Metabolomics: A Global Biochemical Approach to the Study of Central Nervous System Diseases (original) (raw)
INTRODUCTION
Millions of people suffer from mental illnesses or neurodegenerative diseases such as Parkinson's disease (PD), Alzheimer's disease, schizophrenia, depression, addiction, autism, dyslexia, and learning disabilities. These, among other diseases, are in much need for better treatments. Unfortunately, our current understanding of these disorders is limited. In fact it is likely that most of these disorders are not unitary conditions but may be a conglomeration of entities that are yet to be defined. Although some progress has been made in the treatment of psychiatric disorders many patients do not respond to current therapies. We also cannot predict who will respond to which treatment. This inherent variability coupled with intrinsic differences in pharmacology is the underlying factor that affects our inability to predict how an individual patient may respond to a selected therapy. Such uncertainty is distressing for patients and families who engage repeatedly in ‘trial-and-error’ choices in search of ‘the right fit’ and for clinicians thus resorting to widespread switching of medications (Weiden and Buckley, 2007) and polypharmacy (Tranulis et al, 2008). The inordinately high personal and societal burden of inadequate trial-and-error management underscores an urgent need for validated biomarkers that establish diagnosis, guide drug selection, and reliably predict response to treatment (Insel, 2007; Lieberman et al, 2005).
To understand brain function and its complexity new ideas, new approaches, and new technologies are much needed. Efforts to scale-up data production and analysis in neuroscience have been escalating. Genomics including comparative genomics, gene expression atlases and the organization of genome-scale projects, gene microarrays, proteomics, monitoring the activity of individual neurons using multiple-electrode recordings, and imaging studies have all provided powerful approaches to the study of neurological disorders (articles in Nat Neurosci 7, 425, published in 2004). Metabolomics the newest of the omics approaches provides powerful tools for defining perturbations in metabolic pathways and networks in human disease (Kaddurah-Daouk et al, 2008; Kristal et al, 2007a, 2007b; Lindon et al, 2007; Harrigan and Goodacre, 2003). The metabolome defines a metabolic state as regulated by net interactions between gene and environment influences and provides information that can possibly bridge the gap between genotype and phenotype. It provides a missing piece to a systems approach to the study of diseases of the central nervous system (CNS). Metabolic signatures for CNS disorders could result in the identification of biomarkers for disease, for disease progression or for response to therapy. In addition metabolomics provides powerful tools for the process of drug discovery and drug development by providing detailed biochemical knowledge about drug candidates, their mechanism of action, therapeutic potential, and side effects as exemplified in this review.
FROM THE GENOME TO THE METABOLOME IN THE STUDY OF CNS DISORDERS
Advances in genomics, the omics field of DNA sequence analysis, have accelerated the ability to link human disease with its origins in the genome. Examples in the neurosciences include the recognition of genetic risk factors for Alzheimer's disease such as amyloid mutations, APOe4 vs APOe2 or APOe3 (Strittmatter et al, 1993; Corder et al, 1993; Strittmatter et al, 1993); PD synuclein, Parkin, Pink1, DJ-1 (Polymeropoulos et al, 1997; Morris, 2005; Klein et al, 2005; Kitada et al, 1998); Huntington's disease (HD) expanded CAG repeat in the huntingtin gene (Jenkins et al, 2005; Myers et al, 1993; The Huntington's Disease Collaborative Research Group, 1993); amyotrophic lateral sclerosis (ALS) mutations in SOD (Rosen et al, 1993), and the importance of serotonin transporter variation in relation to a number of neuropsychiatric syndromes such as depression, obsessive compulsive disorder, and neuroticism (Serretti et al, 2006; Bloch et al, 2008). Advances in transcriptomics and proteomics might impact our ability to understand neurological disorders pathogenesis.
One of the major missing pieces of the ‘-omics’ revolution was the -omics field of quantitative metabolite analysis known as metabolomics (with alternate names used such as metabonomics and metabolic profiling) (Kaddurah-Daouk et al, 2008; Kristal et al, 2007a, 2007b; Lindon et al, 2007; Harrigan and Goodacre, 2003). In contrast to classical biochemical approaches that focus tightly on single metabolites, single metabolic reactions and their kinetic properties, and/or defined sets of linked (ie, precursor/product, intermediary metabolism) reactions and cycles, metabolomics collects quantitative data on a broader series of metabolites in an attempt to gain an overall picture or understanding of metabolism and/or metabolic shifts associated with conditions of interest (Kristal et al, 2007a, 2007b).
METABOLOMICS: CONCEPT, TOOLS, AND PROCESS
Metabolomics tools enable us to study the metabolome, the repertoire of small molecules present in cells and tissue (Harrigan and Goodacre, 2003; Kaddurah-Daouk et al, 2008; Kristal et al, 2007a, 2007b; Lindon et al, 2007). The identities, concentrations, and fluxes of these substances are the final product of interactions between gene expression, protein expression, and the cellular environment (Figure 1). Unlike earlier, more rudimentary analytical methods, metabolomics today offers analytical instruments that can simultaneously quantitate thousands of substances present in a biological sample of interest and sophisticated mathematical tools that can find a molecular signal amongst millions of pieces of data (Kell, 2004).
Figure 1
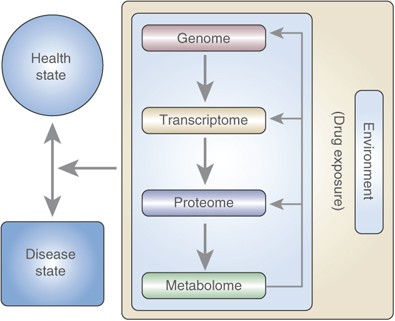
Flow of information from the genetic code (DNA) to proteins, and finally to metabolites. The environment and DNA affect the end products and influence health disease states. In this case drug exposure is the environmental alteration being tested.
Metabolomics utilizes instruments that can simultaneously quantitate thousands of small molecules in a biological sample. This analytical capability must then be joined to sophisticated mathematical tools that can identify a molecular signal for a disease or a drug. Ideally, metabolomics will ultimately contribute a detailed map of the regulation of metabolic pathways, and, therefore, of the interaction of proteins encoded by the genome with environmental factors, including drug exposure. Therefore, the metabolome represents a state function for an individual at a particular point in time or after exposure to a specific environmental stimulus (eg, a specific drug or potentially even a mood state). Many diseases disrupt metabolism and result in changes that are long lasting and that can be captured as metabolic signatures. Initial metabolomic signatures have already been reported for several disease states, including motor neuron disease (MND) (Rozen et al, 2005), depression (Paige et al, 2007), schizophrenia (Holmes et al, 2006; Kaddurah-Daouk, 2006; Kaddurah-Daouk et al, 2007), Alzheimer's disease (Han et al, 2002), cardiovascular and coronary artery disease (Brindle et al, 2002; Sabatine et al, 2005), hypertension (Brindle et al, 2003), subarachnoid hemorrhage (Dunne et al, 2005), preeclampsia (Crocker et al, 2005), type 2 diabetes (Van der Greef et al, 2007; Van Doorn et al, 2007; Wang et al, 2005; Yuan et al, 2007), liver cancer (Yang et al, 2004), ovarian cancer (Odunsi et al, 2005), breast cancer (Fan et al, 2005), and HD (Underwood et al, 2006). These signatures are made up of tens of metabolites that are deregulated, with concentrations that are modified in the disease state or after drug exposure. As a result, analysis of these signatures and their components can potentially provide information with regard to disease pathophysiology.
The choice of metabolomic analytical instrumentation and software is generally goal-specific, as each type of instrument has certain strengths. Liquid chromatography followed by coulometric array detection, for example, has been used in the identification of signatures in ALS (Rozen et al, 2005) and most recently for PD (Bogdanov et al, 2008). It is excellent for mapping neurotransmitter pathways (eg, dopamine (DA) and serotonin) and pathways involved in oxidative stress. Gas chromatography in conjunction with mass spectroscopy is often used in the analysis of lipid subsets (Kaddurah-Daouk et al, 2007; Watkins, 2004; Watson, 2006). Liquid chromatography with mass spectroscopy is often used to obtain the largest possible biochemical profile information subset. It is a sensitive tool that can be used to characterize, identify, and quantify a large number of compounds in a biological sample (Kristal et al, 2007a, 2007b; Fiehn, 2008; Tolstikov et al, 2007) where metabolite concentrations might cover a broad range of information with regard to disease pathophysiology. In addition to popular high-sample-throughput applications, NMR is particularly powerful for metabolite structural determinations, including the atomic positions of isotopic labels (eg, 13C, 15N, or 2H) in different isotopomers generated during stable isotope tracer studies (Fan and Lane, 2007). For example NMR-based high throughput analysis has been used successfully in toxicology studies (Coen et al, 2004; Lindon et al, 2000, 2003). NMR applications provide detailed maps of biochemical pathways or networks, which can also serve as crucial inputs for in silico quantitative flux analysis (Dauner et al, 2001; de Graaf et al, 2000).
