Dysbindin Modulates Prefrontal Cortical Glutamatergic Circuits and Working Memory Function in Mice (original) (raw)
INTRODUCTION
Emerging evidence suggests that a significant number of the gene mechanisms that contribute to risk for schizophrenia modulate complex cognitive functions that depend on the prefrontal cortex and the medial temporal lobe (Arguello and Gogos, 2006; Cannon, 2005; Harrison and Weinberger, 2005; Weinberger et al, 2001). Indeed, some of these genes, including DISC1, DTNBP1, COMT, and NRG1, are expressed normally within the cortical circuitry that subserves cognition (Lipska et al, 2006; Tunbridge et al, 2007; Weickert et al, 2004), and it is believed that polymorphisms within these genes consequently explain variation in cognitive phenotypes, such as declarative memory, attention, and executive functions, within the human population. For example, a rare haplotype within the DISC1 gene associates with the low prefrontal cortical volume and impairments in cognitive function, specifically working memory (Cannon et al, 2005). These genotype–phenotype relationships further underscore the value of cognitive mechanisms as candidate endophenotypes for schizophrenia and related psychotic disorders.
More recently, attention has focused on DTNBP1, the gene that codes for dystrobrevin-binding protein (dysbindin), and its relationship to cognition (Burdick et al, 2007; Burdick et al, 2006; Donohoe et al, 2007; Luciano et al, 2009). In humans, variation within this gene is associated with the risk for developing schizophrenia and impairments in cognitive performance. Moreover, this gene is expressed within cortical neurons, including pyramidal neurons (Talbot et al, 2004), suggesting that it is well-positioned to modulate functions that depend on the cortical excitatory tone (Chen et al, 2008; Numakawa et al, 2004). DTNBP1 haplotypes that represent vulnerability markers for schizophrenia appear to associate with low dysbindin expression (Bray et al, 2005); given the putative role for dysbindin in vesicular trafficking and transmitter exocytosis (Chen et al, 2008; Numakawa et al, 2004; Talbot et al, 2006; Talbot et al, 2004), a reduction in the dysbindin expression may lead to a decrease in the glutamate output (Numakawa et al, 2004), a mechanism that could explain deviant patterns of prefrontal cortical activity in schizophrenia (Manoach, 2003; Weinberger and Berman, 1988; Williamson, 1987).
Neuronal microcircuits, involving glutamatergic pyramidal neurons and local, inhibitory interneurons, within the prefrontal cortex have essential roles in the encoding and maintenance of information in working memory (Fuster, 2001; Goldman-Rakic, 1995; Miller, 2000). In particular, neurons within the prefrontal cortex show increased activity during the maintenance of information within working memory, and this ‘memory-related’ activity has been attributed to glutamate-dependent recurrent excitation within the local circuit (Compte et al, 2000; Compte et al, 2003; Durstewitz and Seamans, 2002). Compromised glutamate release, hypothetically secondary to low dysbindin expression, may be expected to compromise the ability of cortical circuits to maintain recurrent excitation and to ‘fail’ during the maintenance of information held in working memory. These alterations in glutamatergic transmission may relate to the altered patterns of prefrontal activity shown by patients with schizophrenia compared with controls across varying degrees of memory load and, in particular, to the reduced activation of cortical regions during high memory load conditions in these patients (Callicott et al, 2000; Karlsgodt et al, 2007; Karlsgodt et al, 2009; Manoach, 2003).
To examine these relationships more directly, we examined working memory function and excitatory neurotransmission within the prefrontal cortex in the so-called ‘sandy mice’ that possess a large deletion contained wholly within the dtnbp1 gene (Li et al, 2003). Several recent studies involving these null mutant mice have shown that the loss of dysbindin function impedes excitatory neurotransmission (Chen et al, 2008; Numakawa et al, 2004; Talbot et al, 2006), effects that likely correspond to behavioral abnormalities, including social deficits, enhanced stimulant sensitization, and poor performance in certain simple memory tests (Bhardwaj et al, 2009; Cox et al, 2009; Feng et al, 2008; Hattori et al, 2008; Takao et al, 2008). To complement these studies, we sought to gather direct evidence that spatial working memory function, an important endophenotype for schizophrenia (Glahn et al, 2003), is affected by the loss of dysbindin, as well as to determine whether physiological changes in the neural circuitry of working memory are a consequence of this mutation. We hypothesized that null mutation of the gene coding for dysbindin would impair working memory and that this cognitive endophenotype would be associated with a decrease in excitatory neurotransmission within circuits involved in this cognitive process.
MATERIALS AND METHODS
Animals
Dysbindin mutant mouse breeders on the DBA/2J background were obtained from the Roswell Park Cancer Institute (Conyers, GA). Experimental mice were generated by heterozygote crosses, and genotypes were determined by polymerase chain reaction. The wt product (472 bp) was amplified with the following primers: 5′-TGAGCCATTAGGAGATAAGAGCA-3′ and 5′-AGCTCCACCTGCTGAACATT-3′; the _dys_− product (274 bp) was amplified with the following primers: 5′-TCCTTGCTTCGTTCTCTGCT-3′ and 5′-CTTGCCAGCCTTCGTATTGT-3′. The fragments were separated on a 3% agarose gel.
Group-housed male mice were used in the behavioral and electrophysiological studies. The mice in the behavioral experiments were 60–100 days of age during experimentation, whereas the mice used in the recording studies were 45–60 days of age. All experimental protocols were approved by the Chancellor's Animal Research Committee at UCLA or the Medical University of South Carolina Institutional Animal Care and Use Committee.
