Mass Spectrometry Strategies for Clinical Metabolomics and Lipidomics in Psychiatry, Neurology, and Neuro-Oncology (original) (raw)
INTRODUCTION
Metabolomics, including the subfield of lipidomics, involve the analysis of endogenous biomolecules, generally <2000 Da, in complex biological matrices (Nicholson et al, 1999). The metabolome consists of a diverse array of biomolecules that are the ultimate products of transcription, translation, and protein activities (Figure 1). As a result of the chemical diversity of these metabolites, no single analytical platform can sample the entire metabolome/lipidome. Mass spectrometric approaches are some of the most robust analytical platforms that provide both structural and sensitive quantitative data for complex biological samples (Koal and Deigner, 2010; Dudley et al, 2010; Mishur and Rea, 2012). Mass spectrometry is being used to analyze the metabolome (Dudley et al, 2010), lipidome (Postle, 2012), proteome (Al-Ayadhi and Halepoto, 2013), microRNAs (Izumi et al, 2012), epigenetic methylation and acetylation of histones (Britton et al, 2011), and DNA methylation (Hu et al, 2012). In this review, the focus will be metabolomics and lipidomics platforms and their utility in neurology, neuro-oncology, and psychiatry. Specifically, we will focus on sulfur amino-acid metabolism as an example of metabolomics studies and on plasmalogens as an example of lipidomics studies in Alzheimer’s disease.
Figure 1
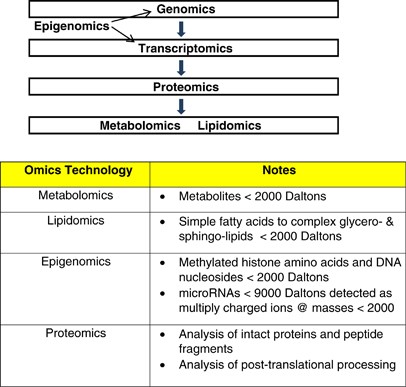
Overview of omics technologies in which mass spectrometry is being utilized.
HISTORICAL BACKGROUND
Mass spectrometry platforms are utilized for the structural elucidation of unidentified biomolecules and to accurately quantitate biomolecules based upon mass selective detection. Advances in methods for the introduction of complex biological extracts into a mass spectrometer, which is under high vacuum, combined with the development of high-resolution and tandem mass spectrometry systems have provided the mass spectrometry platforms used today for metabolomics research. The introduction in 1978 of the first commercial gas chromatograph-quadrupole mass spectrometer with a four-channel PROMIM unit for mass fragmentographic quantiation resulted in a rapid expansion of the uses of gas chromatography–mass spectrometry (GC–MS) in the neurosciences. During the 1970s and 1980s, the laboratories of Erminio Costa, Richard Wyatt, and other early visionaries trained a new generation of scientists in the uses of GC–MS in neuroscience research. A number of general principles were generated during these two decades of research utilizing GC–MS for quantitation of amino acids, biogenic amines, acetylcholine, and psychoactive drugs (see Summary Box: Basic Concepts of Targeted Metabolomics).
Although the majority of studies during this period focused on the targeted analysis of up to 10 metabolites, the first large-scale metabolomics study was published in 1978 (Gates et al, 1978). This study characterized over 150 urinary organic acids in a single GC–MS analysis, using the oxime-TMS derivatization procedure that remains popular today for non-targeted GC–MS metabolomics studies.
The next major advance that dominated the 1990s was the introduction of electrospray ionization (Fenn et al, 1989) for liquid chromatography tandem mass spectrometry (LC-ESI-MS/MS), with triple-quadrupole instruments dominating the field. This technology had the advantage of not requiring sample derivatization and the ability to analyze polar and large-molecular-weight biomolecules that could not be analyzed by GC–MS. This MS platform soon became a major methodology for drug pharmacokinetic and neurochemical analyses. The only disadvantage to LC–MS and LC–MS/MS is the inferior chromatography relative to gas chromatography.

In the 2000s, technical advances led to the development of high-resolution mass spectrometry systems. The first commercial Fourier transform ion cyclotron resonance (Marshall et al, 1998), orbitrap (Hu et al, 2005), and time-of-flight (Emary et al, 1990) mass spectrometers, coupled with HPLC, were introduced into the marketplace. Unit mass resolution, as is obtained with quadrapoles, was replaced by high-resolution (>140 000) mass analyses, a very advantageous feature for the analysis of complex biological matrices.
Mass Spectrometry and the ‘Omics’ Technologies
The aforementioned advances in mass spectrometry (MS) strategies have broadened the scope for metabolomics analyses of human diseases and disease models. The ‘omics’ technologies utilizing MS include epigenomics, metabolomics, lipidomics, and proteomics. The highest value asset of these approaches is that they can readily be applied to patient samples, thus bypassing many of the assumptions that are intrinsic to all animal models of human disease. In the case of the neurosciences, these technologies can be applied to biofluids (the cerebrospinal fluid, plasma, saliva, urine), brain dialysates, tissue biopsies, post-mortem brain analyses, and human cell lines. In metabolomics research, there is increasing usage of plasma, saliva, and lymphoblasts derived from specific patient populations. In all of these sampling paradigms, a fixed snapshot of a metabolite’s steady-state level can be obtained readily.
In clinical studies, a specific patient population is compared with appropriately matched controls in an effort to define previously unrecognized associations between metabolites and clinical end points. Preliminary efforts to define such biomarkers generally utilize non-targeted analyses, with the wealth of data obtained being subjected to extensive deconvolution to identify specific metabolites. The theoretical construct behind this approach is that the analyses are unbiased and will sample a large portion of the metabolome; however, these assumptions have a number of limitations that should not be overlooked:
- 1
Bias . Although non-targeted analytical approaches are termed ‘non-biased’, they are in fact biased to detect molecules that are present in a sample at high concentrations and biomolecules that ionize more efficiently than competing analytes. - 2
Trace Metabolites . Non-targeted analyses often do not possess the sensitivity required to detect/monitor trace metabolites. Targeted metabolomics studies utilizing stable isotope internal standards are the superior approaches for monitoring trace but critical metabolites. - 3
Quantitation . Non-targeted analyses provide relative levels of metabolites, whereas targeted methods generate absolute values. The trade-off is that, in non-targeted analyses, analytical standards are not required, whereas in targeted analyses they are critical. This is the most serious limitation for targeted metabolomics in that there are a large number of metabolites for which analytic standards are not commercially available and even fewer stable isotope internal standards are available. The perfect stable isotope internal standards are 13C, 15N, 34S, and 18O. Deuterated (2H) internal standards are often used because of their lower cost and greater availability; however, they suffer from differences in retention times in chromatographic systems and from differential solubility in solvents, and in some cases deuterium exchange can occur. Stable isotope internal standards are superior to structural analogs in correcting for ionization efficiency, shifts in retention times, and for sample losses in extraction, transfers, and derivatization (Ciccimaro and Blair, 2010). - 4
Platform Limitations . No single analytical platform, targeted or non-targeted, is capable of sampling the entire metabolome/lipidome. Multiple platforms are required and demand validation for different biological matrices if reproducible data are to be published. This validation is essential, such that reported metabolites are not artifacts of sample preparation or sample derivatization as has occurred, for example, with a number of reports of untargeted metabolomics studies utilizing the oxime-TMS derivatization protocol for GC–MS. In our laboratory, we utilize GC–MS/MS (Thermo Quantum triple quadrapole) for small molecules that can be volatilized by sample derivatization and nano-LC high-resolution MS (Thermo Q-Exactive orbitrap) for highly charged and high-molecular-weight biomolecules. High-resolution MS is also utilized in which sample interferences cannot be resolved by GC–MS/MS and for direct infusion (‘shotgun’) analyses. - 5
Matrix Effects . Co-eluting materials can act to mask the true levels of a metabolite during the ionization process in the MS source, thereby biasing non-targeted analyses. This technical problem can be significantly minimized by the use of stable isotope internal standards in targeted analyses. Nano-LC-MS also offers a potential solution to this issue with non-targeted analyses, as the small droplets produced by the reduced flow rates result in reduced ion suppression from matrix components (Emmett et al, 1995; Valaskovic et al, 2006). - 6
Unidentified Metabolites. There is an ever-increasing accumulation of non-identified metabolites in publications of non-targeted research studies. This is an alarming trend in that there is minimal follow-up on the identification of these metabolites. - 7
Identified but not Validated Metabolites . Unfortunately, there is also an increasing number of metabolomics publications in which metabolites are identified by database searches of recorded masses but which are not validated with analytical standards. Further, follow-up with expanded investigations of ancillary metabolites associated with a given metabolite change is not undertaken. This lack of scientific rigor does not provide the foundation required for translational research.