An overview of a ‘typical’ metabolomics study is depicted in Kaddurah-Daouk et al (2008). Samples of interest (eg, plasma, cerebral spinal fluid, tissue biopsy, etc) are collected, then small molecules are extracted from the sample and are analyzed using techniques that separate and quantitate molecules of interest as mentioned above. Combinations of these techniques can be used to augment separations and/or expand the analyte information collected. These data sets must then be collected and curated, a process that can take significant time for the overall experiment yet tools are being developed to make this process faster (Styczynski et al, 2007). After curation, the data are analyzed by one or more software packages designed for studies of data sets that are far too large for human evaluation. A database is then generated for the diseased patients and another for the controls or for the same patients before and after drug therapy. These databases include levels of detectable metabolites and identity (or a description of the properties) of the metabolites (ie, oxidation–reduction potential, mass/charge ratio, etc, whenever known.)
The application of software tools for the analysis of the information contained in a database can: (1) identify signature of a disease (eg, compounds that highlight a disease state); (2) predict class (eg, disease or control, pre- or postdrug exposure); (3) identify unrecognized groups in the data (eg, drug response subgroups); (4) identify interactions between variables; and (5) place these variables within known biochemical pathways.
Metabolomics data can be analyzed with a range of statistical and machine-learning algorithms. These algorithms can be classified within two major classes: unsupervised and supervised (Kell, 2004). They can be useful in the identification of biomarkers (Sajda, 2006; Shin and Markey, 2006). Unsupervised algorithms find patterns in the data without any biases, and are typically driven by the largest changes (variance). Supervised algorithms require that samples be labeled in groups a priori, and they uncover the features (variables) that best discriminate between those groups. Examples of unsupervised methods that have been routinely used in analyzing molecular fingerprinting data include principal component analysis (PCA) and self-organizing maps (Patterson et al, 2008).
METABOLIC CHANGES IN CNS DISORDERS: RATIONALE FOR METABOLOMICS APPROACH
Over the last half a century, neurochemists have identified a series of molecules in the brain which carry important messenger functions such as DA, epinephrine, norepinephrine (NE), serotonin, acetylcholine (Ach), _γ_-amino butyric acid (GABA), substance P, bradykinin, and many more. The identification of these molecules required technically challenging experiments and once these compounds were identified, research shifted to understanding their mechanisms of regulation, synthesis and release, and the pathways that gave rise to them. Notably, however, these are molecules that are primarily thought by most, even today, as individual molecules. The next stage—the consideration of pathways—entered the picture as people asked how molecules were made, and what the limiting factors in their synthesis were. Today, metabolomics provides new and powerful tools to study hundreds of these key molecules simultaneously and quantitatively (Kristal et al, 2007a, 2007b). We can now interrogate almost to entirety metabolites within key neurotransmitter pathways such as DA and serotonin enabling us to move to a far more detailed analysis of implications of these pathways in CNS disorders and for mapping more clearly mechanism of action of drugs that target these pathways.
Abnormalities in several metabolic pathways have been identified in neuropsychiatric disorders. There is increasing evidence linking mitochondrial dysfunction, as well as metabolic abnormalities to neurodegenerative diseases (Thomas and Beal, 2007; Lin and Beal, 2006). Mitochondria are critical regulators of cell death, a key feature of neurodegeneration. There may be a variety of factors, which link mitochondria to neurodegeneration in the various neurodegenerative diseases. For instance, in HD there is recent evidence that suggests that the coactivator PGC1-α, a key regulator of mitochondria biogenesis and respiration, is downregulated. This appears to be the case both in patients, as well as several animal models of HD. In PD the autosomal recessive genes Parkin, DJ1, and PINK1 are all linked to either oxidative stress or mitochondrial dysfunction. In ALS there is strong evidence that mutant superoxide dismutase directly interacts with the outer mitochondrial membrane, as well as the intermembrane space and matrix. A number of therapies that target basic mitochondrial processes such as energy metabolism and free radical generation are under development.
In psychiatric diseases many metabolic changes have been identified and linked to disease pathogenesis. Changes in neurotransmitter pathways such as DA and serotonin seem to be important in disease pathogenesis and response to therapy.
In schizophrenia impairments in neurotransmitter, signal transduction, antioxidant system, membrane composition, and immune functions have been noted among several other changes (Mahadik and Yao, 2006). There is sufficient evidence to suggest that aberrant neurotransmission might play a role in the pathogenesis of schizophrenia. In particular, aberrant dopaminergic, serotonergic, and glutamatergic systems have been noted (Miyamoto et al, 2003). Some data also exist that support the involvement of histamine, and Ach neurotransmitter systems (Feuerstein, 2008). It is unclear, however, to what extent these neurochemical findings reflect primary rather than secondary pathology, compensatory mechanisms, or environmental influences, and it is not clear how these pathways interact in schizophrenia pathogenesis. Evidence exists that phospholipids which play a critical role in the structure and function of membranes are impaired in schizophrenia (Horrobin, 1998; Berger et al, 2002; Skosnik and Yao, 2003; Mahadik and Yao, 2006). Lipids and their constituent fatty acids which provide scaffolding for many key functional systems, including neurotransmitter receptor binding, signal transduction, transmembrane ion channels, prostanoid synthesis, and mitochondrial electron-transport systems could contribute to pathogenesis in schizophrenia (Rotrosen and Wolkin, 1987; Lieberman and Koreen, 1993).
In bipolar disorder (BD) perturbations in neurotransmitters systems including Ach (Janowsky et al, 1972), GABA (Brambilla et al, 2003), norepinephrine (NE) (Schildkraut, 1974), DA (Goodwin and Sack, 1974), glutamate (Glu) (Kugaya and Sanacora, 2005), and serotonin (5HT) (Mahmood and Silverstone, 2001; Sobczak et al, 2002) have been identified. Abnormalities in lipids and increased inflammation and immune activation may also contribute to the pathogenesis of mood disorders, in part by modulating the metabolism of NT systems (Wichers et al, 2005). It is not well understood which biochemical pathways are regulated during the shift from the manic to the depressed states.
In major depressive disorders (MDD) multiple neurotransmitter systems are thought to be deregulated and much attention has been given to the role of impairment in central monoaminergic function. Given that SSRIs are commonly used for the treatment of MDD, serotonin (5HT) abnormities have been traditionally linked to MDD’ pathogenesis (Delgado and Moreno, 2000). Not only serotonergic, but also noradrenergic systems are likely to be involved in antidepressant action, and specific impairments in these systems may also play a role in disease pathogenesis (Delgado and Moreno, 2000). The GABAergic neurotransmitter system has become increasingly implicated in the neurobiology of mood disorders (Brambilla et al, 2003; Sanacora and Saricicek, 2007). GABAergic involvement in the pathophysiology and treatment of mood disorders is supported by several lines of evidence spanning from animal studies showing stress-related changes in GABAergic function to the demonstration of GABAergic abnormalities and genetic associations in depressed patients (Brambilla et al, 2003; Sanacora and Saricicek, 2007). Specifically, the association of lower GABA levels with depression is a consistent finding (Petty and Schlesser, 1981). Indeed, reduced levels have been consistently reported in plasma (Petty and Schlesser, 1981), CSF (Gerner et al, 1984; Kasa et al, 1982), and brain (Bhagwagar et al, 2007; Hasler et al, 2007; Sanacora and Saricicek, 2007) of individuals diagnosed with depression. Likewise, GABA agonists and antagonists have the ability to modulate behavioral models of depression in rodents, existing antidepressant medications have GABAergic effects and there is some evidence for a clinical antidepressant efficacy associated with GABAergic drugs. Glutamatergic abnormalities in MDD patients including decreased CSF Glu and glycine (Frye et al, 2007) but increased levels of Gln have been reported (Levine et al, 2000). Furthermore, antidepressant and mood stabilizing medications have glutamatergic effects and Glu modulating agents in the treatment of depression.
APPLICATIONS OF METABOLOMICS IN THE STUDY OF CNS DISEASES
It is clear that perturbations in a variety of metabolic and signaling pathways could contribute to the pathogenesis of CNS disorders. Metabolomics provides powerful tools to map in greater detail these perturbations and their relationship to disease pathogenesis and response to therapy. Applications of metabolomics for the study of CNS diseases includes: (1) added information about mechanisms of disease; (2) identification of prognostic, diagnostic, and surrogate markers for a disease state; (3) the ability to subclassify disease based on metabolic profiles; (4) identification biomarkers for drug response phenotypes and for development of metabolic side effects (pharmacometabolomics); and (5) added tools in the process of drug discovery and drug development.