Working Memory Testing
Mice were trained and tested using a delayed non-match-to-position test (Marrs et al. 2005; Aarde and Jentsch, 2006) in small aluminum and Plexiglas operant conditioning chambers (Med-Associates, St Albans, VT), fitted with a horizontal array of five nose-poke apertures on one side of the box and a 20-mg pellet delivery magazine on the opposite side. This delayed non-match-to-position task emphasizes retrospective encoding and maintenance of spatial information in a manner analogous to spatial delayed response tests used to measure working memory in rats, monkeys, and humans.
Each trial in the delayed non-match-to-sample task consists of both a sample phase and a choice phase. The inter-trial interval and time-out periods were 5 and 3 s, respectively. In the sample phase, one of the five nose-poke apertures was chosen at random and illuminated for up to 15 s. A response into the illuminated aperture (correct sample-phase response) caused the aperture light to extinguish and the magazine light to be illuminated. The magazine remained lit until an entry into the magazine is detected, after which the choice phase was initiated (see below). In the sample phase, the trial was aborted and a time-out ensues if the mouse makes a response in an unlit nose-poke aperture during stimulus presentation (incorrect sample-phase response) or failed to respond to the illuminated aperture during the 15-s presentation period (sample-phase omission). During the choice phase of the trial, two apertures were illuminated: the sample aperture as well as another, randomly selected aperture (the non-match location). Both of these apertures were illuminated for up to 15 s. A response in any aperture other than that of the non-match location resulted in a time-out, and an incorrect choice-phase response was recorded. Failure to make a response while the apertures were illuminated resulted in a time-out, and a choice-phase omission was recorded. Responses to the non-match location (correct response) triggered magazine illumination and pellet delivery.
During the first 30 days of training, there was no imposed delay, allowing the mice to acquire the ability to carry out the task under relatively low memory load conditions. After this initial training, probe sessions were administered in which minimum delay periods were imposed between a correct sample-phase response and the first response into the magazine that initiated the choice phase. The imposed delays were 0.5, 5, or 10 s and were interpolated in pseudorandom order and at equivalent frequencies across the session. The tasks ended after 75 trials are completed or 60 min passes, whichever comes first.
Incorrect choices and omissions during the sample phase and omissions during the choice phase were calculated and analyzed by one-way analyses of variance (ANOVAs), with genotype as the factor. Accuracy of responding during the choice phase was analyzed by repeated-measures ANOVA with genotype as the factor and delay length as the repeated measure.
Electrophysiological Recordings
Brain slices were prepared from male wt/wt, wt/_dys_−, and _dys_−/_dys_− mice. Subjects were anesthetized with the inhalant isoflurane (Abbott Laboratories). The brain was then removed and coronal slices were cut at 300 μm thickness in ice-cold high-sucrose solution containing (in mM): 200 sucrose, 1.9 KCl, 1.2 Na2HPO4, 33 NaHCO3, 6 MgCl2, 0.5 CaCl2, 10 glucose, 0.4 ascorbic acid. The slices were incubated at 33°C for at least 1 h before recordings; the incubation medium was an artificial cerebrospinal fluid solution containing (in mM): 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 4 MgCl, 1 CaCl, 10 D-glucose, 15 sucrose, 0.4 ascorbic acid, continuously aerated with 5% CO2/95% O2. After incubation, the slices were transferred to a submerged chamber and superfused with oxygenated artificial cerebrospinal fluid (in mM: 125 NaCl, 2.5 KCl, 25 NaHCO3, 2.0 CaCl2, 1.3 MgCl2, 10 glucose and 0.4 ascorbic acid) at room temperature. Recordings were determined using a Multiclamp 700B amplifier (Axon Instruments, Sunnyvale, CA), connected to a computer running Windows XP and Axograph X® software and later analyzed off-line. All recordings were obtained from neurons in layers V or VI of the prelimbic or infralimbic cortex, identified using infrared-differential interference contrast optics and video microscopy.
Current clamp
For current-clamp recordings, thick-walled borosilicate pipettes (3–7 MΩ tip resistance) were filled with (in mM): 125 K+ gluconate, 3 KCl, 2 MgCl2, 10 HEPES, 0.1 EGTA. A series of current steps (1000 ms duration, −100 to +300 pA, at 1 Hz) were injected to evoke spike firing at various steady-state membrane potentials. In between the steps, cells were held as close to −80 mV as possible through DC current injection (referred to as holding current in Table 1). Measures of the intrinsic membrane excitability (rheobase current, holding current, the number of evoked spikes, input resistance, action potential threshold, amplitude, and half-width) were compared across genotypes.
Table 1 Membrane Properties of Prefrontal Cortex Neurons Recorded in Wild-Type and Heterozygous or Homozygous Dysbindin Null Mutant Mice
Voltage clamp
For voltage-clamp recordings, electrodes (3–7 M resistance in situ) were filled with a solution containing (in mM): 135 CsCl, 10 HEPES, 2 MgCl2, 1 EGTA, 4 NaCl, 2 Na-ATP, 0.3 Tris-GTP, 1 QX-314, 10 phosphocreatine; 285 mosmol. All the voltage-clamp experiments were carried out in the presence of 100 μM picrotoxin. Series resistances (10–20 MΩ) and input resistances were continually monitored throughout the experiment through a −1 mV (100 ms) hyperpolarizing pulse. Evoked EPSCs (eEPSCs) were elicited by applying low-intensity, square-wave pulses (50–150 μA; 100 μs in duration) using a bipolar concentric electrode placed within 200 μm of the recording electrode. The eEPSC amplitude was defined as the mean amplitude during a 1- to2-ms window at the peak of the EPSC minus the amplitude during a similar window immediately before the stimulus artifact. Pulses were administered every 30 s and peak eEPSC amplitude was measured. Stimulus intensity was gradually increased in order to construct an input–output curve from 10 μA up to 500 μA (in some cases), until maximum amplitude was reached, meaning that response amplitude remained consistent regardless of increasing stimulation intensity. The responses included in the analysis were limited to those measured at 75% of maximum amplitude. Paired pulse stimuli were delivered at a frequency of 0.3 Hz, with an ISI of 50 ms.