Direct Infusion (‘Shotgun’) Analyses
The increased specificity provided by tandem MS and high-resolution MS has stimulated a new trend of constant infusion MS with no chromatography, an approach that has also been termed ‘shotgun’ MS. This now has also been extended to targeted studies utilizing constant infusion tandem MS multiple reaction monitoring (CI-MRM) of lipids (Han, 2005) and metabolites (Sun et al, 2007). These approaches are useful in a number of bioanalytical situations but demand increased vigilance in demonstrating specificity, as these methods cannot distinguish between confounding geometric isomers (eg, L-serine and D-serine), isobars (eg bis(monoacylglycero)phosphate and phosphatidylglycerol), and structural isomers (eg, alanine, _β_-alanine, and sarcosine).
Targeted Metabolomics Platforms: Steady-State Measurements
Targeted metabolomics assay platforms are extremely useful for hypothesis testing. These experiments require extensive planning to define the key metabolites in biochemical pathways of interest. Rate-limiting precursors, end-product pools, critical intermediates, and potential alternate precursors or products need to be incorporated into an assay platform. As no single analytical platform can sample all metabolites, this generally requires the design of multiple platforms that provide the greatest selectivity and sensitivity for specific chemotypes. Attempting to sample a broad scope of the metabolome with a single platform inevitably results in limitations for the quantitation of some metabolites and the risk of publishing incorrect data. In the design of targeted metabolomics platforms, the use of transcriptomics and proteomics findings, along with previous findings from non-targeted studies, greatly improves the utility of individualized assay platforms. Intelligent design of targeted assay platforms allows investigators to rationally interrogate defined biochemical pathways in-depth.
Biofluid Compartments
Biopsy and post-mortem tissues are often utilized to generate metabolomics profiles that can define strategies for sampling patient biofluids. In the case of brain studies, biopsy samples are mainly limited to cancer patients. Autopsy tissues are valuable resources when the post-mortem sampling time, storage conditions, and patient clinical information are all accurately recorded.
The optimal biofluid for metabolomics studies of neurological or psychiatric disorders is the cerebrospinal fluid (CSF), which is in intimate contact with brain structures. However, CSF studies are complicated in that they generally involve sampling of the spinal CSF, which is far removed from rostral brain structures where it is formed, and also by the dynamics of CSF export to the bloodstream.
Brain microdialysis is also used to perfuse local brain areas and obtain a profile of localized metabolism (Bianchi et al, 2004; Wibom et al, 2010). However, CSF collection and brain microdialysis are both invasive procedures. Plasma is the alternate biofluid of choice, as it is more easily sampled and interacts with all tissues, providing an integrated output of changes in the homeostasis of this central compartment. Saliva metabolomics is also an area of increasing investigation because of the ease of obtaining this biofuid (Zhang et al, 2009). Investigation of patient plasma cell populations (erythrocytes, platelets, and lymphocytes) also provides useful information of cellular metabolism in relation to plasma measurements:
- 1
Erythrocytes : This is a circulating cell population that lacks organelles such that all proteins have to be imported. A number of transporter functions can be characterized in erythrocytes: for example, erythrocytes accumulate ergothioneine via the SLC22A4 transporter (Gründemann et al, 2005); erythrocytes are also useful for assessing the glutathione status as has been done in metabolic studies of schizophrenia (Altuntas et al, 2000). - 2
Platelets : Platelets synthesize a large number of complex mediators that can be quantitated via metabolomic approaches. These cells also possess a large array of transporters that can be assessed, as has been done for the dopamine transporter (Frankhauser et al, 2006) - 3
Lymphocytes : Mediators of immune function can be monitored in this complex cell population. Lymphocytes have been used to assess cellular glutathione status in autism (Suh et al, 2008) and mechanisms of DNA methylation in schizophrenia (Zhubia et al, 2009).
A further major resource is the availability of lymphoblasts from different disease populations (Coriell Cell Repository; Autism Genetics Resource Exchange). In addition to steady-state measurements of metabolites, transporter function and metabolic flux rates can be monitored with these cells in culture (Wood et al, 2011).
Although flux rates can be determined in biofluids via stable isotope precursor labeling to evaluate precursor–product relationships (Creek et al, 2012), such clinical studies are rarely conducted at this time. Alternate approaches are to measure levels of rate-limiting precursors in defined pathways as well as stable end-product pools. Measurements of precursor/product ratios provide valuable indices of metabolic dynamics/perturbations (Krone et al, 2010), and in cases of differential compartmentation of the precursor and product pools further information regarding the balance between product synthesis and transport rates can be obtained.
Translational Research: Moving Metabolomics Tools to the Clinic
The metabolome is the ultimate product of gene, mRNA, and protein activities (Figure 1). As such, metabolomics has the potential for disease diagnosis, for stratification of patients in a heterogeneous patient population, for monitoring therapeutic efficacy and disease progression, and for defining new therapeutic targets (Nordström and Lewensohn, 2010; Abu-Asab et al, 2011). All of these are important properties for neurological and psychiatric diseases, almost all of which are multifactorial conditions not involving a single-gene mutation.
Despite great expectations for translational research in metabolomics, only a limited number of biomarkers have been validated via this approach to date. A number of factors have contributed to this void:
- A large number of metabolomics investigators are oblivious to the complicated issue of heterogeneity in patient populations (Abu-Asab et al, 2011). This heterogeneity demands that the normal range for metabolites be established as a part of a comprehensive metabolomics study. Subsequent subtyping of patients within a given disease diagnosis will ultimately provide superior diagnostic tools to support personalized patient care.