Psychiatry and neurology have long posed unique difficulties for the development of biomarkers because of the lack of access to relevant tissues. Initial biomarkers studies have relied on studies of platelet markers, spinal fluid metabolites, tissue obtained at autopsy from suicide victims or those who died of natural causes, as well as use of imaging techniques ranging from EEG, CT, MRI, MRS, and PET scans. All of these techniques have limitations especially given that our disease classification systems have also evolved and matured over the years. Although spinal fluid studies of numerous metabolites have been conducted in the study of CNS diseases, to date there exists no standard ‘normative’ reference level for most of these metabolites. This is because individual studies could measure only a few metabolites at a time and measurements are usually performed only in diseased individuals and relatively few controls are studied. Thus, there is a wide variation from study to study in the ‘normal’ values. Ventricular CSF and lumber CSF metabolites are not identical and it is not totally clear which correlates better with brain biochemistry. The correlations between plasma and CSF have also not been well established for many of these metabolites. In addition, the high complexity of the brain (compared to say the muscle or the heart) has made the identification of reliable biomarkers much more difficult. Thus, these issues argue for a systematic correlative study of plasma and spinal fluid metabolites using a technology such as metabolomics that can provide assessment of global metabolite signatures. Further, the establishment of ‘brain banks’ with rapid autopsy procedures at many centers has now made available tissue samples that can be analyzed in disorders such as Alzheimer's, major depression, schizophrenia, Lewy body dementia, frontotemporal dementia, and other conditions. Many psychiatric clinical trials also routinely collect samples from well-characterized patients before and after treatment with drugs or placebo. It is of interest to note that metabolomics can provide means to study variation in biochemical profiles among patients and could result in more effective ways to sub classify patients and provide well-defined intermediate phenotype that can describe clinical phenotype being observed by clinicians. This could have a significant impact on clinical trials design and outcome as we can start to evaluate how subpopulations of patients respond to therapy and select drugs that are more effective for each grouping. The metabolic signatures of antidepressants (SSRI class) are being mapped in good and poor responders (Metabolomics Network for Drug Response Phenotypes funded by NIH) to obtain information that can lead to a deeper understanding of pathways implicated in response and variation in response to these therapies. Mapping of signatures associated with response and of signatures associated with development of metabolic adverse events can lead to development of useful biomarkers that can help select the right drug for each patients resulting in personalizing therapy. Indeed, a new field is emerging which is called Pharmacometabolomis (of Pharmacometabonomics) than can compliment the field of pharmacogenomics.
Signatures of disease can also provide clues for drug targets. For example if a particular signature is related to a disorder then one could trace back potential targets in that metabolic framework for drug development. In addition a signature for a CNS disorder that suggests that certain metabolites are low or missing, may suggest potential ways of replacing or altering production of these metabolites as modes of treatment in line with replacement of DA in PD.
EARLY FINDINGS FROM METABOLOMICS STUDIES OF CNS DISORDERS
We have started to explore metabolic perturbations in neurodegenerative and psychiatric diseases. We attempt to understand unique and commonly perturbed pathways that contribute to the death of neurons in degenerative diseases such as ALS, PD, and HD. In psychiatric disorders such as depression, bipolar and schizophrenia we attempt to identify biomarkers for disease, disease progression, and response to therapy and define pathways implicated. Below we summarize initial findings from metabolomics studies of such CNS disorders.
METABOLOMIC ANALYSIS OF NEURODEGENERATIVE DISEASES
Motor Neuron Disease
MNDs are a heterogeneous group of rare disorders that affect motor neurons and cause diverse signs and symptoms. These are fatal diseases that are poorly understood and treatments are mostly nonexistent or supportive at best.
ALS affects both lower and upper motor neurons (Rowland and Shneider, 2001). Whether ALS is a single disease entity or a syndrome caused by different conditions remains unknown. In addition, it is not clear whether MNDs are closely related but distinct disorders or whether they represent different points on the spectrum of a single disease.
We hypothesized that diseases such as ALS might produce characteristic perturbations of the metabolome. We used HPLC followed by electrochemical detection to profile plasma from 28 patients with MND and 30 healthy controls (Rozen et al, 2005). Using multivariate regression techniques we were able to distinguish ALS patients from controls (Figures 2 and 3) and also separate ALS patients on Riluzole or off Riluzole. Please note that in Figure 2 left panel is control subjects whereas central panels and right panel are patients. Each column is a subject and each raw is a metabolite. Red are metabolites that are upregulated and blue downregulated. A sub-signature for patients with LMND and slow disease progression has been identified (Figure 2, right panel of four patients). In addition, a signature for the drug riluzole (used to treat patients with ALS) that includes endogenously induced metabolites has been defined (Figure 2, panel labeled MND and riluzole). In a subsequent study of 19 subjects with MND who were not taking riluzole and 33 healthy control subjects, six compounds were significantly elevated in MND, whereas the number of compounds with decreased concentration was similar to the initial study (Rozen et al, 2005).
Figure 2
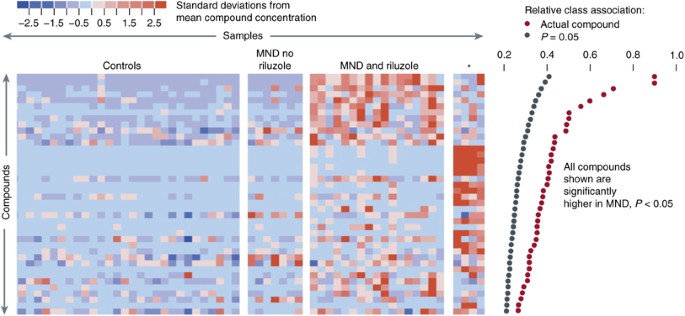
Metabolites with significantly different concentrations in normal and motor neuron disease (MND) plasmas in the first study. (a) Metabolites higher in MND patients than in controls. Each row represents a metabolite, each column represents a healthy control or a patient, and each colored square represents the relative concentration of a single metabolite in a single person. Compounds are sorted by decreasing association with MND. Significant association measures at _P_=0.05 are indicated by black dots at the right. The association measures for the actual data are indicated by red dots. The metabolites that are high in MND define three subgroups. These consist of patients not taking riluzole, patients taking riluzole, and four patients with a distinctive signature (indicated by an asterisk). Three of these patients had LMN disease. Figure presented with permission from Metabolomics Journal. Rozen et al, 2005.
Figure 3
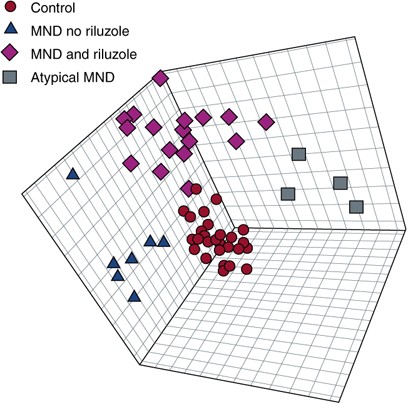
Partial least squares discriminant analysis (PLS-DA) distinguished subgroups of motor neuron disease (MND). Models using projections into three dimensions provided statistically significant separations between subgroups (P<0.01 by permutation test—random assignment of samples to subgroups). Red are controls; purple are MND patients on riluzole; blue are MND patients off riluzole and black are atypical MND patients. Figure presented with permission from Metabolomics Journal. Rozen et al, 2005.
In both data sets, it was possible to separate MND patients from controls using multivariate regression techniques. These data suggested that metabolomic studies can be used to detect signatures for some CNS disorders peripherally, could identify sub-groupings within a disease state, and that they can also detect drug exposure signatures. Elucidation of the structures of signature molecules in ALS and other forms of MND might provide insight into aberrant biochemical pathways that could lead to diagnostic markers and targets for drug design. A program funded by the ALS Association (collaboration between MGH, Metabolon Inc. and network of academic institutions) has been initiated to collect samples from MND patients nationally to better dissect MNDs using a variety of metabolomics platforms including LC–MS and gas chromatography–mass spectrometry (GC–MS). We are defining metabolic signatures for UMNDs, LMNDs, and ALS to determine the relatedness of these diseases at the metabolic level and to define biochemical markers for disease and progression. Although clinicians can classify these diseases in a rough way, we believe that metabolomics can provide a more objective classification.
Metabolomic Analysis of Parkinson's Disease
A recent study was conducted by Bogdanov et al (2008) to look for biomarkers in plasma useful for the diagnosis of PD. Metabolomic profiling using high-performance liquid chromatography coupled with electrochemical coulometric array detection was used to evaluate 25 controls and 66 PD patients. Initial analysis of the metabolomic profiles from plasma demonstrated a clear differentiation between PD patients and control subjects (P<0.01 by permutation test). A partial least squares discriminant analysis (PLS-DA) scores plot based on complete digital maps is shown in Figure 4. Both unmedicated PD patients and the patients on different types of antiparkinsonian medication (Sinemet, DA receptor agonists, and combination of both Sinemet and the agonists) were included in the initial analysis. To evaluate the possibility that the observed separation between the patients and controls is due to medications that patients are on and not due to disease state plasma from 15 unmedicated PD patients were profiled and compared to profiles of 25 control subjects. A PLS-DA scores plot based on the analysis of complete digital maps for unmedicated PD patients and control subjects showed a complete and significant separation of these two groups (P<0.01 by permutation test; Figure 5). The metabolites 8-OHdG and glutathione were significantly increased in PD patients whereas the level of uric acid was significantly reduced. Both glutathione and uric acid are well-known antioxidants. Higher uric acid levels lower risk for PD and slow the progression of the illness (de Lau et al, 2005; Ascherio et al, 2006). Glutathione levels are reduced in the substantia nigra of PD postmortem brain tissue. Increased oxidized glutathione was found in plasma of PD patients (Younes-Mhenni et al, 2007). Changes in levels and ratio of oxidized to reduced glutathione probably reflect a response to oxidative damage.