Miniature EPSCs (mEPSCs) were obtained from 50–200 sweeps per cell; TTX (1 μM) was added to the recording buffer when mEPSCs were assessed. Amplitude and frequencies were calculated using MiniAnalysis software® with a detection threshold of 8 pA, events were then manually checked for accuracy.
Statistical Analysis
Parametric ANOVAs were used to examine main effects (eg, genotype) and interactions with repeated measures, when appropriate. Significant main effects or interactions were followed up using post hoc tests. In some cases, paired _t_-tests were used to evaluate within subject, a priori hypothesized effects. All figures present data as mean±SEM.
RESULTS
Spatial Working Memory
We first sought to determine whether null mutation of the dtnbp1 gene associates with working memory dysfunction in mice. Working memory performance of wt/wt, wt/_dys_−, and _dys_−/_dys_− mice was evaluated using an operant delayed-non-match-to-position task (Figure 1). During each trial in this procedure, mice first respond to a single illuminated aperture (the sample), experience a delay during which no cues are presented, and then must make a response into one of two apertures: the sample stimulus and a non-match stimulus. Across an initial 30-day training period in which the conditional rules of the task were trained, all groups acquired the ability and carried out the task equivalently, showing above chance levels of choice accuracy. Probe sessions were then delivered in which delays of 0.5–10 s were introduced; as expected, a main effect of delay was found for choice accuracy (F(2,56)=4.9, _p_=0.01), indicating that performance was sensitive to the length of the retention interval. Considering all completed trials, choice accuracy was significantly affected by genotype (Figure 1; F(2,28)=4.2, _p_=0.02), but the genotype × delay interaction did not reach significance; post hoc Tukey tests confirmed that mutants showed poorer choice accuracy than did controls (p<0.01), but that the difference between wild-type and heterozygous mice only reached the trend level (_p_=0.07).
Figure 1
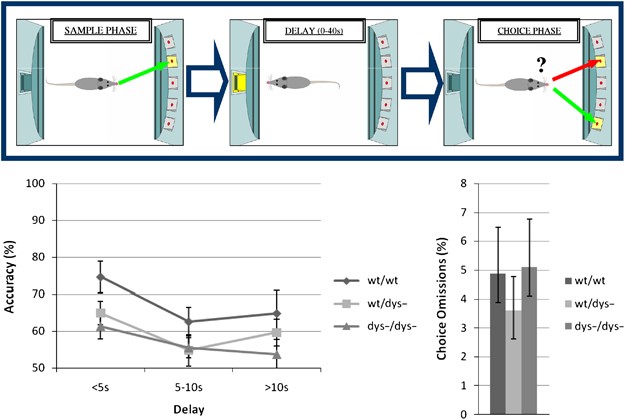
An operant delayed non-match-to-position test (schematized in the top panel) was used to assess the ability of wild-type (wt/wt) and heterozygous and homozygous null mutant dysbindin mice (wt/_dys_− and _dys_−/_dys_−, respectively) to maintain information about the spatial location of a target across a brief delay. Heterozygous and homozygous null mutant mice made fewer incorrect choices (bottom left panel) but not more omissions (bottom right panel) as compared with wild-type littermates, suggestive of a spatial working memory deficit. Data represent means±SEM.
To assess whether sensorimotor phenotypes could contribute to the observed impairments in _dys_−/_dys_− mice, we examined other aspects of task performance. We first calculated correct responding in the sample phase that only requires animals to respond to a single illuminated light; this measure was not affected by genotype (F(2,28)=0.04, _p_=0.95), indicating that the two groups were equally able to process the sensory properties of the stimuli and to emit a conditionally appropriate response. Furthermore, omission rates did not differ between the two groups for either the sample (F(2,28)=1.4, _p_=0.26) or choice phases (Figure 1; F(2.28)=0.3, _p_=0.7), indicating that both groups were equally motivated to carry out the task and were equally capable of responding within the time constraints of the two components of each trial. Therefore, the poor response accuracy in mutants observed during the non-matching phase of the trial appears to depend on the need to encode and retain information about the sample cue across the delay period.
Physiological Properties of Prefrontal Cortical Pyramidal Neurons
Given the important role for delay-related, glutamate-dependent activity of prefrontal cortical pyramidal neuron networks in the representation of visuospatial information maintained in working memory (Goldman-Rakic, 1995), we next examined whether genetic dysbindin deficiency altered glutamatergic transmission in the prefrontal cortex, using in vitro recordings from deep layer pyramidal neurons within the medial frontal cortex in wt/wt, wt_/dys_−, and _dys_−/_dys_− mice.