- The vast majority of published metabolomics studies are statistically under-powered, limiting the ability to draw firm conclusions. There are numerous reports of patient populations with an N of 20, which is entirely insufficient to evaluate heterogeneous patient groups.
- There is an alarming trend to present large heat maps of metabolomics data without reporting the essential/critical metabolites that are changed and the magnitude of their perturbation. This is analogous to a fisherman returning home and presenting the fish echogram but not presenting the fish he/she caught.
Familial vs Sporadic Disease Populations
Patients with neurological disorders are predominantly those with sporadic disease. However, there are also smaller numbers of familial disease populations, as is observed in Alzheimer’s disease (AD) and amyotrophic lateral sclerosis (ALS). The intuitive assumption is that heterogeneity is less in a familial disease population. This has in fact been clearly validated with metabolomics studies of ALS-cerebrospinal fluid in sporadic and familial patients (Wuolikainen et al, 2011). However, heterogeneity in both familial and sporadic patient populations involves a number of factors that need to be considered in the analysis of metabolomics data sets:
- Differing rates of disease progression
- Environmental and lifestyle factors
- Differing phenotypic profiles of gastrointestinal microflora
- Co-existing pathologies
- Potential of alternate pathways to result in a final common pathology
Tiered Analyses of Biological Samples
In our studies of clinical samples, we utilize a tiered approach to identify points of interest in biochemical dysfunction and rapidly generate quantitative data. First we obtain broad-range, high-resolution (140 000 @ 200), negative and positive ion spectra (200 to 2000 daltons) with direct infusion ESI (Schumann et al, 2012). For these studies, we analyze both methyl-tert butyl ether (Wood et al, 2010) and acetonitrile/methanol/formic acid extracts (Wood et al, 2011), with each extraction solution containing a number of stable isotope internal standards across the analysis range of molecular weights. When biochemical differences are detected in a defined patient population, we next expand the analysis of that pathway and interconnected pathways and implement chromatographic methods to validate the observations. Once a biochemical change is better characterized, we next evaluate circulating blood cells (erythrocytes and platelets), and, if available, we utilize patient lymphoblasts for precursor-labeling studies. This tiered and integrated approach rapidly yields data that define the potential etiology and consequences of detected metabolic changes and define which analytical standards are required to purchase, thereby containing operational costs.
Metabolomics in Psychiatric Disorders: Sulfur Amino-Acid (SAA) Metabolism in Autism and Schizophrenia
The NIMH Research Domain Criteria (RDoC) initiative has been formulated to advance research focused on defining the etiologies of mental health disorders (Morris and Cuthbert, 2012). As mental illness is considered to be the result of dysfunctional brain circuits, metabolomics has considerable value in contributing to this initiative. Non-targeted metabolomics may reveal unexpected biochemical observations, whereas targeted metabolomics platforms, designed to investigate focused hypotheses of neuronal dysfunction, are more likely to generate the data required to discount or validate hypothetical constructs of biochemical deficits or excesses in mental illness.
In multifactorial diseases, a patient’s phenotype is dependent upon metabolic dysfunctions in multiple pathways; therefore, a given metabolic pathway may be involved in more than one disease population and may demonstrate heterogeneous expression within a given disease population. To exemplify this approach, we will review the current status of sulfur amino-acid metabolomics in schizophrenia and autism, two psychiatric neurodevelopmental disorders that are characterized by significant heterogeneity in phenotypic expression. A key principle of modern metabolomics is that, if a significant perturbation in a metabolic pathway is detected in a non-targeted analysis, then other metabolites in that pathway, or in alternate pathways where metabolites can be diverted, should also be detected in a more targeted metabolomics analysis. In the case of thiol pathways, the interrelationships of many complex and compartmentalized metabolites make this targeted analysis difficult but relevant, as thiols subserve a diverse array of metabolic and epigenetic functions.
In both patient populations, sulfur amino-acid metabolism is altered in a significant subset of patients. These observations are sometimes considered disturbing; however, such a view is not appropriate as this type of heterogeneity is consistent with the multifactorial basis of these diseases.
As presented in Figure 2, SAA metabolism is involved in a diverse array of biochemical pathways that regulate the following:
- 1
Endogenous antioxidant pools of glutathione (GSH). - 2
Epigenetic methylation of DNA and methylation of numerous substrates. _S_-adenosymethionine (SAM) is the methyl donor for a diverse array of methyltransferases. - 3
Polyamine synthesis, which requires SAM also as an aminopropyl donor. - 4
The synthesis of 3-amino-3-carboxypropyl, which is involved in the post-translational modification of histidine in protein synthesis of elongation factor 2 and in the synthesis of wybutosine, a tRNA tricyclic nucleotide Lin (2011). - 5
Sulfation pathways involving 3’-phosphoadenosine-5’-phosphosulfate (PAPS)-dependent sulfonation of sphingolipids, cholesterol, steroids, neurotransmitters, bile acids, peptides, proteins, and xenobiotics. - 6
Enzymes dependent on molybdopterin cofactor (Moco). Cysteine metabolism by cysteine desulfurase (EC 2.8.1.7) is a critical step in the biosynthesis of Moco (Rouault, 2012), in which molybdenum is covalently bound to the dithiolate moiety of molybdopterin (Havemayer et al, 2011). There are a number of Moco-dependent enzymes that could affect neuronal function in schizophrenia and in autism. These include mitochondrial amidoxime-reducing component (mARC), an enzyme complex dependent upon molybdopterin cofactor, sulfite oxidase (EC 1.8.2.1, 1.8.3.1), and aldehyde oxidase (EC 1.2.3.1). mARC also is responsible for the reduction of Nω-hydroxy-arginine to arginine (Kotthaus et al, 2011).
Figure 2
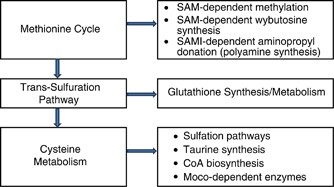
Schematic presentation of thiol metabolic pathways. CoA, coenzyme A; Moco, molybdopterin cofactor; SAM, _S_-adenosylmethionine; SAMI, decarboxylated S-adenosylmethionine.
Metabolomics evaluations of plasma in schizophrenia (Wood and Wood, 2013) and autism have demonstrated a number of common decrements in sulfur amino acids, including a decreased antioxidant capacity (decreased glutathione). However, the unique differences in SAA metabolism include increased methionine in schizophrenia and decreased inorganic sulfate in autism (Table 1).
Table 1 Plasma Sulfur Amino-Acid Metabolomics in Autism Spectrum Disorders (ASD) and in Schizophrenia Patients (SP)
The increases in methionine provide the capacity to synthesize greater amounts of SAM, which in turn can lead to altered methylation status of a number of metabolites and macromolecules like DNA. In this regard, increased levels of SAM have been measured in the prefrontal cortex in schizophrenia (Vuksan-Ćusa et al, 2011), a brain area characterized by abnormal GABAergic neuronal development, potentially as a result of GABAergic promoter hypermethylation (Costa et al, 2009). Pathways dependent upon aminopropyl or 3-amino-3-carboxypropyl donation via SAM-dependent mechanisms remain to be explored but could be responsible for a number of complex alterations in genetic regulation in schizophrenia.