Figure 4
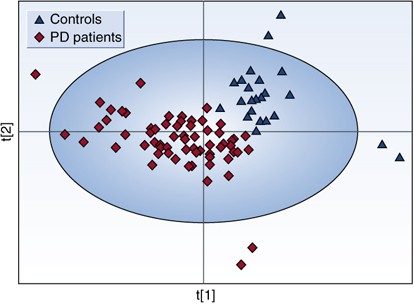
Partial least squares discriminant analysis (PLS-DA) scores plot showing a significant separation (P<0.01 by permutation test—random assignment of samples to different groups) between control subjects (_n_=25) and all Parkinson's disease (PD) patients (medicated and unmedicated, _n_=66) using complete digital maps. Figure presented with permission from Brain. Bogdanov et al, 2008.
Figure 5
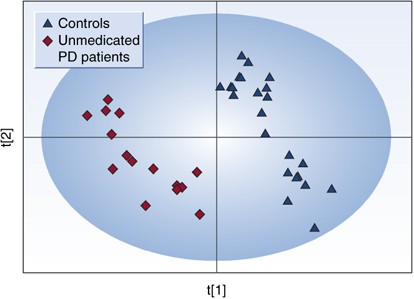
Partial least squares discriminant analysis (PLS-DA) scores plot showing a significant separation (P<0.01 by permutation test) between control subjects (_n_=25) and unmedicated PD patients (_n_=15) using complete digital maps. Figure presented with permission from Brain. Bogdanov et al, 2008.
It is of interest to note that drugs used to treat PD such as Sinemet, DA receptor agonists, and combination of both Sinemet and the agonists do induce partial reversal of metabolic profiles in PD patients (Bogdanov et al, 2008) where they become more similar to controls. Few patients’ metabolic profiles did not revert and it is of interest to try to correlate in the future reversal of metabolic profiles with reversal of clinical symptoms.
This PD study demonstrates the promise of metabolomics in the development of biomarkers for PD. Diagnostic markers could have great utility in accelerating clinical trials and might enable sub-stratification of patients and identification of homogenous population of PD subjects not contaminated by other Parkinsonian syndromes. A disease progression marker if identified and validated in longitudinal studies could serve as a surrogate endpoint, which might be more quantitative than clinical scales. Lastly, if one could identify early biochemical changes and signatures that are unique to patients at the earliest stages of illness, or even pre-symptomatically it would be of great importance. One could then screen first-degree relatives, patients with hyposmia and patients carrying genetic risk factors. A number of other manifestations of systemic illness may increase risk for PD. If these patients can be identified early one could carry out primary prevention trials.
Metabolomics in the Study of Huntington's Disease
HD is a devastating autosomal-dominant neurodegenerative condition that manifests with movement disorder, behavioral disturbance, and cognitive deterioration. Although it can present at any age, the median age of onset is 40 years, and death typically follows some 15–20 years after symptom onset. The HD mutation is a (CAG)n trinucleotide repeat expansion at the 5′ end of the transcript encoding HD. The (CAG)n repeats are translated into a polyglutamine tract. Disease is caused by >35 CAG repeats and the age at onset correlates inversely with CAG repeat number. Considerable progress has been made with modeling Huntington's pathogenesis in cell and animal models. The development of biomarkers that better sub classify patients or that correlate with disease and disease progression could provide very valuable tools as clinical trials are costly and time consuming due to the slow disease course, insidious onset and patient-to-patient variability. The validation of animal models which are used to select candidate therapies and an evaluation of their relevance to human disease is of great importance as they could yield models that are more effective in predicting therapeutic outcome in patients.
In a study conducted by Underwood et al (2006) metabolic profiling of human HD patients and a transgenic mouse model for HD by gas chromatography–time-of-flight–mass spectrometry (GC–TOF–MS) revealed clear differences in metabolic profiles. This trend was seen in both transgenic mice and wild-type littermates, and human patients compared to control subjects. The metabolic signatures in mice and humans are indicative of a change to a procatabolic phenotype in early HD preceding symptom onset, with changes in various markers of fatty acid breakdown (including glycerol and malonate) and also in certain aliphatic amino acids. The data raise the prospect of a robust molecular definition of progression of HD before symptom onset, and if validated in a genuinely prospective fashion these biomarker trajectories could facilitate the development of useful therapies for this disease as well as biomarkers for disease and its progression.
Metabolomic Analysis of a Mouse Model of Batten Disease
Neuronal ceroid lipofuscinoses (NCLs) are a series of autosomal recessive diseases that collectively constitute the most common cause of childhood neurodegeneration with an incidence of 1 in 12 500 (Goebel and Sharp, 1998; Banerjee et al, 1992). The disorders are typified by their progressive nature with symptoms including visual disturbances, psychomotor deterioration, mental impairment, worsening seizures, blindness, and, ultimately, premature death (Gardiner, 2002; Hofmann et al, 2002; Mitchison et al, 2004; Mitchison and Mole, 2001; Wisniewski et al, 2001). Although six of the causative genes have been characterized, the underlying disease pathogenesis for this family of disorders is unknown. The Cln3 gene has been identified as responsible for the juvenile NCL (Batten disease) and an animal model has been generated for this disease. Using a metabolomics approach based on high resolution 1H NMR spectroscopy of the cortex, cerebellum, and remaining regions of the brain in conjunction with statistical pattern recognition, Pears et al (2005) evaluated metabolic deficits in Cln3 null mutant mice aged 1–6 months. They defined increased Glu concentration and a decrease in GABA concentration in aqueous extracts from the three regions of the brain. These changes are consistent with the reported altered expression of genes involved in Glu metabolism in older mice and imply a change in neurotransmitter cycling between Glu/glutamine and the production of GABA. Further variations in _myo_-inositol, creatine, and _N_-acetyl-aspartate were also identified. These metabolic changes were distinct from the normal aging/developmental process. Together, these changes represent the first documented presymptomatic symptoms of the Cln3 mouse at 1 month of age and demonstrate the versatility of 1H NMR spectroscopy as a tool for phenotyping mouse models of disease.
METABOLOMIC ANALYSIS IN PSYCHIATRIC DISORDERS
We have been using random and targeted metabolomics approaches for the study of psychiatric disorders. We are testing existing hypothesis around the impairment of neurotransmitter pathways such as DA and serotonin and lipid pathways and we are also generating new hypothesis about pathways that might be implicated in disease and disease progression. Below we provide early findings for such studies.
Metabolic Signatures in Depression
Given the body of evidence suggesting that abnormalities in the metabolome do exist both centrally and in the periphery a recently published metabolomics study was conducted in plasma as a first step towards mapping biomarkers for depression. Metabolomic analysis of blood plasma was performed on 9 depressed, 11 remitted, and 10 never-depressed older adults (Paige et al, 2007). Hundreds of metabolites were measured using GC–MS. Metabolite identification was based on a combination of chromatographic properties and mass spectra. A library of 800 commercially available human metabolite standards (Metabolon Inc.) has been assembled and analyzed on an MS platform that helped in compound identification.
Concentrations of each metabolite were normalized to mean scaled values of two recovery standards that represent hydrophobic and hydrophilic compounds. Univariate _t_-tests (Welch's two-sample _t_-tests) were performed for each metabolite to determine metabolic differences between healthy controls, and individuals who are depressed or in remission. False discovery rate (FDR) was accounted for and the multiple testing and _q_-value were estimated FDR for every possible list of ‘significant’ metabolites. Metabolites that were altered in currently depressed patients when compared with controls included several fatty acids, glycerol, and GABA. Analyses comparing concentrations in remitted and currently depressed patients revealed a pattern of metabolite alterations similar to the control vs currently depressed analyses. One difference observed in the remitted patients relative to the depressed patients was elevation of the concentration of the ketone 3-hydroxybutanoic acid (Figure 6).
Figure 6
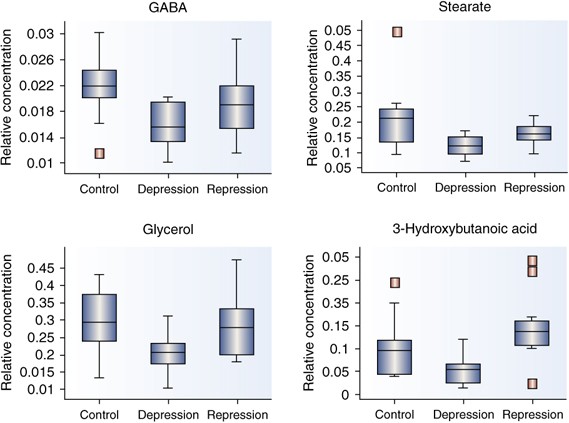
Metabolites significantly different in the depressed state when compared to remitted state or healthy nondepressed controls. Figure presented with permission from Int J Geriatric Psychiatry. Paige et al, 2007.
These preliminary results will need to be examined and validated in larger longitudinal cohorts. However, these findings suggest that the depressed state may be associated with alterations in the metabolism of lipids and neurotransmitters, and that treatment with antidepressants adjusts some of the aberrant pathways in disease so that the patients in remission have a metabolic profile more similar to controls than to the depressed population. An evaluation of such changes in CSF samples is needed to establish how closely these findings are to central changes.