We first measured basic membrane properties in pyramidal neurons from all three genotypes and found that while spike amplitude, spike half-width, input resistance, and cell capacitance showed no significant effect of genotype, rheobase and spike threshold were significantly affected (Table 1). Rheobase current was significantly affected by genotype (F(2,24)=5.1; _p_=0.009, Table 1), with _dys_−/_dys_− mice showing an ∼50% reduction compared with wt/wt mice. This change in rheobase current can be attributed to a significant reduction in spike threshold in neurons recorded from _dys_−/_dys_− mice (−56.7±1.5 mV) compared with wt/wt mice (−47.2±2.0 mV; F(2,24)=5.6, _p_=0.006), but not to differences in the amount of holding current injected to maintain a membrane potential of −80 mV (Table 1), suggesting that resting membrane potentials were not different between genotypes. For rheobase and membrane threshold, the corresponding values for heterozygous mice were intermediate between wt/wt and _dys_−/_dys_− mice. These data suggest that dysbindin deletion or reduction may increase the excitability of pyramidal neurons.
We next sought to examine whether null mutation of the dtnbp1 gene affected excitatory transmission within the local circuitry of the prefrontal cortex; our a priori hypothesis, stemming from recently published study (Chen et al, 2008; Numakawa et al, 2004; Talbot et al, 2004), was that dysbindin deletion would impair excitatory synaptic transmission. An assessment of eEPSC amplitude showed a significant difference among the three genotypes (Figure 2a; F(2,44)=3.2, p<0.05), with a significant reduction in evoked responses in both wt/_dys_− and _dys_−/ _dys_− mice, relative to wt/wt controls (p<0.05). These initial data suggested that evoked synaptic responses were lower in mutant, as compared with wild-type animals.
Figure 2
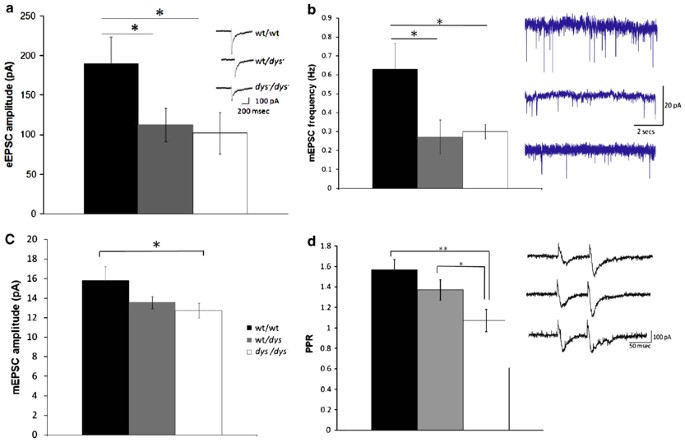
Whole cell recordings of prefrontal cortical neuron in wt/wt, wt/_dys_− and _dys_−/_dys_− mice were conducted. Null mutation of the dysbindin gene affected the amplitdue of EPSCs evoked by electrical stimulation (panel a), the frequency and amplitude of miniature EPSCs (panels b and c, respectively) and the degree of paired pulse facilitation (PPF) evoked by trains of electrical stimulations (panel d). Collectively, these results indicate a defect in pre-synaptic glutamate release from cortical neurons. Data represent means±SEM. Significantly different from wt/wt: *p<0.05, **p<0.01.
To further investigate changes in synaptic transmission, mEPSCs were recorded from pyramidal neurons in the presence of picrotoxin (100 μM) and TTX (1 μM), and the frequency of mEPSCs across a broad range of amplitudes (8–100 pA) was analyzed. We observed an overall significantly lower frequency of mEPSCs (main effect of genotype: F(2,33)=4.1, _p_=0.01) in _dys_−/_dys_− (0.3±0.03 Hz; _n_=10) and wt/_dys_− mice (0.27±0.1 Hz; _n_=7) compared with wt/wt mice (0.6±0.1 Hz; _n_=16, Figure 2b); these genotypic differences were most obvious for events in the range of 20–30 pA. In addition, mEPSC amplitude (F(2,33)=2.2, _p_=0.04) was also significantly reduced in _dys_−/_dys_− (12.7±0.8; _n_=16) compared with wt/wt neurons (15.8±1.4; _n_=10; Figure 2c). These data suggested that reduced dysbindin expression leads to significant decreases in excitatory transmission within the prefrontal cortex.
Owing of the extant data indicating that dysbindin is expressed within glutamatergic terminals and interacts with vesicle-trafficking proteins, we hypothesized that the reduction in mEPSCs may result from the decreased pre-synaptic release of glutamate in dysbindin mutants. Therefore, we next examined paired-pulse facilitation. The overall ANOVA examining the effects of genotype on the paired-pulse ratio showed a significant main effect of genotype (F(2,26)=7.4, _p_=0.0009; Figure 2d) that was largely attributable to a reduction in the paired-pulse ratio in dys_−/dys_− mice (ratio=1.1±0.06; _n_=9) compared with wt/wt controls (ratio=1.6±0.1; _n_=8). These results further support the idea that dysbindin deletion or reduction impairs cortical glutamate release.
DISCUSSION
Null mutation of the gene encoding dysbindin led to specific changes in performance of a delayed non-match-to-position test of spatial working memory and the associated, underlying neural mechanisms within the prefrontal cortex believed to mediate this cognitive function. Although dysbindin null subjects were as capable as wild-type controls in processing and responding in a conditionally appropriate way to visual cues, they were unable to respond correctly in a delayed non-match-to-position task, indicating that they had difficulty encoding or maintaining the item in spatial working memory. These data suggest that poor working memory maintenance is a phenotypic consequence to the loss of dysbindin expression.
