents die during this epidemic? This pattern of susceptibility, so dramatically illustrated among the immunologically naive population of Alaska, was also seen in other parts of the world. The general trend was that children between the ages of 4 and 12 showed a substantially decreased mortality rate during the 1918 pandemic (Fig. 1 and Table 1). It should be emphasized that these children were not protected from infection, but, for reasons that are as mysterious today as they were in 1918, they were able to cope with the disease much better than their adult counterparts.
Figure 3: The Flu Orphans.

Children in the remote Alaskan village of Nushagak survived the 1918–1919 influenza pandemic. However, most of their parents and grandparents succumbed to the 1918 pandemic virus, probably because they had not been exposed to an earlier H1-like influenza virus as a result of their geographic isolation. The photograph was taken in the summer of 1919. Printed with permission from the Alaska State Library, Core: Nushagak-People-4, Alaskan Packers Association, PCA 01-2432.
Table 1 The 1918 influenza pandemic: age distribution, immune status and disease susceptibility of the human population
This pattern of disease susceptibility, where children fare better than adults, is not unique to influenza virus and is also seen in other infections. A classic example is that of tuberculosis, where it is well documented that children between the ages of 5 and 14 have a lower clinical case rate compared with any other segment of the population (Fig. 4)10,29. In fact, in the older German literature, this age period (5–14 years of age) is referred to as the 'favorable school age period'. Similarly, morbidity and mortality to several viruses, such as mumps, measles, Varicella-Zoster virus (chicken pox, VZV), poliomyelitis, Epstein Barr virus (EBV) and hepatitis E virus (HEV), are much more pronounced if the infection is acquired for the first time as an adult (or during adolescence) than they are if the infection is acquired as a child29,[30](/articles/ni1530#ref-CR30 "Svitil, K.A. Discover dialogue: virologist David Baltimore. The danger of getting sick from this disease in the United States is trivial. Discover, 1 August 2003 ( http://discovermagazine.com/2003/aug/featdialogue
)."). The severe manifestations of EBV infection (for example, infectious mononucleosis) are rarely, if ever, seen in children. Also, chicken pox is a relatively mild disease, but it can be disfiguring and even life threatening if the infection is first acquired as an adult. It is also worth noting that in the 2003 severe acute respiratory syndrome (SARS) epidemic, the death rate was much lower in children than in adults[31](/articles/ni1530#ref-CR31 "Stadler, K. et al. SARS—beginning to understand a new virus. Nat. Rev. Microbiol. 1, 209–218 (2003)."),[32](/articles/ni1530#ref-CR32 "Finlay, B.B., See, R.H. & Brunham, R.C. Rapid response research to emerging infectious diseases: lessons from SARS. Nat. Rev. Microbiol. 2, 602–607 (2004).").Figure 4: The honeymoon period of tuberculosis.
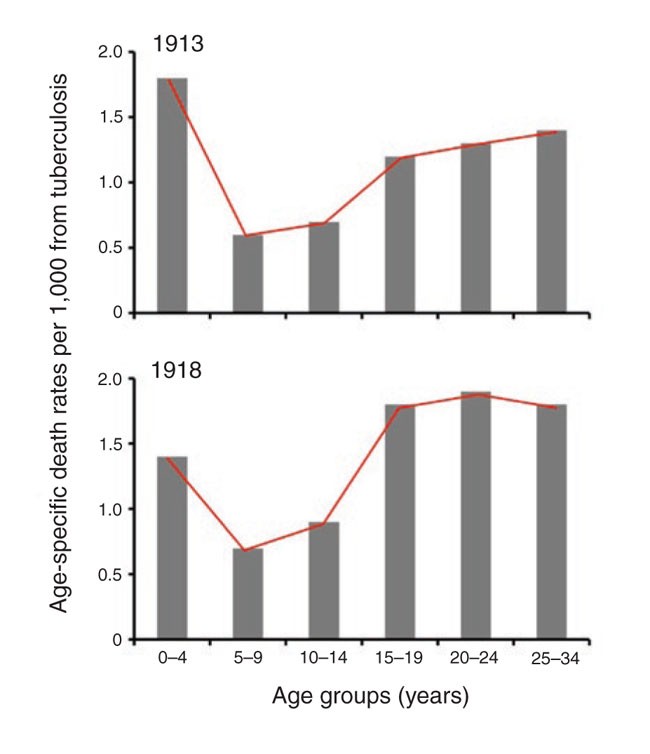
Age-specific death rates from tuberculosis (all forms) in England and Wales for 1913 and 1918. Note that the 5–14-year-old group had a lower mortality rate than the other age groups (data from ref. 10).
Why do children cope with these various infectious agents better than adults? What are the reasons for this honeymoon period with infectious diseases? Disease is usually the result of direct damage to the host by the pathogen. However, the immune response generated against the pathogen can end up causing immunopathological damage and exacerbating the disease33,34. There is a delicate balance between the protective and pathogenic aspects of an immune response, and it is possible that the regulation of this critical balance is different between children and adults such that beneficial responses are favored over harmful ones in children. Given the complex nature of these immune interactions and the fine balance between protective and pathogenic responses, it is possible that even subtle differences in regulation could have profound effects on the clinical outcome.
It would be interesting to see whether there are differences in the generation of regulatory T cells, in the expression of inhibitory receptors such as PD-1, or in the production of cytokines such as IL-10 or IL-17 that modulate immune responses to pathogens in children versus adults following infection35,36,37,38,39,40,41,42. It is also possible that children fare better against infectious diseases because they have a greater regenerative capacity for the immune system—their thymuses and bone marrow more actively produce immune cells—and also for other tissues, thereby resulting in faster repair of damaged organs. It is important to note that this change in disease susceptibility occurs around the time of puberty and it is possible that sex-associated hormones are involved in this transition43.
The outcome of viral infections is greatly influenced by early innate responses: in particular, the production of type 1 interferons that not only provide a critical early check on viral growth, but also activate natural killer cells and enhance the development of specific immune responses44,45. It is conceivable that the type 1 interferon response after viral infection is more efficient in children than in adults. From this perspective, it would be interesting to examine Toll-like receptor or MDA-5/RIG-I expression on dendritic cells from children versus adults and to look at the numbers and function of plasmacytoid dendritic cells, the major interferon-producing cells46,47. Also, it would be useful to quantitate antigen-specific T and B cell responses and to determine if the quality of these specific responses is different between adults and children. In fact, a recent study analyzing the immune response to the human papilloma virus vaccine has shown that pre-adolescent girls (9–12 years old) made higher antibody responses than 18–23-year-old young women48,49 (Fig. 5).
Figure 5: Antibody responses to the human papillomavirus (HPV) vaccine.

Note that 9–12-year-old girls made higher antibody responses than young women (18–23 year olds). The data shown in this figure are for HPV type 6 at 7 months after vaccination. A similar trend in the antibody responses was seen for the other three HPV types present in the vaccine48,49.
Although the phenomenon of the honeymoon period has been recognized for nearly a hundred years, there have been few, if any, studies directly addressing this issue. It is important to try and understand the underlying mechanisms of this pattern of disease susceptibility. It should be possible to address some of the questions directly in human studies, but it will also be necessary to start developing small animal models to carry out more mechanistic studies. Also, valuable information and insight will come from studies in nonhuman primates using the same pathogens that have shown a difference in their ability to cause disease in children versus adults. The knowledge gained from these studies will provide a better understanding of host-pathogen interactions and better prepare us for dealing with future epidemics and emerging infections.