Primer-site SNPs mask mutations (original) (raw)
To the editor:
Sanger-sequencing of diploid DNA from PCR amplicons is the standard resequencing method for targeted mutation detection1. Heterozygosity at polymorphisms within the PCR primer sites causes preferential amplification of the matched chromosome2 and suppresses the signal from an alternate allele on the mismatched chromosome. Analysis of ten resequenced Encyclopedia of DNA Elements (ENCODE) regions3,4 shows that this phenomenon is responsible for a large fraction of missed mutations, most of which can be recovered by doubling PCR amplicon coverage.
The resequencing traces produced by the HapMap project from ten ENCODE regions, together with the chip-based genotypes for many of the discovered single-nucleotide polymorphisms (SNPs) therein3, are a useful reference for assessing the missed heterozygote rate (MHR) of trace-based mutation detection software. While testing our SNP-discovery program POLYBAYES5 on this data set, we uncovered many SNPs with gross discrepancies between the chip-based genotypes and the traces of the same individuals. Specifically, these individuals were heterozygous according to the chip-based genotypes yet only one of the two SNP alleles was detectable in the traces (Supplementary Fig. 1 online). Often, the same SNP was also sequenced from a second, overlapping amplicon, where the traces confirmed the heterozygous genotype. We noticed that a disproportionate fraction of the discre-pant traces were from amplicons whose primers overlapped other SNPs (primerSNPs). To ensure that these discrepancies were not artifacts of our new software, we confirmed the results with another SNP discovery program, POLYPHRED6.
Of all amplicons studied, 15.4% had primerSNPs, and in these amplicons, the missed heterozygote rate (the uncalled heterozygotes in the traces as a fraction of all heterozygous genotypes) was 22.2% (Supplementary Data online and Supplementary Methods online). This is more than twice the 9.1% overall MHR in the entire data set and nearly four times the 6.1% rate in amplicons without a primerSNP. Moreover, amplicons with primerSNPs account for half of all missed heterozygotes. One plausible explanation is that the missed heterozygous individuals are also heterozygous at a primerSNP. If so, the PCR primer would preferentially anneal to and amplify the chromosome with the perfectly matched allele. In the resulting sequence trace, the signal for the allele on the preferentially amplified chromosome will dominate the alternate allele (Fig. 1a). Indeed, the MHR was 58.4% in individuals heterozygous at a primerSNP, nearly ten times the 6.1% rate in individuals homozygous at the same primerSNP. Furthermore, if our explanation is correct, the single SNP allele present in the trace will be on the same chromosome as the perfectly matched primerSNP allele. Using phased haplotype data from the HapMap project, together with the primer sequence, we were able to correctly predict the identity of the missed SNP allele 93.1% of the time. These findings suggest that primerSNP heterozygosity is a major cause of missed heterozygotes.
Figure 1: Heterozygosity at a primerSNP.