Encoding and maintenance of visuospatial information in working memory is believed to involve the selective recruitment of the prefrontal cortical neuronal circuits, which show persistent activity during the active maintenance of the information (Compte et al, 2000; Compte et al, 2003; Durstewitz et al, 2000; Seamans et al, 2003). As dysbindin has been shown to modulate the glutamate outflow (Chen et al, 2008; Numakawa et al, 2004), we hypothesized that the poor working memory shown in dysbindin mice was due to a compromised glutamate transmission. Electrophysiological recordings from deep-layer pyramidal neurons in the medial prefrontal cortex confirmed this hypothesis by revealing that the amplitude of eEPSCs and the frequency of miniature EPSCs were decreased; moreover, paired-pulse facilitation was not present in dysbindin mutant mice. Collectively, these data support the conclusion that the pre-synaptic release of glutamate is impaired in dysbindin-deficient mice.
Of importance, hippocampal circuits also contribute to the performance of delayed match/non-match-to-sample tasks such as the one used here (Deadwyler et al, 1996; Hampson and Deadwyler, 1996, 2000; Hampson et al, 1993); hence, neural dysfunction within the hippocampus could also contribute to the working memory phenotype in dysbindin mutant mice. Other behavioral studies in dysbindin null mutant mice have found deficits in tasks that predominantly measure hippocampal function (Bhardwaj et al, 2009; Cox et al, 2009; Takao et al, 2008); hence, the current behavioral effects may not rely upon prefrontal cortical dysfunction alone. Future studies directly assessing the hippocampal circuitry, using electrophysiology and gene expression, are underway.
Relationship to Cognitive Endophenotypes for Schizophrenia
The ability to maintain and manipulate information held in working memory appears to be a quantitative trait, and the heritability of working memory performance among unaffected relatives of individuals diagnosed with schizophrenia supports the conclusion that this aspect of cognition is an endophenotype and/or marker of genetic liability for the disorder (Glahn et al, 2002; Glahn et al, 2003). Genetic variation in other schizophrenia risk genes, most notably DISC1, associates with spatial working memory function in humans (Burdick et al, 2005; Cannon et al, 2005; Koike et al, 2006; Kvajo et al, 2008; Li et al, 2007), and studies in mouse genetic models has further supported the linkages between these particular genes and cognitive performance (Cox et al, 2009; Takao et al, 2008). Further to that, the current results support a role for dysbindin in the modulation of spatial working memory and possibly other prefrontal cortical-dependent cognitive functions. It should be noted that, however, dysbindin is expressed in brain regions other than the prefrontal cortex; its considerable expression in the hippocampus suggests that it could (through cellular actions similar to those reported here to occur in the prefrontal cortex) strongly affect synaptic plasticity and associated memory functions (Cox et al, 2009; Takao et al, 2008).
Dysbindin and its Relation to Glutamatergic Mechanisms
Earlier findings regarding the function of dysbindin suggested either no, or an inverse, correlation between levels of dysbindin and VGlutT-1 protein levels in the dentate gyrus of hippocampus in humans (Talbot et al, 2004) and in younger animals (<30 days old) (Chen et al, 2008). However, recent studies using more selective antibodies suggest that there is a positive correlation between dysbindin-1 and VGluT-1 levels in older animals, such as the ones used in this study (45–60 days of age) as well as in humans (Talbot, personal communication). Therefore, it is likely that glutamate release is reduced in the dys_−/dys_− and wt/_dys_− animals due to an impairment of the cells’ ability to package glutamate into vesicles; this hypothesis is supported by our observation of reduced amplitude of eEPSCs and frequency of mEPSCs in vitro. In addition, we observed a significant decrease in the amplitude of mEPSCs in the dys_−/dys_− animals, suggesting that dysbindin deletion may further impair glutamatergic transmission through postsynaptic mechanisms, as well. This issue requires further investigation in order to specify the relevant mechanisms involved.
Interestingly, although dysbindin deletion appears to decrease glutamate release at the axon terminal, it appears that the absence of dysbindin may also result in an increase in excitability, as shown by a decrease in the amount of intra-somatic current injection required to evoke an action potential (rheobase). When the basic physiological properties of prefrontal cortical pyramidal neurons were examined, significant difference between the genotypes were found in spike threshold and rehobase current, as well as a decrease of more than 50% in cell capacitance in the wt/_dys_− and dys_−/dys_− groups. As _C_=1/R, a decrease in capacitance will result in an increase in resistance, therefore rendering the cell more excitable. Changes in capacitance may result from a reduction in cell size, neuronal dendritic branching, or spine density. Another possibility is that the loss of dysbindin may affect GABAergic interneurons, eliciting concomitant reductions in GABA release and resulting in a disinhibition of pyramidal cells, a possibility that deserves future exploration. Regardless of the mechanisms underlying this increase in excitability, it appears that any effects that might result from an increase in the generation of action potentials in the dys_−/dys_− mice may be counteracted by the relevant impairments in glutamate release. Future experiments will investigate changes in dendritic branching as well as alterations in GABA release and interneuron function in dysbindin-deficient mice.