In addition to its role in glutathione synthesis, cysteine is oxidized to form sulfate, the essential sulfur function required for the synthesis of 3′-phosphoadenosine-5′-phosphosulfate (PAPS), the major sulfur donor in sulfation reactions (Figure 2). Decreases in circulating sulfate levels in autism (Waring and Klovrza, 2000; Geier et al, 2009; Adams et al, 2011; Kern et al, 2011; Bowling et al, 2012) are further reflected by decreased levels of dehydroepiandrosterone sulfate (DHEA-S) in autistic adults (Strous et al, 2005) and decreased ability to sulfonate acetaminophen in children (Alberti et al, 1999). More in-depth evaluation of sulfonation mechanisms and metabolites is clearly needed in autistic individuals, as this potentially includes steroids, neurotransmitters, bile acids, peptides, and xenobiotics.
In summary, a number of alterations in sulfur amino-acid metabolism have been reported for both autism and schizophrenia (Table 1). The data are sufficient to warrant implementation of focused metabolomics platforms to evaluate these complex changes in larger cohorts of patients.
Metabolomics in Neurology: SAA Metabolism in Amyotrophic Lateral Sclerosis (ALS)
There have been a limited number of metabolomics studies in ALS. In a non-targeted metabolomics evaluation of ALS plasma, decrements in catechol sulfate and 4-vinylphenol sulfate were measured (Lawton et al, 2012). These are microbial-host metabolites in that catechol and vinylphenol are microbial metabolites from gastrointestinal flora, although the sulfation of these metabolites is performed in the human liver. These data suggest that, as in autism, decreased sulfation capacity may occur in ALS. However, in contrast to autism, plasma inorganic sulfate is not decreased but may be slightly elevated (Pean et al, 1994; Woolsey, 2008). Plasma cysteine (Pean et al, 1994; Woolsey, 2008) and pantothenic acid (Lawton et al, 2012) also appear to be elevated in ALS, which are further indicators of abnormal SAA metabolism in this devastating disorder.
Non-targeted metabolomics evaluations of ALS cerebrospinal fluid (CSF) further support abnormal SAA metabolism in ALS, with decrements in both methionine and putrescine being reported (Wuolikainen et al, 2011). These data are suggestive of abnormal SAM function in ALS, a suggestion that can be examined using targeted SAA metabolomics platforms.
In summary, as in autism and schizophrenia, alterations in sulfur amino-acid metabolism in ALS suggest that targeted metabolomics studies should now follow preliminary non-targeted findings.
Metabolomics in Neurology: Plasmalogens in Alzheimer’s Disease (AD)
In the search for biomarkers of AD, the focus has been extensively confined to the evaluation of proteins in plaques and tangles, hallmark features of AD pathology at autopsy. However, these proteins accumulate in the brain with normal aging and are present at levels comparable to those in AD patients, in non-agenarians who demonstrate minimal cognitive deficit (Erten-Lyons et al, 2009; Maarouf et al, 2011). These factors render these proteins and their peptide metabolites of limited value in the diagnosis of AD (Bazenet and Lovestone, 2012).
Similarly, metabolomics studies have not generated reliable biomarkers of AD. In contrast, a number of lipidomics alterations in AD have been demonstrated in post-mortem brain and in the plasma of AD patients (Florent-Béchard et al, (2009); Astarita et al., 2010; Wood, 2012). The most robust alterations are decrements in brain sulfatides (Han et al, 2002) and decreases in ethanolamine plasmalogens (PlsEtn) in both brain (Ginsberg et al, 1995; Han et al, 2001; Han 2005) and plasma (Goodenowe et al, 2007; Wood et al, 2010). These lipid classes are complex structural lipids that also serve as reservoirs for lipid mediators of signal transduction.
Sulfatides and white matter ethanolamine plasmalogens (eg, PlsEtn18:0/18:1) are decreased early in the disease process, supporting MRI imaging studies that have demonstrated fiber tract disruption in mild cognitive impairment (MCI) and in the early phases of AD (Stricker et al, 2009). In contrast, gray matter ethanolamine plasmalogens (eg PlsEtn18:0/22:6) decrease in a disease severity-dependent manner. Decreases in plasmalogen synthesis have been demonstrated in both the AD liver (Astarita et al, 2010) and brain (Kou et al, 2011; Grimm et al, 2011).
Decreases in AD cortical sulfatides are paralleled by large increases in cortical (Han et al, 2002, Cutler et al, 2004; He et al, 2010), CSF (Satoi et al, 2005), and plasma (Han et al, 2011) ceramides, which are precursors/degradation products of sulfatides. In contrast, another study has reported no differences in plasma ceramide levels in AD (Mielke et al, 2010) but decreased plasma levels in MCI. Immunohistochemistry studies suggest that the increases in cortical ceramides are mainly in astrocytes (Satoi et al, 2005). The potential signaling effects of alterations in brain ceramides in AD remain to be determined.
In summary, brain glycerophospholipid and sphingolipid metabolisms are dramatically affected in AD white matter early in the disease process. Further, decrements in myelin sulfatides and PlsEtn support imaging studies, demonstrating dysmyelination in MCI and AD patients. The utility of measuring decrements in plasma PlsEtn as a biomarker of AD requires further validation in larger cohorts of AD patients.
Metabolomics in Neuro-Oncology: SAA Metabolism in Glioblastoma
Glioblastoma is a devastating cancer with incredibly aggressive growth rates and an associated poor prognosis. Although a number of gene changes have been reported in glioblastoma, there have been limited metabolomics studies conducted. However, there are a number of consistent changes in SAA metabolism that may contribute to tumor progression. For example, hypotaurine and its metabolic end product taurine are elevated in glioblastoma microdialysates in GBM patients (Bianchi et al, 2004, Wibom et al, 2010), suggesting increased synthesis and release of taurine by GBM cells. This conclusion is also congruent with observations of decreased blood levels of microRNAs (Roth et al, 2011) that regulate cysteamine dioxygenase (EC 1.13.11.19; cysteamine→hypotaurine), the rate-limiting enzyme involved in the synthesis of hypotaurine.
Further evidence of abnormal sulfur amino-acid metabolism includes increased cysteine and decreased cystathionine in tumor microdialysates in GBM patients (Wibom et al, 2010). Decrements in blood levels of microRNAs (Roth et al, 2011) regulating cystathionine γ-lyase (EC 4.4.1.1; cystathionine→cysteine) also are consistent with these observations.
_S_-methylcysteine metabolism has not been studied extensively, but this SAA is avidly accumulated by glioblastoma tumors (Deng et al, 2011) and is increased in tumor microdialysates (Wibom et al, 2010). The metabolic sources of _S_-methylcysteine remain to be defined, but alterations in DNA repair by o-6-methylguanine-DNA methyltransferase (EC 2.1.1.63; DNA-6-o-methylguanine+protein-cysteine→DNA+protein S-methylcysteine) is one potential source, suggesting that this may represent a unique biomarker of dysregulation of DNA repair mechanisms in GBM.