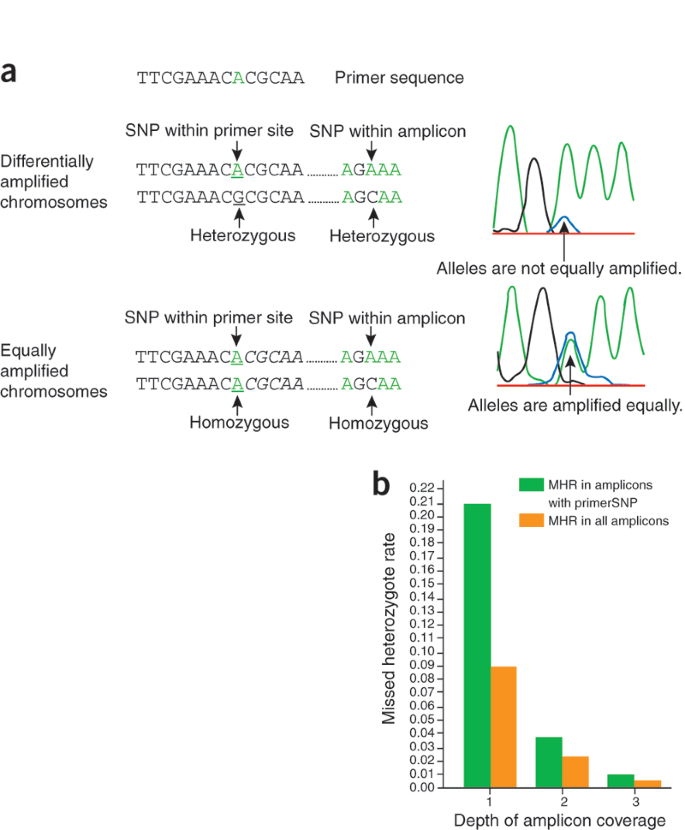
(a) For an individual heterozygous at a primerSNP (top), the chromosome matching the primer sequence is amplified more efficiently than the chromosome containing the mismatched allele. Sequencing results in reduced color intensity for the allele on the mismatched chromosome. For an individual homozygous at a primerSNP (bottom), amplification and allele-specific color intensities are balanced. (b) The MHR decreases with increasing amplicon coverage both in amplicons with primerSNPs and in all amplicons.
A typical focus of medical resequencing projects is the detection of rare SNPs or individual mutations, normally present as a single heterozygote within the resequenced cohort. In amplicons with primerSNPs, such variants are missed 25% of the time (as compared to the 14.5% rate in all amplicons). For such rare SNPs, when the single heterozygote is missed, the SNP is not discovered. For common SNPs, missing a fraction of the heterozygotes results in the reduction of heterozygous genotype frequencies from Hardy-Weinberg expectations and can force quality-control procedures3 to discard the SNP (Supplementary Fig. 2 online).
Many resequencing protocols already have provisions for avoiding primerSNPs, especially toward the 3′ end of the primer sequence where imperfect annealing owing to allelic mismatch is assumed to inhibit polymerase binding. We found that SNPs anywhere within the primer can cause missed heterozygotes and therefore must be avoided (Supplementary Fig. 3 online). Unfortunately, previously unknown SNPs cannot be avoided. We find, however, that their impact can be dramatically reduced by sequencing from more than a single amplicon. In amplicons with a primerSNP, sequencing from one additional amplicon reduces the MHR fivefold, from 20.9% to 3.7% (Fig. 1b), and from a third amplicon, to below 1%. Sequencing from increased amplicon coverage also reduces the overall MHR, and mitigates other sequencing errors and software limitations.
All ENCODE data used in this analysis including the resequencing traces, anchored assemblies and POLYPHRED candidate SNP mark-ups are available online (http://bioinformatics.bc.edu/marthlab/pcrbias).
Note: Supplementary information is available on the Nature Methods website.
References
- Sjoblom, T. et al. Science 314, 268–274 (2006).
Article Google Scholar - Ikegawa, S., et al. Hum. Genet. 110, 606–608 (2002).
Article CAS Google Scholar - The International HapMap Consortium. Nature 437, 1299–1320 (2005).
- The ENCODE Project Consortium. Science 306, 636–640 (2004).
- Marth, G.T. et al. Nat. Genet. 23, 452–456. (1999).
Article CAS Google Scholar - Stephens, M., et al. Nat. Genet. 38, 375–381 (2006).
Article CAS Google Scholar
Author information
Authors and Affiliations
- Department of Biology, Boston College, Chestnut Hill, 02467, Massachusetts, USA
Aaron R Quinlan & Gabor T Marth
Authors
- Aaron R Quinlan
You can also search for this author inPubMed Google Scholar - Gabor T Marth
You can also search for this author inPubMed Google Scholar
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
About this article
Cite this article
Quinlan, A., Marth, G. Primer-site SNPs mask mutations.Nat Methods 4, 192 (2007). https://doi.org/10.1038/nmeth0307-192
- Issue Date: March 2007
- DOI: https://doi.org/10.1038/nmeth0307-192