Recordings during performance of delayed alternation tasks have shown an increase in pyramidal cell firing during the delay period of the task (Fuster and Alexander, 1971; Aarde and Jentsch, 2006; Goldman-Rakic, 1995; Miller, et al, 1996). This recurrent excitatory activity has been proposed to be the cellular correlate for working memory, suggesting that decreases in glutamatergic transmission, such as the decreases reported here in the dys_−/dys_−and wt/_dys_− mice, may underlie the cognitive deficits observed in some neuropsychiatric disorders such as schizophrenia and bipolar disorder. A promising new avenue for pharmacological treatments is aimed at targeting modulators of NMDA receptors, such as the glycine transporter, or more recently, positive allosteric modulators of the mGluR5 glutamate receptor. Several studies have shown that mGluR5 antagonists not only decrease burst firing of PFC pyramidal neurons but also impair spatial working memory (Ballard et al, 2005; Homayoun and Moghaddam, 2006; Homayoun et al, 2004; Locchi et al, 2007), suggesting that mGluR5 agonists could restore normal levels of activity in dysfunctional cortical circuits affected in schizophrenia.
References
- Aarde SM, Jentsch JD (2006). Haploinsufficiency of the arginine-vasopressin gene is associated with poor spatial working memory performance in rats. Hormones Behav 49: 501–508.
Article CAS Google Scholar - Arguello PA, Gogos JA (2006). Modeling madness in mice: one piece at a time. Neuron 52: 179–196.
Article CAS Google Scholar - Ballard TM, Woolley ML, Prinssen E, Huwyler J, Porter R, Spooren W (2005). The effect of the mGlu5 receptor antagonist MPEP in rodent tests of anxiety and cognition: a comparison. Psychopharmacology (Berl) 179: 218–229.
Article CAS Google Scholar - Bhardwaj SK, Baharnoori M, Sharif-Askari B, Kamath A, Williams S, Srivastava LK (2009). Behavioral characterization of dysbindin-1 deficient sandy mice. Behav Brain Res 197: 435–441.
Article CAS Google Scholar - Bhardwaj SK, Baharnoori M, Sharif-Askari B, Kamath A, Williams S, Srivastava LK (2009). Behavioral characterization of dysbindin-1 deficient sandy mice. Behav Brain Res 197: 435–441.
Article CAS Google Scholar - Bray NJ, Preece A, Williams NM, Moskvina V, Buckland PR, Owen MJ et al (2005). Haplotypes at the dystrobrevin binding protein 1 (DTNBP1) gene locus mediate risk for schizophrenia through reduced DTNBP1 expression. Hum Mol Genet 14: 1947–1954.
Article CAS Google Scholar - Burdick KE, Goldberg TE, Funke B, Bates JA, Lencz T, Kucherlapati R et al (2007). DTNBP1 genotype influences cognitive decline in schizophrenia. Schizophr Res 89: 169–172.
Article Google Scholar - Burdick KE, Hodgkinson CA, Szeszko PR, Lencz T, Ekholm JM, Kane JM et al (2005). DISC1 and neurocognitive function in schizophrenia. Neuroreport 16: 1399–1402.
Article Google Scholar - Burdick KE, Lencz T, Funke B, Finn CT, Szeszko PR, Kane JM et al (2006). Genetic variation in DTNBP1 influences general cognitive ability. Hum Mol Genet 15: 1563–1568.
Article CAS Google Scholar - Callicott JH, Bertolino A, Mattay VS, Langheim FJ, Duyn J, Coppola R et al (2000). Physiological dysfunction of the dorsolateral prefrontal cortex in schizophrenia revisited. Cereb Cortex 10: 1078–1092.
Article CAS Google Scholar - Cannon TD (2005). The inheritance of intermediate phenotypes for schizophrenia. Curr Opin Psychiatry 18: 135–140.
Article Google Scholar - Cannon TD, Hennah W, van Erp TG, Thompson PM, Lonnqvist J, Huttunen M et al (2005). Association of DISC1/TRAX haplotypes with schizophrenia, reduced prefrontal gray matter, and impaired short- and long-term memory. Arch Gen Psychiatry 62: 1205–1213.
Article CAS Google Scholar - Chen XW, Feng YQ, Hao CJ, Guo XL, He X, Zhou ZY et al (2008). DTNBP1, a schizophrenia susceptibility gene, affects kinetics of transmitter release. J Cell Biol 181: 791–801.
Article CAS Google Scholar - Compte A, Brunel N, Goldman-Rakic PS, Wang XJ (2000). Synaptic mechanisms and network dynamics underlying spatial working memory in a cortical network model. Cereb Cortex 10: 910–923.
Article CAS Google Scholar - Compte A, Constantinidis C, Tegner J, Raghavachari S, Chafee MV, Goldman-Rakic PS et al (2003). Temporally irregular mnemonic persistent activity in prefrontal neurons of monkeys during a delayed response task. J Neurophysiol 90: 3441–3454.
Article Google Scholar - Cox MM, Tucker AM, Tang J, Talbot K, Richer DC, Yeh L et al (2009). Neurobehavioral abnormalities in the dysbindin-1 mutant, sandy, on a C57BL/6J genetic background. Genes Brain Behav 8: 390–397.
Article CAS Google Scholar - Deadwyler SA, Bunn T, Hampson RE (1996). Hippocampal ensemble activity during spatial delayed-nonmatch-to-sample performance in rats. J Neurosci 16: 354–372.
Article CAS Google Scholar - Donohoe G, Morris DW, Clarke S, McGhee KA, Schwaiger S, Nangle JM et al (2007). Variance in neurocognitive performance is associated with dysbindin-1 in schizophrenia: a preliminary study. Neuropsychologia 45: 454–458.
Article Google Scholar - Durstewitz D, Seamans JK (2002). The computational role of dopamine D1 receptors in working memory. Neural Netw 15: 561–572.
Article Google Scholar - Durstewitz D, Seamans JK, Sejnowski TJ (2000). Dopamine-mediated stabilization of delay-period activity in a network model of prefrontal cortex. J Neurophysiol 83: 1733–1750.