In summary, sulfur amino-acid metabolism is dysfunctional in GBM patients and more in-depth studies of SAA pathways are warranted.
Overview
Advancements in mass spectrometry have revolutionized our abilities to analyze complex biological matrices. The pitfalls regarding the specificity of colorimetric and ELISA assays and the use of non-selective detectors in chromatographic systems have been overcome by mass selective detection and quantitation of biomolecules. Although there is currently a rapid expansion of metabolomics data in the literature, based upon mass spectrometric techniques, the next critical step is to move this wealth of information into focused translational research studies such that new clinical tools are realized within the next 10 years. Funding agencies are aggressively seeking translational research proposals in metabolomics. The complexity that these agencies, as well as potential investigators, have to consider is that biomarker discovery and validation (ie, a validated assay for reporting absolute patient values) requires a multidisciplinary team to achieve success. The analytical team includes synthetic organic chemists (analytical standards), analytical chemists, biochemists, statisticians, and regulatory expertise. The clinical team requires clinical researchers, practicing physicians, phlebotomists, nurses, research assistants, and their office staff. Additionally, access to multiple clinical sites is required to attain sufficient numbers of subjects to define potential subsets of patients in a multifactorial disease. In essence, a great deal of planning, collaboration, and expertise will need to be coordinated to advance metabolomics findings into clinical practice.
FUNDING AND DISCLOSURE
The author declares no conflict of interest and no funding to write this review.
References
- Abu-Asab MS, Chaouchi M, Alesci S, Galli S, Laassri M, Cheema AK et al (2011). Biomarkers in the age of omics: time for a systems biology approach. OMICS 15: 105–112 Clear overview of omics approaches.
CAS PubMed PubMed Central Google Scholar - Adams JB, Audhya T, McDonough-Means S, Rubin RA, Quig D, Geis E et al (2011). Nutritional and metabolic status of children with autism vs. neurotypical children, and the association with autism severity. Nutr Metab (Lond) 8: 34 In-depth evaluation of sulfur amino acids in autism.
PubMed Central Google Scholar - Alberti A, Pirrone P, Elia M, Waring RH, Romano C (1999). Sulphation deficit in ‘low-functioning’ autistic children: a pilot study. Biol Psychiatry 46: 420–424.
CAS PubMed Google Scholar - Al-Ayadhi L, Halepoto DM (2013). Role of proteomics in the discovery of autism biomarkers. J Coll Physicians Surg Pak 23: 137–143.
PubMed Google Scholar - Altuntas I, Aksoy H, Coskun I, Cayköylü A, Akçay F (2000). Erythrocyte superoxide dismutase and glutathione peroxidase activities, and malondialdehyde and reduced glutathione levels in schizophrenic patients. Clin Chem Lab Med 38: 1277–1281.
CAS PubMed Google Scholar - Astarita G, Jung KM, Berchtold NC, Nguyen VQ, Gillen DL, Head E et al (2010). Deficient liver biosynthesis of docosahexaenoic acid correlates with cognitive impairment in Alzheimer's disease. PLoS ONE 5: e12538. Demonstration of a liver peroxisomal deficit in Alzheimers Disease.
PubMed PubMed Central Google Scholar - Bazenet C, Lovestone S (2012). Plasma biomarkers for Alzheimer's disease: much needed but tough to find. Biomark Med 6: 441–454 Comprehensive review of the current status of biomarker research for Alzheimer's disease.
CAS PubMed Google Scholar - Bianchi L, De Micheli E, Bricolo A, Ballini C, Fattori M, Venturi C et al (2004). Extracellular levels of amino acids and choline in human high grade gliomas: an intraoperative microdialysis study. Neurochem Res 29: 325–334.
CAS PubMed Google Scholar - Bjerkenstedt L, Edman G, Hagenfeldt L, Sedvall G, Wiesel FA (1985). Plasma amino acids in relation to cerebrospinal fluid monoamine metabolites in schizophrenic patients and healthy controls. Br J Psychiatry 147: 276–282.
CAS PubMed Google Scholar - Bowling FG, Heussler HS, McWhinney A, Dawson PA (2012). Plasma and urinary sulfate determination in a cohort with autism. Biochem Genet 51: 147–153.
PubMed Google Scholar - Britton LM, Gonzales-Cope M, Zee BM, Garcia BA (2011). Breaking the histone code with quantitative mass spectrometry. Expert Rev Proteomics 8: 631–643.
CAS PubMed PubMed Central Google Scholar - Bullinger D, Neubauer H, Fehm T, Laufer S, Gleiter CH, Kammerer B (2007). Metabolic signature of breast cancer cell line MCF-7: profiling of modified nucleosides via LC-IT MS coupling. BMC Biochem 8: 25.
PubMed PubMed Central Google Scholar - Ciccimaro E, Blair IA (2010). Stable-isotope dilution LC–MS for quantitative biomarker analysis. Bioanalysis 2: 311–341 Excellent overview of the utility of stable isotope internal standards in quantitative mass spectrometry.
CAS PubMed Google Scholar - Costa E, Chen Y, Dong E, Grayson DR, Kundakovic M, Maloku E et al (2009). GABAergic promoter hypermethylation as a model to study the neurochemistry of schizophrenia vulnerability. Expert Rev Neurother 9: 87–98.
CAS PubMed Google Scholar - Creek DJ, Chokkathukalam A, Jankevics A, Burgess KE, Breitling R, Barrett MP (2012). Stable isotope-assisted metabolomics for network-wide metabolic pathway elucidation. Anal Chem 84: 8442–8447.
CAS PubMed PubMed Central Google Scholar - Cutler RG, Kelly J, Storie K, Pedersen WA, Tammara A, Hatanpaa K et al (2004). Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer's disease. Proc Natl Acad Sci USA 101: 2070–2075.
CAS PubMed PubMed Central Google Scholar - Deng H, Tang X, Wang H, Tang G, Wen F, Shi X et al (2011). S-11C-methyl-L-cysteine: a new amino acid PET tracer for cancer imaging. J Nucl Med 52: 287–293.
CAS PubMed Google Scholar - Dietrich-Muszalska A, Malinowska J, Olas B, Głowacki R, Bald E, Wachowicz B et al (2012). The oxidative stress may be induced by the elevated homocysteine in schizophrenic patients. Neurochem Res 37: 1057–1062.
CAS PubMed PubMed Central Google Scholar - Dudley E, Yousef M, Wang Y, Griffiths WJ (2010). Targeted metabolomics and mass spectrometry. Adv Protein Chem Struct Biol 80: 45–83 Outstanding review of targeted metabolomics.
CAS PubMed Google Scholar - Emary WB, Lys I, Cotter RJ, Simpson R, Hoffman A (1990). Liquid chromatography/time-of-flight mass spectrometry with high-speed integrated transient recording. Anal Chem 62: 1319–1324.
CAS PubMed Google Scholar - Emmett MR, Andrén PE, Caprioli RM (1995). Specific molecular mass detection of endogenously released neuropeptides using in vivo microdialysis/mass spectrometry. J Neurosci Methods 62: 141–147 The first demonstration of the utility of nano-electrospray for quantitation of neuropeptides.