Metabolic Signatures in Schizophrenia and Its Treatment
Several metabolomics studies have recently been conducted in an attempt to better define pathways modified in schizophrenia and its treatment (Holmes et al, 2006; Kaddurah-Daouk et al, 2007; Tkachev et al, 2007; Tsang et al, 2006). In one study we used a specialized lipidomics platform and measured more than 300 polar and nonpolar lipid metabolites (structural and energetic lipids) across 7 lipid classes to evaluate global lipid changes in schizophrenia before and after treatment with three commonly prescribed atypical antipsychotics, olanzapine, risperidone, and aripiprazole (Kaddurah-Daouk et al, 2007).
Lipidomics is a branch of metabolomics that specifically focuses on comprehensive assessment of lipid biochemistry (German et al, 2007; Watson, 2006; Wiest and Watkins, 2007). In this particular study, lipid profiles were obtained for 50 patients with schizophrenia before and after 2–3 weeks of treatment with olanzapine, risperidone, or aripiprazole. Data are presented in heat maps where column headers indicate fatty acid metabolites as they appear in each distinct lipid class (rows). Metabolites that are upregulated are shown in red whereas those that are downregulated are shown in green. Figure 7a shows differences in individual lipid metabolites in the plasma of patients with schizophrenia as compared to controls whereas Figure 7b shows effect of three drugs olanzapine, risperidone, and aripiprazole. Figure 7c shows the most significantly modified lipid metabolites in plasma of patients treated with olanzapine and highlights which of these metabolites are also modified upon treatment with risperidone or aripiprazole.
Figure 7
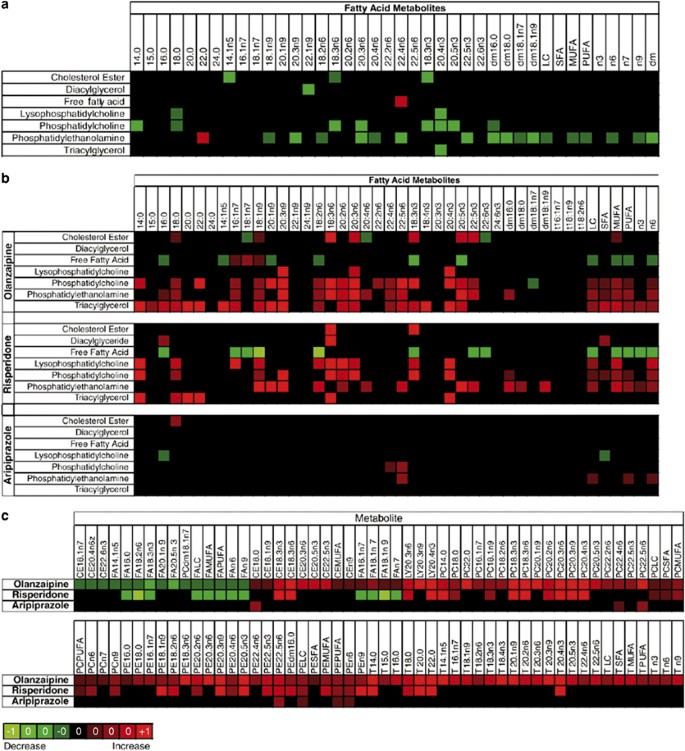
Heat map showing differences in individual lipid metabolites in the plasma of patients with schizophrenia as compared to controls (a), in the plasma of schizophrenic patients post-treatment as compared to pretreatment (b) with olanzapine, risperidone, and aripiprazole. (c) The most significantly modified lipid metabolites in plasma of patients treated with olanzapine and highlights which of these metabolites are also modified upon treatment with risperidone or aripiprazole. The column headers indicate fatty acid metabolites as they appear in each distinct lipid class (rows). In (a), lipids whose percent levels were higher in patients vs controls are shown in red whereas those with decreased level are shown in green. In (b) and (c), the percent increase in any lipid upon treatment with drug is shown in red squares and decrease in green squares. The brightness of each color corresponded to the magnitude of the difference in quartiles. The brighter the square is the larger the difference. Figure presented with permission from Molecular Psychiatry. Kaddurah-Daouk et al, 2007.
At baseline, and before drug treatment, major changes were noted in two phospholipids classes, phosphotidylethanolamine (PE) and phosphotidylcholine (PC), suggesting that phospholipids that are important in proper membrane structure and function seem to be impaired in patients with schizophrenia (Figure 7; Kaddurah-Daouk et al, 2007). This confirmed previous observations but establishes a far more detailed biochemical map for sites of perturbations. Some of the biochemical perturbations were seen within the _ω_-3 and _ω_-6 subclasses in PE and PC. In addition shifts between saturated and ployunsaturated fatty acids were noted (Figure 7; Kaddurah-Daouk et al, 2007).
The effects of three antipsychotic drugs, olanzapine, risperidone, and aripiprazole, on lipid biochemical pathways were then evaluated by comparing metabolic profiles at baseline to post-treatment (Kaddurah-Daouk et al, 2007). It was of interest that each of the three drugs studied had a unique signature suggesting that although these drugs share some effects, they also have many effects that are unique for each. PE concentrations that were suppressed at baseline in patients with schizophrenia were elevated after treatment with all three drugs. However, olanzapine and risperidone affected a much broader range of lipid classes than did aripiprazole, with approximately 50 lipids that were increased after exposure to these drugs, but not after aripiprazole therapy (Figure 7). On balance, aripiprazole induced minimal changes in the lipidome (Figure 7), consistent with its limited metabolic side effects. There were also increased concentrations of triacylglycerols and decreased free fatty acid concentrations after both olanzapine and risperidone, but not after aripiprazole therapy (Figure 7; Kaddurah-Daouk et al, 2007). All of these changes suggest peripheral effects that might be related to the metabolic side effects that have been reported for this class of drugs and highlights lipases in the liver as possibly targets for these drugs. Finally, a PCA identified baseline lipid alterations that seemed to correlate with acute treatment response (Kaddurah-Daouk et al, 2007).
Collectively, these results raised the possibility that a more definitive long-term randomized study of these drugs in which global lipid changes would be correlated with clinical outcomes might yield biomarkers related to response and development of side effects. This study of atypical antipsychotic drugs illustrates the way in which metabolomics might contribute to our understanding of drug response phenotypes and how it provides tools to analyze pathways implicated in variation to response for this class of drugs. In addition it illustrates how metabolomics could be a very valuable tool for biomarker discovery. Studies carried by our newly established Schizophrenia Metabolomics Network are generating data about global changes in neurotransmitter pathways, purine pathways, one carbon metabolism-related pathways, intermediary metabolic pathways, and other pathways and will provide a far better chance at defining a truly comprehensive signature for schizophrenia.
Additional metabolomics studies in schizophrenia include a (1)H NMR spectroscopy-based metabonomic analysis plasma where samples from 21 pairs of monozygotic twins discordant for schizophrenia and 8 pairs of age-matched healthy twins demonstrated alterations in lipid profiles of both affected and unaffected schizophrenia twins (Tsang et al, 2006). In another study (1)H NMR profiling of CSF samples from drug-naïve patients with first-onset schizophrenia, suggested alterations in glucose regulation an abnormality that seems to get corrected by early treatment with antipsychotics (Holmes et al, 2006). Finally, an interesting metabolomic study on post-mortem tissue provides support to the notion that abnormalities at the level of glutamatergic neurotransmission and myelin synthesis may be important in schizophrenia (Tkachev et al, 2007).
Metabolic Signatures in Bipolar Disease
Bipolar affective disorder (BD) is a severe and debilitating psychiatric condition characterized by the alternating mood states of mania and depression. The pathophysiology of the disorder and the mechanism of action of therapies used for its treatment remain poorly understood. Lan et al, 2008 used 1H NMR spectroscopy-based metabonomic analysis to identify molecular changes in post-mortem brain tissue (dorsolateral prefrontal cortex) of patients with a history of BD. In an animal model they also evaluated signatures of lithium and valproate two drugs used to treat BD. Several metabolites were found to be concordantly altered in both the animal and human tissues. Glu levels were increased in post-mortem bipolar brain, whereas the Glu/glutamine ratio was decreased following valproate treatment, and GABA levels were increased after lithium treatment, suggesting that the balance of excitatory/inhibitory neurotransmission is central to the disorder.
In another study and using GC–TOF–MS we measured 246 metabolite signals in plasma of patients with BD pre and post-treatment with lithium. Using 89 metabolites identified with chemical structures we were able to achieve clear separation between pre- and post-treatment samples using PCA and PLS plots (RKD). Treatment was associated with a significant change in levels of 20 metabolites. Some of these metabolites included molecules belonging to pathways postulated to be responsible for lithium's therapeutic effects and some were previously unidentified (unpublished data). We are trying to define which sets of metabolites at baseline correlate with positive therapeutic outcome.
Collectively, data demonstrate the high potential of metabolomics for the identification of biomarkers and metabolic signatures of treatment response. Replication studies are underway to validate our findings.
Metabolomics in Addictive Disorder
Many experiments are underway to evaluate signatures in addicts who use cocaine or opioids. Early findings suggest that neurotransmitter pathways, purine pathways, and pathway implicated in oxidative stress all seem to be affected by these addictive substances. Further validation of these findings could provide new insights about addiction mechanisms and ways to try to intervene with that.