Article CAS Google Scholar - Feng YQ, Zhou ZY, He X, Wang H, Guo XL, Hao CJ et al (2008). Dysbindin deficiency in sandy mice causes reduction of snapin and displays behaviors related to schizophrenia. Schizophr Res 106: 218–228.
Article Google Scholar - Fuster J, Alexander GE (1971). Neuron activity related to short-term memory. Science 173: 652–654.
Article CAS Google Scholar - Fuster JM (2001). The prefrontal cortex—an update. Neuron 30: 319–333.
Article CAS Google Scholar - Glahn DC, Kim J, Cohen MS, Poutanen VP, Therman S, Bava S et al (2002). Maintenance and manipulation in spatial working memory: dissociations in the prefrontal cortex. NeuroImage 17: 201–213.
Article CAS Google Scholar - Glahn DC, Therman S, Manninen M, Huttunen M, Kaprio J, Lonnqvist J et al (2003). Spatial working memory as an endophenotype for schizophrenia. Biol Psychiatry 53: 624–626.
Article Google Scholar - Goldman-Rakic PS (1995). Cellular basis of working memory. Neuron 14: 477–485.
Article CAS Google Scholar - Hampson RE, Deadwyler SA (1996). Ensemble codes involving hippocampal neurons are at risk during delayed performance tests. Proc Natl Acad Sci USA 93: 13487–13493.
Article CAS Google Scholar - Hampson RE, Deadwyler SA (2000). Cannabinoids reveal the necessity of hippocampal neural encoding for short-term memory in rats. J Neurosci 20: 8932–8942.
Article CAS Google Scholar - Hampson RE, Heyser CJ, Deadwyler SA (1993). Hippocampal cell firing correlates of delayed-match-to-sample performance in the rat. Behav Neurosci 107: 715–739.
Article CAS Google Scholar - Harrison PJ, Weinberger DR (2005). Schizophrenia genes, gene expression, and neuropathology: on the matter of their convergence. Mol Psychiatry 10: 40–68; image 45.
Article CAS Google Scholar - Hattori S, Murotani T, Matsuzaki S, Ishizuka T, Kumamoto N, Takeda M et al (2008). Behavioral abnormalities and dopamine reductions in sdy mutant mice with a deletion in Dtnbp1, a susceptibility gene for schizophrenia. Biochem Biophys Res Commun 373: 298–302.
Article CAS Google Scholar - Homayoun H, Moghaddam B (2006). Bursting of prefrontal cortex neurons in awake rats is regulated by metabotropic glutamate 5 (mGlu5) receptors: rate-dependent influence and interaction with NMDA receptors. Cereb Cortex 16: 93–105.
Article Google Scholar - Homayoun H, Stefani MR, Adams BW, Tamagan GD, Moghaddam B (2004). Functional interaction between NMDA and mGlu5 receptors: effects on working memory, instrumental learning, motor behaviors, and dopamine release. Neuropsychopharmacology 29: 1259–1269.
Article CAS Google Scholar - Karlsgodt KH, Glahn DC, van Erp TG, Therman S, Huttunen M, Manninen M et al (2007). The relationship between performance and fMRI signal during working memory in patients with schizophrenia, unaffected co-twins, and control subjects. Schizophr Res 89: 191–197.
Article Google Scholar - Karlsgodt KH, Sanz J, van Erp TG, Bearden CE, Nuechterlein KH, Cannon TD (2009). Re-evaluating dorsolateral prefrontal cortex activation during working memory in schizophrenia. Schizophr Res 108: 143–150.
Article Google Scholar - Koike H, Arguello PA, Kvajo M, Karayiorgou M, Gogos JA (2006). Disc1 is mutated in the 129S6/SvEv strain and modulates working memory in mice. Proc Natl Acad Sci USA 103: 3693–3697.
Article CAS Google Scholar - Kvajo M, McKellar H, Arguello PA, Drew LJ, Moore H, MacDermott AB et al (2008). A mutation in mouse Disc1 that models a schizophrenia risk allele leads to specific alterations in neuronal architecture and cognition. Proc Natl Acad Sci USA 105: 7076–7081.
Article CAS Google Scholar - Li W, Zhang Q, Oiso N, Novak EK, Gautam R, O’Brien EP et al (2003). Hermansky–Pudlak syndrome type 7 (HPS-7) results from mutant dysbindin, a member of the biogenesis of lysosome-related organelles complex 1 (BLOC-1). Nat Genet 35: 84–89.
Article CAS Google Scholar - Li W, Zhou Y, Jentsch JD, Brown RA, Tian X, Ehninger D et al (2007). Specific developmental disruption of disrupted-in-schizophrenia-1 function results in schizophrenia-related phenotypes in mice. Proc Natl Acad Sci USA 104: 18280–18285.
Article CAS Google Scholar - Lipska BK, Peters T, Hyde TM, Halim N, Horowitz C, Mitkus S et al (2006). Expression of DISC1 binding partners is reduced in schizophrenia and associated with DISC1 SNPs. Hum Mol Genet 15: 1245–1258.
Article CAS Google Scholar - Locchi F, Dall’Olio R, Gandolfi O, Rimondini R (2007). Water T-maze, an improved method to assess spatial working memory in rats: pharmacological validation. Neurosci Lett 422: 213–216.
Article CAS Google Scholar - Luciano M, Miyajima F, Lind PA, Bates TC, Horan M, Harris SE et al (2009). Variation in the Dysbindin gene and normal cognitive function in three independent population samples. Genes Brain Behav 8: 218–227.