CAS PubMed Google Scholar - Erten-Lyons D, Woltjer RL, Dodge H, Nixon R, Vorobik R, Calvert JF et al (2009). Factors associated with resistance to dementia despite high Alzheimer disease pathology. Neurology 72: 354–360.
CAS PubMed PubMed Central Google Scholar - Fenn JB, Mann M, Meng CK, Wong SF, Whitehouse CM (1989). Electrospray ionization for mass spectrometry of large biomolecules. Science 246: 64–71 The introduction of electrospray ionization for characterization of biomolecules.
CAS PubMed Google Scholar - Florent-Béchard S, Desbène C, Garcia P, Allouche A, Youssef I, Escanyé MC et al (2009). The essential role of lipids in Alzheimer's disease. Biochimie 91: 804–809.
PubMed Google Scholar - Frankhauser P, Grimmer Y, Bugert P, Deuschle M, Schmidt M, Schloss P (2006). Characterization of the neuronal dopamine transporter DAT in human blood platelets. Neurosci Lett 399: 197–201.
CAS PubMed Google Scholar - Gates SC, Dendramis N, Sweeley CC (1978). Automated metabolic profiling of organic acids in human urine. I. Description of methods. Clin Chem 24: 1674–1679 One of the first metabolomics studies characterizing over 150 organic acids in urine.
CAS PubMed Google Scholar - Gates SC, Sweeley CC, Krivit W, DeWitt D, Blaisdell BE (1978). Automated metabolic profiling of organic acids in human urine. II. Analysis of urine samples from ‘healthy’ adults, sick children, and children with neuroblastoma. Clin Chem 24: 1663–1673.
CAS PubMed Google Scholar - Geier DA, Geier MR (2006). A clinical and laboratory evaluation of methionine cycle-transsulfuration and androgen pathway markers in children with autistic disorders. Horm Res 66: 182–188.
CAS PubMed Google Scholar - Geier DA, Kern JK, Garver CR, Adams JB, Audhya T, Geier MR (2009). A prospective study of transsulfuration biomarkers in autistic disorders. Neurochem Res 34: 386–393.
CAS PubMed Google Scholar - Ginsberg L, Rafique S, Xuereb JH, Rapoport SI, Gershfeld NL (1995). Disease and anatomic specificity of ethanolamine plasmalogen deficiency in Alzheimer's disease brain. Brain Res 698: 223–226.
CAS PubMed Google Scholar - Goodenowe DB, Cook LL, Liu J, Lu Y, Jayasinghe DA, Ahiahonu PW et al (2007). Peripheral ethanolamine plasmalogen deficiency: a logical causative factor in Alzheimer's disease and dementia. J Lipid Res 48: 2485–2498 The first demonstration of plasmalogen deficiency in the serum of Alzheimer’s patients.
CAS PubMed Google Scholar - Grimm MO, Kuchenbecker J, Rothhaar TL, Grösgen S, Hundsdörfer B, Burg VK et al (2011). Plasmalogen synthesis is regulated via alkyl-dihydroxyacetonephosphate-synthase by amyloid precursor protein processing and is affected in Alzheimer's disease. J Neurochem 116: 916–925.
CAS PubMed Google Scholar - Gründemann D, Harlfinger S, Golz S, Geerts A, Lazar A, Berkels R et al (2005). Discovery of the ergothioneine transporter. Proc Natl Acad Sci USA 102: 5256–5261.
PubMed PubMed Central Google Scholar - Gysin R, Kraftsik R, Boulat O, Bovet P, Conus P, Comte-Krieger E et al (2011). Genetic dysregulation of glutathione synthesis predicts alteration of plasma thiol redox status in schizophrenia. Antioxid Redox Signal 15: 2003–2010.
CAS PubMed Google Scholar - Han X, Holtzman DM, McKeel DW Jr (2001). Plasmalogen deficiency in early Alzheimer's disease subjects and in animal models: molecular characterization using electrospray ionization mass spectrometry. J Neurochem 77: 1168–1180 In depth characterization of the deficiency of brain plasmalogens in Alzheimer’s disease, utilizing tandem quadrapole mass spectrometry.
CAS PubMed Google Scholar - Han X, M Holtzman D, McKeel DW Jr, Kelley J, Morris JC (2002). Substantial sulfatide deficiency and ceramide elevation in very early Alzheimer's disease: potential role in disease pathogenesis. J Neurochem 82: 809–818.
CAS PubMed Google Scholar - Han X (2005). Lipid alterations in the earliest clinically recognizable stage of Alzheimer's disease: implication of the role of lipids in the pathogenesis of Alzheimer's disease. Curr Alzheimer Res 2: 65–77.
CAS PubMed Google Scholar - Han X, Rozen S, Boyle SH, Hellegers C, Cheng H, Burke JR et al (2011). Metabolomics in early Alzheimer's disease: identification of altered plasma sphingolipidome using shotgun lipidomics. PLoS ONE 6: e21643.
CAS PubMed PubMed Central Google Scholar - Havemeyer A, Lang J, Clement B (2011). The fourth mammalian molybdenum enzyme mARC: current state of research. Drug Metab Rev 43: 524–539.
CAS PubMed Google Scholar - He X, Huang Y, Li B, Gong CX, Schuchman EH (2010). Deregulation of sphingolipid metabolism in Alzheimer's disease. Neurobiol Aging 31: 398–408.
CAS PubMed Google Scholar - Hu Q, Noll RJ, Li H, Makarov A, Hardman M, Graham Cooks R (2005). The Orbitrap: a new mass spectrometer. J Mass Spectrom 40: 430–443 Excellent review of orbitrap mass spectrometry.
CAS PubMed Google Scholar - Hu CW, Liu HH, Li YJ, Chao MR (2012). Direct analysis of 5-methylcytosine and 5-methyl-2'-deoxycytidine in human urine by isotope dilution LC-MS/MS: correlations with N-methylated purines and oxidized DNA lesions. Chem Res Toxicol 25: 462–470.
CAS PubMed Google Scholar - Izumi Y, Takimura S, Yamaguchi S, Iida J, Bamba T, Fukusaki E (2012). Application of electrospray ionization ion trap/time-of-flight mass spectrometry for chemically-synthesized small RNAs. J Biosci Bioeng 113: 412–419.
CAS PubMed Google Scholar - James SJ, Cutler P, Melnyk S, Jernigan S, Janak L, Gaylor DW et al (2004). Metabolic biomarkers of increased oxidative stress and impaired methylation capacity in children with autism. Am J Clin Nutr 80: 1611–1617.
CAS PubMed Google Scholar - Kern JK, Geier DA, Adams JB, Garver CR, Audhya T, Geier MR (2011). A clinical trial of glutathione supplementation in autism spectrum disorders. Med Sci Monit 17: CR677–CR682.
CAS PubMed PubMed Central Google Scholar - Kim TH, Moon SW (2011). Serum homocysteine and folate levels in korean schizophrenic patients. Psychiatry Investig 8: 134–140.
CAS PubMed PubMed Central Google Scholar - Koal T, Deigner HP (2010). Challenges in mass spectrometry based targeted metabolomics. Curr Mol Med 10: 216–226 Outstanding review of issues in translational research for biomarkers.