FUTURE DIRECTIONS AND CLINICAL IMPLICATIONS
The study of metabolism at the global or ‘-omics’ level, referred to as metabolomics, is a new but rapidly growing field that has the potential to impact our understanding of molecular mechanisms of disease. Disease states like schizophrenia disrupt metabolism and result in metabolic signatures that are long lasting and that can be defined using complimentary metabolomics platforms. A deeper understanding of global perturbations in biochemical pathways in complex diseases such as schizophrenia, depression, Parkinson's, and Alzheimer's disease and upon treatment with drugs could provide valuable insights about mechanisms of disease, drug effects, and variation in drug response and provide needed prognostic, diagnostic and surrogate biomarkers. In addition, metabolomics could provide biochemical labels to the diverse clinical manifestations of CNS diseases leading to sub-classification of disease based on different etiologies and biochemical perturbations. This could stream line clinical trials and improve outcomes. It should be noted that research at this stage represents first steps towards the development of a metabolic signature as biomarkers for disease or its treatment. We are still learning how to deal with confounding factors and our sample sizes so far are relatively small. Proper matching of patients and controls for age gender ethnic background and many other factors should be considered carefully. Close monitoring of diet and exercise and possible confounding effects of other medications and disease states have to be considered. Ideally more than one site should be used for recruitment of patients and controls. Longitudinal studies are needed to confirm and expand on these initial findings. Replication studies and blinded studies are needed to validate markers identified. Connecting central and peripheral changes in CNS disorders is key towards defining if and how biochemical changes in plasma are related to changes in the brain. Combining metabolomics with imaging approaches and other omics approaches might be powerful ways to achieve these goals. Metabolomics has the potential to enable mapping of early biochemical changes in disease and hence provide an opportunity to develop predictive biomarkers that can trigger earlier interventions. The inclusion of metabolomics in all steps of drug discovery and drug development we believe will become a routine as biochemistry is basic to understanding how drugs work and what their safety and efficacy profiles are.
References
- Ascherio A, LeWitt PA, Watts A, Kieburtz K, Rudolph A, Schwid SR, et al, and on behalf of the PSG DATA TOP investigators (2006). CSF as well as serum urate are predictors of Parkinson's disease progression. Movement Disorders Society International Congress on Parkinson's Disease and Movement Disorders, Kyoto (Abstract no. LB-2).
- Banerjee P, Dasgupta A, Siakotas A, Dawson G (1992). Evidence for lipase abnormality: high levels of free and triacylglycerol forms of unsaturated fatty acids in neuronal ceroid-lipofuscinoses. Am J Med Genet 42: 549–554.
CAS PubMed Google Scholar - Berger GE, Wood SJ, Pantelis C, Velakoulis D, Wellard RM, McGorry PD (2002). Implications of lipid biology for the pathogenesis of schizophrenia. Aust N Z J Psychiatry 36: 355–366.
PubMed Google Scholar - Bhagwagar Z, Wylezinska M, Jezzard P, Evans J, Boorman E, M Matthews P et al (2007). Low GABA concentrations in occipital cortex and anterior cingulate cortex in medication-free, recovered depressed patients. Int J Neuropsychopharmacol 11: 255–260.
PubMed Google Scholar - Bloch MH, Landeros-Weisenberger A, Sen S, Dombrowski P, Kelmendi B, Coric V et al (2008). Association of the serotonin transporter polymorphism and obsessive-compulsive disorder: Systematic Review. Am J Med Genet B Neuropsychiatr Genet 147B: 850–858.
PubMed Google Scholar - Bogdanov M, Matson W, Wang L, Matson T, Saunders-Pullman R, Bressman S et al (2008). Metabolomic profiling to develop blood biomarkers for Parkinson's disease. Brain 131: 389–396.
PubMed Google Scholar - Brambilla P, Perez J, Barale F, Schettini G, Soares JC (2003). GABAergic dysfunction in mood disorders. Mol Psychiatry 8: 721–737, 715.
CAS PubMed Google Scholar - Brindle JT, Antti H, Holmes E, Tranter G, Nicholson JK, Bethell HW et al (2002). Rapid and noninvasive diagnosis of the presence and severity of coronary heart disease using 1H-NMR-based metabonomics. Nat Med 8: 1439–1444.
CAS PubMed Google Scholar - Brindle JT, Nicholson JK, Schofield PM, Grainger DJ, Holmes E (2003). Application of chemometrics to 1H NMR spectroscopic data to investigate a relationship between human serum metabolic profiles and hypertension. Analyst 128: 32–36.
CAS PubMed Google Scholar - Coen M, Ruepp SU, Lindon JC, Nicholson JK, Pognan F, Lenz EM et al (2004). Integrated application of transcriptomics and metabonomics yields new insight into the toxicity due to paracetamol in the mouse. J Pharm Biomed Anal 35: 93–105.
CAS PubMed Google Scholar - Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW et al (1993). Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science 261: 921–923.
CAS PubMed Google Scholar - Crocker IP, Kenny LC, Thornton WA, Szabo C, Baker PN (2005). Excessive stimulation of poly(ADP-ribosyl)ation contributes to endothelial dysfunction in pre-eclampsia. Br J Pharmacol 144: 772–780.
CAS PubMed Google Scholar - Dauner M, Bailey JE, Sauer U (2001). Metabolic flux analysis with a comprehensive isotopomer model in Bacillus subtilis. Biotechnol Bioeng 76: 144–156.
CAS PubMed Google Scholar - de Graaf AA, Mahle M, Mollney M (2000). Determination of full 13C isotopomer distributions for metabolic flux analysis using heteronuclear spin echo difference NMR spectroscopy. J Biotechnol 77: 25035.
Google Scholar - de Lau LM, Koudtall PJ, Hofman A, Breteler MM (2005). Serum uric cid levels and the risk of Parkinson disease. Ann Neurol 58: 797–800.
CAS PubMed Google Scholar - Delgado PL, Moreno FA (2000). Role of norepinephrine in depression. J Clin Psychiatry 61 (Suppl 1): 5–12.
CAS PubMed Google Scholar - Dunne VG, Bhattachayya S, Besser M, Rae C, Griffin JL (2005). Metabolites from cerebrospinal fluid in aneurysmal subarachnoid haemorrhage correlate with vasospasm and clinical outcome: a pattern-recognition 1H NMR study. NMR Biomed 18: 24–33.
CAS PubMed Google Scholar - Fan TW-M, Lane AN (2007). Structure-based profiling of metabolites and isotopomers by NMR, Mol Cancer 6: 77.
Google Scholar - Fan X, Bai J, Shen P (2005). Diagnosis of breast cancer using HPLC metabonomics fingerprints coupled with computational methods. Conf Proc IEEE Eng Med Biol Soc 6: 6081–6084.
Google Scholar - Feuerstein TJ (2008). Presynaptic receptors for dopamine, histamine, and serotonin. Handb Exp Pharmacol 184: 289–338.
CAS Google Scholar - Fiehn O (2008). Extending the breadth of metabolite profiling by gas chromatography coupled to mass spectrometry. Trends Analyt Chem 27: 261–269.
CAS PubMed PubMed Central Google Scholar - Frye MA, Tsai GE, Huggins T, Coyle JT, Post RM (2007). Low cerebrospinal fluid glutamate and glycine in refractory affective disorder. Biol Psychiatry 61: 162–166.
CAS PubMed Google Scholar - Gardiner RM (2002). Clinical features and molecular genetics basis of the neuronal ceroid lipofuscinoses. Adv Neurol 89: 211–215.
PubMed Google Scholar - German JB, Gillies LA, Smilowitz JT, Zivkovic AM, Watkins SM (2007). Lipidomics and lipid profiling in metabolomics. Curr Opin Lipidol 18: 66–71.
CAS PubMed Google Scholar - Gerner RH, Fairbanks L, Anderson GM, Young JG, Scheinin M, Linnoila M et al (1984). CSF neurochemistry in depressed, manic, and schizophrenic patients compared with that of normal controls. Am J Psychiatry 141: 1533–1540.
CAS PubMed Google Scholar - Goebel HH, Sharp JD (1998). The neuronal ceroid-lipofuscinoses. Recent advances. Brain Pathol 8: 151–162.
CAS PubMed Google Scholar - Goodwin FK, Sack RL (1974). Central dopamine function in affective illness: evidence from precursors, enzyme inhibitors, and studies of central dopamine turnover. Adv Biochem Psychopharmacol 12: 261–279.
CAS PubMed Google Scholar - Han X, M Holtzman D, McKeel Jr DW, Kelley J, Morris JC (2002). Substantial sulfatide deficiency and ceramide elevation in very early Alzheimer's disease: potential role in disease pathogenesis. J Neurochem 82: 809–818.
CAS PubMed Google Scholar - Harrigan G, Goodacre R (2003). Metabolic Profiling: Its Role in Biomarker Discovery and Gene Function Analysis. Kluwer Acad. Publ: Boston.
Google Scholar - Hasler G, van der Veen JW, Tumonis T, Meyers N, Shen J, Drevets WC (2007). Reduced prefrontal glutamate/glutamine and gamma-aminobutyric acid levels in major depression determined using proton magnetic resonance spectroscopy. Arch Gen Psychiatry 64: 193–200.