Article CAS Google Scholar - Manoach DS (2003). Prefrontal cortex dysfunction during working memory performance in schizophrenia: reconciling discrepant findings. Schizophr Res 60: 285–298.
Article Google Scholar - Marrs W, Kuperman J, Avedian T, Roth RH, Jentsch JD (2005). Alpha-2 adrenoceptor activation inhibits phencyclidine-induced deficits of spatial working memory in rats. Neuropsychopharmacol 30: 1500–1510.
Article CAS Google Scholar - Miller EK (2000). The prefrontal cortex and cognitive control. Nat Rev Neurosci 1: 59–65.
Article CAS Google Scholar - Miller EK, Erickson CA, Desimone R (1996). Neural mechanisms of visual working memory in prefrontal cortex of the macaque. J Neurosci 16: 5154–5167.
Article CAS Google Scholar - Numakawa T, Yagasaki Y, Ishimoto T, Okada T, Suzuki T, Iwata N et al (2004). Evidence of novel neuronal functions of dysbindin, a susceptibility gene for schizophrenia. Hum Mol Genet 13: 2699–2708.
Article CAS Google Scholar - Seamans JK, Nogueira L, Lavin A (2003). Synaptic basis of persistent activity in prefrontal cortex in vivo and in organotypic cultures. Cereb Cortex 13: 1242–1250.
Article Google Scholar - Takao K, Toyama K, Nakanishi K, Hattori S, Takamura H, Takeda M et al (2008). Impaired long-term memory retention and working memory in sdy mutant mice with a deletion in Dtnbp1, a susceptibility gene for schizophrenia. Mol Brain 1: 11.
Article Google Scholar - Talbot K, Cho DS, Ong WY, Benson MA, Han LY, Kazi HA et al (2006). Dysbindin-1 is a synaptic and microtubular protein that binds brain snapin. Hum Mol Genet 15: 3041–3054.
Article CAS Google Scholar - Talbot K, Eidem WL, Tinsley CL, Benson MA, Thompson EW, Smith RJ et al (2004). Dysbindin-1 is reduced in intrinsic, glutamatergic terminals of the hippocampal formation in schizophrenia. J Clin Invest 113: 1353–1363.
Article CAS Google Scholar - Tunbridge EM, Weickert CS, Kleinman JE, Herman MM, Chen J, Kolachana BS et al (2007). Catechol-_o_-methyltransferase enzyme activity and protein expression in human prefrontal cortex across the postnatal lifespan. Cereb Cortex 17: 1206–1212.
Article CAS Google Scholar - Weickert CS, Straub RE, McClintock BW, Matsumoto M, Hashimoto R, Hyde TM et al (2004). Human dysbindin (DTNBP1) gene expression in normal brain and in schizophrenic prefrontal cortex and midbrain. Arch Gen Psychiatry 61: 544–555.
Article CAS Google Scholar - Weinberger DR, Berman KF (1988). Speculation on the meaning of cerebral metabolic hypofrontality in schizophrenia. Schizophr Bull 14: 157–168.
Article CAS Google Scholar - Weinberger DR, Egan MF, Bertolino A, Callicott JH, Mattay VS, Lipska BK et al (2001). Prefrontal neurons and the genetics of schizophrenia. Biol Psychiatry 50: 825–844.
Article CAS Google Scholar - Williamson P (1987). Hypofrontality in schizophrenia: a review of the evidence. Can J Psychiatry 32: 399–404.
Article CAS Google Scholar
Acknowledgements
This work was supported by PHS Grants MH-83269 (TDC, JDJ, and AL) and K12-GM081265 (HT-D) and an NARSAD Distinguished Investigator Award (TDC).
Author information
Authors and Affiliations
- Department of Psychology, University of California, Los Angeles, CA, USA
James David Jentsch, Corey Jairl, Matthew Tinsley & Tyrone D Cannon - Staglin Family Music Festival Center for Cognitive Neuroscience, Semel Institute for Neuroscience and Human Behavior, University of California, Los Angeles, CA, USA
James David Jentsch & Tyrone D Cannon - Consortium for Neuropsychiatric Phenomics, University of California, Los Angeles, CA, USA
James David Jentsch, Tyrone D Cannon & Antonieta Lavin - Department of Neurosciences, Medical University of South Carolina, Charleston, SC, USA
Heather Trantham-Davidson & Antonieta Lavin
Authors
- James David Jentsch
- Heather Trantham-Davidson
- Corey Jairl
- Matthew Tinsley
- Tyrone D Cannon
- Antonieta Lavin
Corresponding author
Correspondence toJames David Jentsch.
Additional information
DISCLOSURE/CONFLICT OF INTEREST
Dr Jentsch has received compensation as a consultant for Merck Research Laboratories. The other author(s) declare that, except for income received from their primary employer, no financial support or compensation has been received from any individual or corporate entity over the past 3 years for research or professional service, and there are no personal financial holdings that could be perceived as constituting a potential conflict of interest.
Rights and permissions
About this article
Cite this article
Jentsch, J., Trantham-Davidson, H., Jairl, C. et al. Dysbindin Modulates Prefrontal Cortical Glutamatergic Circuits and Working Memory Function in Mice.Neuropsychopharmacol 34, 2601–2608 (2009). https://doi.org/10.1038/npp.2009.90
- Received: 11 April 2009
- Revised: 14 June 2009
- Accepted: 17 June 2009
- Published: 29 July 2009
- Issue Date: November 2009
- DOI: https://doi.org/10.1038/npp.2009.90