CAS PubMed Google Scholar - Kotthaus J, Wahl B, Havemeyer A, Kotthaus J, Schade D, Garbe-Schönberg D et al (2011). Reduction of N(ω)-hydroxy-L-arginine by the mitochondrial amidoxime reducing component (mARC). Biochem J 433: 383–391.
CAS PubMed Google Scholar - Kou J, Kovacs GG, Höftberger R, Kulik W, Brodde A, Forss-Petter S et al (2011). Peroxisomal alterations in Alzheimer's disease. Acta Neuropathol 122: 271–283.
CAS PubMed PubMed Central Google Scholar - Krone N, Hughes BA, Lavery GG, Stewart PM, Arlt W, Shackleton CH (2010). Gas chromatography/mass spectrometry (GC/MS) remains a pre-eminent discovery tool in clinical steroid investigations even in the era of fast liquid chromatography tandem mass spectrometry (LC/MS/MS). J Steroid Biochem Mol Biol 121: 496–504.
CAS PubMed PubMed Central Google Scholar - Lawton KA, Cudkowicz ME, Brown MV, Alexander D, Caffrey R, Wulff JE et al (2012). Biochemical alterations associated with ALS. Amyotroph Lateral Scler 13: 110–118.
CAS PubMed Google Scholar - Lin H (2011). S-Adenosylmethionine-dependent alkylation reactions: when are radical reactions used? Bioorg Chem 39: 161–170.
CAS PubMed PubMed Central Google Scholar - Maarouf CL, Daugs ID, Kokjohn TA, Walker DG, Hunter JM, Kruchowsky JC et al (2011). Alzheimer's disease and non-demented high pathology control nonagenarians: comparing and contrasting the biochemistry of cognitively successful aging. PLoS ONE 6: e27291.
CAS PubMed PubMed Central Google Scholar - Marshall AG, Hendrickson CL, Jackson GS (1998). Fourier transform ion cyclotron resonance mass spectrometry: a primer. Mass Spectrom Rev 17: 1–35.
CAS PubMed Google Scholar - McLafferty FW (1981). Tandem mass spectrometry. Science 214: 280–287 Outstanding early review of tandem mass spectrometry.
CAS PubMed Google Scholar - Mielke MM, Haughey NJ, Ratnam Bandaru VV, Schech S, Carrick R, Carlson MC et al (2010). Plasma ceramides are altered in mild cognitive impairment and predict cognitive decline and hippocampal volume loss. Alzheimers Dement 6: 378–385.
CAS PubMed PubMed Central Google Scholar - Mishur RJ, Rea SL (2012). Applications of mass spectrometry to metabolomics and metabonomics: detection of biomarkers of aging and of age-related diseases. Mass Spectrom Rev 31: 70–95.
CAS PubMed Google Scholar - Morris SE, Cuthbert BN (2012). Research Domain Criteria: cognitive systems, neural circuits, and dimensions of behavior. Dialogues Clin Neurosci 14: 29–37.
PubMed PubMed Central Google Scholar - Muntjewerff JW, Kahn RS, Blom HJ, den Heijer M (2006). Homocysteine, methylenetetrahydrofolate reductase and risk of schizophrenia: a meta-analysis. Mol Psychiatry 11: 143–149.
CAS PubMed Google Scholar - Nicholson JK, Lindon JC, Holmes E (1999). 'Metabonomics': understanding the metabolic responses of living systems to pathophysiological stimuli via multivariate statistical analysis of biological NMR spectroscopic data. Xenobiotica 29: 1181–1189.
CAS PubMed Google Scholar - Nordström A, Lewensohn R (2010). Metabolomics: moving to the clinic. J Neuroimmune Pharmacol 5: 4–17.
PubMed Google Scholar - Orešič M, Tang J, Seppänen-Laakso T, Mattila I, Saarni SE, Saarni SI et al (2011). Metabolome in schizophrenia and other psychotic disorders: a general population-based study. Genome Med 3: 19 An excellent example of the utility of metabolomics research in a defined clinical population.
PubMed PubMed Central Google Scholar - Paşca SP, Dronca E, Kaucsár T, Craciun EC, Endreffy E, Ferencz BK et al (2009). One carbon metabolism disturbances and the C677T MTHFR gene polymorphism in children with autism spectrum disorders. J Cell Mol Med 13: 4229–4238.
PubMed Google Scholar - Pastural E, Ritchie S, Lu Y, Jin W, Kavianpour A, Khine Su-Myat K et al (2009). Novel plasma phospholipid biomarkers of autism: mitochondrial dysfunction as a putative causative mechanism. Prostaglandins Leukot Essent Fatty Acids 81: 253–264.
CAS PubMed Google Scholar - Pean A, Steventon GB, Waring RH, Foster H, Sturman S, Williams AC (1994). Pathways of cysteine metabolism in MND/ALS. J Neurol Sci 124 (Suppl): 59–61.
CAS PubMed Google Scholar - Postle AD (2012). Lipidomics. Curr Opin Clin Nutr Metab Care 15: 127–133.
CAS PubMed Google Scholar - Regland B (2005). Schizophrenia and single-carbon metabolism. Prog. Neuropsychopharmacol Biol Psychiatry 29: 1124–1132.
CAS PubMed Google Scholar - Roth P, Wischhusen J, Happold C, Chandran PA, Hofer S, Eisele G et al (2011). A specific miRNA signature in the peripheral blood of glioblastoma patients. J Neurochem 118: 449–457.
CAS PubMed Google Scholar - Rouault TA (2012). Biogenesis of iron-sulfur clusters in mammalian cells: new insights and relevance to human disease. Dis Model Mech 5: 155–164.
CAS PubMed PubMed Central Google Scholar - Satoi H, Tomimoto H, Ohtani R, Kitano T, Kondo T, Watanabe M et al (2005). Astroglial expression of ceramide in Alzheimer's disease brains: a role during neuronal apoptosis. Neuroscience 130: 657–666.
CAS PubMed Google Scholar - Schuhmann K, Almeida R, Baumert M, Herzog R, Bornstein SR, Shevchenko A (2012). Shotgun lipidomics on a LTQ Orbitrap mass spectrometer by successive switching between acquisition polarity modes. J Mass Spectrom 47: 96–104 Excellent overview of the utility of orbitrap MS in shotgun (constant infusion) lipidomics.
CAS PubMed Google Scholar - Stricker NH, Schweinsburg BC, Delano-Wood L, Wierenga CE, Bangen KJ, Haaland KY et al (2009). Decreased white matter integrity in late-myelinating fiber pathways in Alzheimer's disease supports retrogenesis. Neuroimage 45: 10–16.
CAS PubMed Google Scholar - Strous RD, Golubchik P, Maayan R, Mozes T, Tuati-Werner D, Weizman A et al (2005). Lowered DHEA-S plasma levels in adult individuals with autistic disorder. Eur Neuropsychopharmacol 15: 305–309.
CAS PubMed Google Scholar - Suh JH, Walsh WJ, McGinnis WR, Lewis A, Ames BN (2008). Altered sulfur amino acid metabolism in immune cells of children diagnosed with autism. Am J Biochem Biotech 4: 105–113.