CAS PubMed Google Scholar - Hofmann SL, Atashband A, Cho SK, Das AK, Gupta P, Lu JY (2002). Neuronol ceroid lipofuscinoses caused by defects in soluble lysosomal enzymes (CLN1 and CLN2). Curr Mol Med 2: 423–437.
CAS PubMed Google Scholar - Holmes E, Tsang TM, Huang JT, Leweke FM, Koethe D, Gerth CW et al (2006). Metabolic profiling of CSF: evidence that early intervention may impact on disease progression and outcome in schizophrenia. PLoS Med 3: e327.
PubMed PubMed Central Google Scholar - Horrobin DF (1998). The membrane phospholipid hypothesis as a biochemical basis for the neurodevelopmental concept of schizophrenia. Schizophr Res 30: 193–208.
CAS PubMed Google Scholar - Insel TM (2007). Devising prevention and treatment strategies for the nation's diverse populations with mental illness. Psychiatr Serv 58: 395.
PubMed PubMed Central Google Scholar - Jenkins BG, Andreassen OA, Dedeoglu A, Leavitt B, Hayden M, Borchelt D et al (2005). Effects of CAG repeat length, HTT protein length and protein context on cerebral metabolism measured using magnetic resonance spectroscopy in transgenic mouse models of Huntington's disease. J Neurochem 95: 553–562.
CAS PubMed Google Scholar - Kaddurah-Daouk R (2006). Metabolic profiling of patients with schizophrenia. PLoS Med 3: e363.
PubMed PubMed Central Google Scholar - Kaddurah-Daouk R, Kristal BS, Weinshilboum RM (2008). Metabolomics: a global biochemical approach to drug response and disease. Annu Rev Pharmacol Toxicol 48: 653–683.
CAS PubMed Google Scholar - Kaddurah-Daouk R, McEvoy J, Baillie RA, Lee D, Yao JK, Doraiswamy PM et al (2007). Metabolomic mapping of atypical antipsychotic effects in schizophrenia. Mol Psychiatry 12: 934–945.
CAS PubMed Google Scholar - Kasa K, Otsuki S, Yamamoto M, Sato M, Kuroda H, Ogawa N (1982). Cerebrospinal fluid gamma-aminobutyric acid and homovanillic acid in depressive disorders. Biol Psychiatry 17: 877–883.
CAS PubMed Google Scholar - Kell DB (2004). Metabolomics and systems biology: making sense of the soup. Curr Opin Microbiol 7: 296–307.
CAS PubMed Google Scholar - Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S et al (1998). Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392: 605–608.
CAS PubMed Google Scholar - Klein C, Djarmati A, Hedrich K, Schafer N, Scaglione C, Marchese R et al (2005). PINK1, Parkin, and DJ-1 mutations in Italian patients with early-onset parkinsonism. Eur J Hum Genet 13: 1086–1093.
CAS PubMed Google Scholar - Kristal BS, Kaddurah-Daouk R, Beal MF, Matson WR (2007a). Metabolomics: concept and potential neuroscience application. In: Handbook of Neurochemistry and Molecular Neurobiology: Brain Energetics. Integration of Molecular and Cellular Processes. Springer: New York. pp 889–912. 3rd Edition by editor Abel Lajtha.
Google Scholar - Kristal BS, Shurubor YI, Kaddurah-Daouk R, Matson WR (2007b). High-performance liquid chromatography separations coupled with coulometric electrode array detectors: a unique approach to metabolomics. Methods Mol Biol 358: 159–174.
CAS PubMed Google Scholar - Kugaya A, Sanacora G (2005). Beyond monoamines: glutamatergic function in mood disorders. CNS Spectr 10: 808–819.
PubMed Google Scholar - Lan MJ, McLoughlin GA, Griffin JL, Tsang TM, Huang JT, Yuan P et al (2008). Metabonomic analysis identifies molecular changes associated with the pathophysiology and drug reatment of bipolar disorder. Mol Psychiatry [E pub ahead of print].
- Levine J, Panchalingam K, Rapoport A, Gershon S, McClure RJ, Pettegrew JW (2000). Increased cerebrospinal fluid glutamine levels in depressed patients. Biol Psychiatry 47: 586–593.
CAS PubMed Google Scholar - Lieberman JA, Koreen AR (1993). Neurochemistry and neuroendocrinology of schizophrenia, a selective review. Schizophr Bull 19: 371–429.
CAS PubMed Google Scholar - Lieberman JA, Stroup TS, McEvoy JP, Swartz MS, Rosenheck RA, Perkins DO et al (2005). Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med 353: 1209–1223.
CAS PubMed Google Scholar - Lin MT, Beal MF (2006). Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443: 787–795.
CAS PubMed Google Scholar - Lindon JC, Nicholson JK, Holmes E, Everett JR (2000). Metabonomics: metabolic processes studied by NMR spectroscopy of biofluids. Concepts Magn Reson 12: 289–320.
CAS Google Scholar - Lindon JC, Nicholson JK, Holmes E, Antti H, Bollard ME, Keun H et al (2003). Contemporary issues in toxicology the role of metabonomics in toxicology and its evaluation by the COMET project. Toxicol Appl Pharmacol 187: 137–146.
CAS PubMed Google Scholar - Lindon JC, Holmes E, Nicholson JK (2007). Metabonomics in pharmaceutical R&D. FEBS J 274: 1149–1151.
Google Scholar - Mahadik SP, Yao JK, Phospholipids in schizophrenia (2006). In: Lieberman JA, Stroup TS, Perkins DO (eds). Textbook of Schizophrenia. American Psychiatric Publishing: Washington DC. pp 117–135.
Google Scholar - Mahmood T, Silverstone T (2001). Serotonin and bipolar disorder. J Affect Disord 66: 1–11.
CAS PubMed Google Scholar - Mitchison HM, Lim MJ, Cooper JD (2004). Selectivity and types of cell death in the neuronal ceroid lipofuscinoses. Brain Pathol 14: 86–96.
CAS PubMed Google Scholar - Mitchison HM, Mole SE (2001). Neurodegenerative disease: the neuronal ceroid lipofuscinoses (Batten disease). Curr Opin Neurol 14: 795–803.
CAS PubMed Google Scholar - Miyamoto S, LaMantia AS, Duncan GE, Sullivan P, Gilmore JH, Lieberman JA (2003). Recent advances in the neurobiology of schizophrenia. Mol Interv 3: 27–39.
PubMed Google Scholar - Morris HR (2005). Genetics of Parkinson's disease. Ann Med 37: 86–96.
CAS PubMed Google Scholar - Myers RH, MacDonald ME, Koroshetz WJ, Duyao MP, Ambrose CM, Taylor SA et al (1993). De novo expansion of a (CAG)n repeat in sporadic Huntington's disease. Nat Genet 5: 168–173.
CAS PubMed Google Scholar - Odunsi K, Wollman RM, Ambrosone CB, Hutson A, McCann SE, Tammela J et al (2005). Detection of epithelial ovarian cancer using 1H-NMR-based metabonomics. Int J Cancer 113: 782–788.
CAS PubMed Google Scholar - Paige LA, Mitchell MW, Krishnan KR, Kaddurah-Daouk R, Steffens DC (2007). A preliminary metabolomic analysis of older adults with and without depression. Int J Geriatr Psychiatry 22: 418–423.
PubMed Google Scholar - Patterson AD, Li H, Eichler GS, Krausz KW, Weinstein JN, Fornace Jr AJ et al (2008). UPLC-ESI-TOFMS-based metabolomics and gene expression dynamics inspector self-organizing metabolomic maps as tools for understanding the cellular response to ionizing radiation. Anal Chem 80: 665–674.
CAS PubMed PubMed Central Google Scholar - Pears MR, Cooper JD, Mitchison HM, Mortishire-Smith RJ, Pearce DA, Griffin JL (2005). High resolution 1H NMR-based metabolomics indicates a neurotransmitter cycling deficit in cerebral tissue from a mouse model of Batten disease. J Biol Chem 280: 42508–42514.
CAS PubMed Google Scholar - Petty F, Schlesser MA (1981). Plasma GABA in affective illness. A preliminary investigation. J Affect Disord 3: 339–343.
CAS PubMed Google Scholar - Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A et al (1997). Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science 276: 2045–2047.
CAS PubMed Google Scholar - Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A et al (1993). Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362: 59–62.
CAS PubMed Google Scholar - Rotrosen J, Wolkin A (1987). Phospholipid and prostaglandin hypotheses of schizophrenia. In: Meltzer, NY(ed). Psychopharmacology, The Third Generation on Progress. Raven Press: New York. pp 759–764.
Google Scholar - Rowland L, Shneider N (2001). Amyotrophic lateral scerosis. N Engl J Med 344: 1688–1700.
CAS PubMed Google Scholar - Rozen S, Cudkowicz ME, Bogdanov M, Matson WR, Kristal BS, Beecher C et al (2005). Metabolomic analysis and signatures in motor neuron disease. Metabolomics 1: 101–108.