CAS Google Scholar - Sun G, Yang K, Zhao Z, Guan S, Han X, Gross RW (2007). Shotgun metabolomics approach for the analysis of negatively charged water-soluble cellular metabolites from mouse heart tissue Shotgun metabolomics approach for the analysis of negatively charged water-soluble cellular metabolites from mouse heart tissue. Anal Chem 79: 6629–6640.
CAS PubMed PubMed Central Google Scholar - Valaskovic GA, Utley L, Lee MS, Wu JT. (2006). Ultra-low flow nanospray for the normalization of conventional liquid chromatography/mass spectrometry through equimolar response: standard-free quantitative estimation of metabolite levels in drug discovery. Rapid Commun Mass Spectrom 20: 1087–1096.
CAS PubMed Google Scholar - Vuksan-Ćusa B, Jakovljević M, Sagud M, Mihaljević Peleš A, Marčinko D, Topić R et al (2011). Metabolic syndrome and serum homocysteine in patients with bipolar disorder and schizophrenia treated with second generation antipsychotics. Psychiatry Res 189: 21–25.
PubMed Google Scholar - Waring RH, Klovrza LV (2000). Sulphur metabolism in autism. J Nutrit Envir Med 10: 25–32.
CAS Google Scholar - Wibom C, Surowiec I, Mörén L, Bergström P, Johansson M, Antti H et al (2010). Metabolomic patterns in glioblastoma and changes during radiotherapy: a clinical microdialysis study. J Proteome Res 9: 2909–2919.
CAS PubMed Google Scholar - Wood PL (1982). A selected ion monitoring assay for dopamine and its metabolites using negative chemical ionization. Biomed Mass Spectrom 9: 302–306.
CAS PubMed Google Scholar - Wood PL, Péloquin A (1982). ncreases in choline levels in rat brain elicited by meclofenoxate ncreases in choline levels in rat brain elicited by meclofenoxate. Neuropharmacology 21: 349–354.
CAS PubMed Google Scholar - Wood PL, Kim HS, Altar CA (1987). In vivo assessment of dopamine and norepinephrine release in rat neocortex: gas chromatography-mass spectrometry measurement of 3-methoxytyramine and normetanephrine. J Neurochem 48: 574–579.
CAS PubMed Google Scholar - Wood PL, Kim HS, Cheney DL, Cosi C, Marien M, Rao TS et al (1988). Constant infusion of [13C6]glucose: simultaneous measurement of turnover of GABA and glutamate in defined regions of the brain of individual animals. Neuropharmacology 27: 669–676.
CAS PubMed Google Scholar - Wood PL, Rao TS (1989). GC–MS and GC–MS/MS of monoamines, indoles, trace amines and their associated precursors and metabolites. In: McEwen CN, Larsen BS (eds.) Mass Spectrometry of Biological Materials. Marcel Dekker: New York. pp 197–214.
Google Scholar - Wood PL, Khan MA, Moskal JR (2006). Neurochemical analysis of amino acids, polyamines and carboxylic acids: GC–MS quantitation of tBDMS derivatives using ammonia positive chemical ionization. J Chromatogr B Analyt Technol Biomed Life Sci 831: 313–319.
CAS PubMed Google Scholar - Wood PL, Mankidy R, Ritchie S, Heath D, Wood JA, Flax J et al (2010). Circulating plasmalogen levels and Alzheimer Disease Assessment Scale-Cognitive scores in Alzheimer patients. J Psychiatry Neurosci 35: 59–62.
PubMed PubMed Central Google Scholar - Wood PL, Khan MA, Smith T, Ehrmantraut G, Jin W, Cui W et al (2011). In vitro and in vivo plasmalogen replacement evaluations in rhizomelic chrondrodysplasia punctata and Pelizaeus-Merzbacher disease using PPI-1011, an ether lipid plasmalogen precursor. Lipids Health Dis 10: 182.
CAS PubMed PubMed Central Google Scholar - Wood PL, Smith T, Pelzer L, Goodenowe DB (2011). Targeted metabolomic analyses of cellular models of pelizaeus-merzbacher disease reveal plasmalogen and myo-inositol solute carrier dysfunction. Lipids Health Dis 10: 102. An example of the utility of targeted metabolomics in defining metabolic defects in cell lines.
PubMed PubMed Central Google Scholar - Wood PL (2012). Lipidomics of Alzheimer's disease: current status. Alzheimers Res Ther 4: 5. Comprehensive review of the current status of lipidomics research in Alzheimer’s disease.
PubMed PubMed Central Google Scholar - Wood PL, Wood JA (2013). Thiol metabolism in schizophrenia: Current status. Current Psychiatry Rev 9: 136–147 Comprehensive review of the current status of thiol metabolomics research in schizophrenia.
CAS Google Scholar - Woolsey PB (2008). Cysteine, sulfite, and glutamate toxicity: a cause of ALS? J Altern Complement Med 14: 1159–1164.
PubMed Google Scholar - Wuolikainen A, Moritz T, Marklund SL, Antti H, Andersen PM (2011). Disease-related changes in the cerebrospinal fluid metabolome in amyotrophic lateral sclerosis detected by GC/TOFMS. PLoS One 6: e17947.
CAS PubMed PubMed Central Google Scholar - Yang J, Chen T, Sun L, Zhao Z, Qi X, Zhou K et al (2013). Potential metabolite markers of schizophrenia. Mol Psychiatry 2011: 1–12.
Google Scholar - Zhang L, Xiao H, Wong DT (2009). Salivary biomarkers for clinical applications. Mol Diagn Ther 13: 245–259 Review of the current status of the metabolomics and proteomics of human saliva.
CAS PubMed Google Scholar - Zhubi A, Veldic M, Puri NV, Kadriu B, Caruncho H, Loza I et al (2009). An upregulation of DNA-methyltransferase 1 and 3a expressed in telencephalic GABAergic neurons of schizophrenia patients is also detected in peripheral blood lymphocytes. Schizophr Res 111: 115–122.
CAS PubMed PubMed Central Google Scholar
Acknowledgements
I thank Dr Erminio Costa and Dr Darwin Cheney who introduced me to mass spectrometry during my postdoctoral fellowship and the many collaborators I have worked with over the last 30 years utilizing mass spectrometry to interrogate biological systems.
Author information
Authors and Affiliations
- Department of Physiology and Pharmacology, Metabolomics Unit, DeBusk College of Osteopathic Medicine, Lincoln Memorial University, Math and Science 435, Harrogate, 37752, TN, USA
Paul L Wood
Authors
- Paul L Wood
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toPaul L Wood.
PowerPoint slides
Rights and permissions
About this article
Cite this article
Wood, P. Mass Spectrometry Strategies for Clinical Metabolomics and Lipidomics in Psychiatry, Neurology, and Neuro-Oncology.Neuropsychopharmacol 39, 24–33 (2014). https://doi.org/10.1038/npp.2013.167
- Received: 03 July 2013
- Accepted: 04 July 2013
- Published: 11 July 2013
- Issue Date: January 2014
- DOI: https://doi.org/10.1038/npp.2013.167