CAS PubMed PubMed Central Google Scholar - Sabatine MS, Liu E, Morrow DA, Heller E, McCarroll R, Wiegand R et al (2005). Metabolomic identification of novel biomarkers of myocardial ischemia. Circulation 112: 3868–3875.
CAS PubMed Google Scholar - Sajda P (2006). Machine learning for detection and diagnosis of disease. Annu Rev Biomed Eng 8: 537–565.
CAS PubMed Google Scholar - Sanacora G, Saricicek A (2007). GABAergic contributions to the pathophysiology of depression and the mechanism of antidepressant action. CNS Neurol Disord Drug Targets 6: 127–140.
CAS PubMed Google Scholar - Serretti A, Calati R, Mandelli L, De Ronchi D (2006). Serotonin transporter gene variants and behavior: a comprehensive review. Curr Drug Targets 7: 1659–1669.
CAS PubMed Google Scholar - Schildkraut JJ (1974). Biogenic amines and affective disorders. Annu Rev Med 25: 333–348.
CAS PubMed Google Scholar - Shin H, Markey MK (2006). A machine learning perspective on the development of clinical decision support systems utilizing mass spectra of blood samples. J Biomed Inform 39: 227–248.
CAS PubMed Google Scholar - Skosnik PD, Yao JK (2003). From membrane phospholipid defects to altered neurotransmission: is arachidonic acid a nexus in the pathophysiology of schizophrenia? Prostaglandins Leukot Essent Fatty Acids 69: 367–384.
CAS PubMed Google Scholar - Sobczak S, Honig A, van Duinen MA, Riedel WJ (2002). Serotonergic dysregulation in bipolar disorders: a literature review of serotonergic challenge studies. Bipolar Disord 4: 347–356.
CAS PubMed Google Scholar - Strittmatter WJ, Weisgraber KH, Huang DY, Dong LM, Salvesen GS, Pericak-Vance M et al (1993). Binding of human apolipoprotein E to synthetic amyloid beta peptide: isoform-specific effects and implications for late onset Alzheimer disease. Proc Natl Acad Sci USA 90: 8098–8102.
CAS PubMed PubMed Central Google Scholar - Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS et al (1993). Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci USA 90: 1977–1981.
CAS PubMed PubMed Central Google Scholar - Styczynski MP, Moxley JF, Tong LV, Walther JL, Jensen KL, Stephanopoulos GN (2007). Systematic identification of conserved metabolites in GC/MS data for metabolomics and biomarker discovery. Anal Chem 79: 966–973.
CAS PubMed Google Scholar - The Huntington's Disease Collaborative Research Group (1993). A novel gene containing a trinucleatide repeat that is expanded and unstable on Huntington's Disease chromosomes. Cell 72: 971–983.
Google Scholar - Thomas B, Beal MF (2007). Parkinson's disease. Hum Mol Genet 16: R183–R194.
CAS PubMed Google Scholar - Tkachev D, Mimmack ML, Huffaker SJ, Ryan M, Bahn S (2007). Further evidence for altered myelin biosynthesis and glutamatergic dysfunction in schizophrenia. Int J Neuropsychopharmacol 10: 557–563.
CAS PubMed Google Scholar - Tolstikov VV, Fiehn O, Tanaka N (2007). Application of liquid chromatography-mass spectrometry analysis in metabolomics: reversed-phase monolithic capillary chromatography and hydrophilic chromatography coupled to electrospray ionization-mass spectrometry. Methods Mol Biol 358: 141–155.
CAS PubMed Google Scholar - Tranulis C, Skalli L, Lalonde P, Nicole L, Stip E (2008). Benefits and risks of antipsychotic polypharmacy: an evidence-based review of the literature. Drug Saf 31: 7–20.
CAS PubMed Google Scholar - Tsang TM, Huang JT, Holmes E, Bahn S (2006). Metabolic profiling of plasma from discordant schizophrenia twins: correlation between lipid signals and global functioning in female schizophrenia patients. J Proteome Res 5: 756–760.
CAS PubMed Google Scholar - Underwood B, Broadhurst D, Dunn WB, Ellis DI, Michell AW, Vacher C et al (2006). Huntington disease patients and transgenic mice have similar pro-catabolic serum metabolite profiles. Brain 129: 877–886.
PubMed Google Scholar - van der Greef J, Martin S, Juhasz P, Adourian A, Plasterer T, Verheij ER et al (2007). The art and practice of systems biology in medicine: mapping patterns of relationships. J Proteome Res 6: 1540–1559.
CAS PubMed Google Scholar - van Doorn M, Vogels J, Tas A, van Hoogdalem EJ, Burggraaf J, Cohen A et al (2007). Evaluation of metabolite profiles as biomarkers for the pharmacological effects of thiazolidinediones in type 2 diabetes mellitus patients and healthy volunteers. Br J Clin Pharmacol 63: 562–574.
CAS PubMed Google Scholar - Wang C, Kong H, Guan Y, Yang J, Gu J, Yang S et al (2005). Plasma phospholipid metabolic profiling and biomarkers of type 2 diabetes mellitus based on high-performance liquid chromatography/electrospray mass spectrometry and multivariate statistical analysis. Anal Chem 77: 4108–4116.
CAS PubMed Google Scholar - Watkins SM (2004). Lipomic profiling in drug discovery, development and clinical trial evaluation. Curr Opin Drug Discov Devel 7: 112–117.
CAS PubMed Google Scholar - Watson AD (2006). Thematic review series: systems biology approaches to metabolic and cardiovascular disorders. Lipidomics: a global approach to lipid analysis in biological systems. J Lipid Res 47: 2101–2111.
CAS PubMed Google Scholar - Weiden PJ, Buckley PF (2007). Reducing the burden of side effects during long term antipsychotic therapy: the role of switching medications. J Clin Psychiatry 68: 14–23.
CAS PubMed Google Scholar - Wichers MC, Koek GH, Robaeys G, Verkerk R, Scharpé S, Maes M (2005). IDO and interferon-alpha-induced depressive symptoms: a shift in hypothesis from tryptophan depletion to neurotoxicity. Mol Psychiatry 10: 538–544.
CAS PubMed Google Scholar - Wiest MM, Watkins SM (2007). Biomarker discovery using high-dimensional lipid analysis. Curr Opin Lipidol 18: 181–186.
CAS PubMed Google Scholar - Wisniewski KE, Zhong N, Philippart M (2001). Pheno/genotypic correlations of neuronal ceroid lipofuscinoses. Neurology 57: 576–581.
CAS PubMed Google Scholar - Yang J, Xu G, Zheng Y, Kong H, Pang T, Lv S et al (2004). Diagnosis of liver cancer using HPLC-based metabonomics avoiding false-positive result from hepatitis and hepatocirrhosis diseases. J Chromatogr B Analyt Technol Biomed Life Sci 813: 59–65.
CAS PubMed Google Scholar - Younes-Mhenni S, Frih-Ayed M, Kerkeni A, Bost M, Chazot G (2007). Peripheral blood markers of oxidative stress in Parkinson's disease. Eur Neurol 58: 78–83.
CAS PubMed Google Scholar - Yuan K, Kong H, Guan Y, Yang J, Xu G (2007). A GC-based metabonomics investigation of type 2 diabetes by organic acids metabolic profile. J Chromatogr B Analyt Technol Biomed Life Sci 850: 236–240.
CAS PubMed Google Scholar
Acknowledgements
We thank Katie Jeffress for her assistance with the preparation of this paper. Supported in part by funding from the National Institutes of Health grants R24 GM078233, ‘The Metabolomics Research Network’ (RK-D); National Institute of Neurological Disorders and Stroke and National Institute of Aging R01NS054008 (RK-D), the nonprofit organizations SMRI (RK-D), NARSAD (RK-D).
Author information
Authors and Affiliations
- Department of Psychiatry and Behavioral Sciences, Duke University Medical Center, Durham, NC, USA
Rima Kaddurah-Daouk & K Ranga Rama Krishnan
Authors
- Rima Kaddurah-Daouk
You can also search for this author inPubMed Google Scholar - K Ranga Rama Krishnan
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toRima Kaddurah-Daouk.
Additional information
DISCLOSURE/CONFLICT OF INTEREST
Dr Kaddurah-Daouk is equity holder in Metabolon Inc., a biotechnology company in the metabolomics domain, and also holds IP interest in this field. She has received funding or consultancy fees for BMS, Pfizer, Astra Zeneca, and Lundbeck. Dr Krishnan has served as consultant for Amgen, Bristol-Myer Squibb, CeNeRx, Corcept, GlaxoSmithKline, Johnson and Johnson, Lundbeck, Merck, Organon, Pfizer, Sepracor, Wyeth. In addition he is an inventor on patents in the metabolomics field.
PowerPoint slides
Rights and permissions
About this article
Cite this article
Kaddurah-Daouk, R., Krishnan, K. Metabolomics: A Global Biochemical Approach to the Study of Central Nervous System Diseases.Neuropsychopharmacol 34, 173–186 (2009). https://doi.org/10.1038/npp.2008.174
- Received: 30 July 2008
- Revised: 31 August 2008
- Accepted: 31 August 2008
- Published: 08 October 2008
- Issue Date: January 2009
- DOI: https://doi.org/10.1038/npp.2008.174