When Synaptic Proteins Meet the Genome: Transcriptional Regulation in Cell Death and Plasticity by the Synapto–Nuclear Messenger Jacob (original) (raw)
Neurons express more genes than any other cell type and it is therefore unlikely that synapto–nuclear Ca2+ signaling alone can explain the specific genomic response to the plethora of extracellular stimuli that control gene expression. Possible candidates for encoding of signals at the origin and later decoding at a nuclear destination are synapto–nuclear protein messenger. These proteins translocate to the nucleus in a stimulus-dependent manner, where they can regulate via their nuclear target interactions very specific aspects of gene expression (Jordan and Kreutz, 2009). However, this type of signaling is challenged by space and time constraints. Synapses are localized far away from the nucleus and synaptic signals are very labile and rapidly degraded. On top the minute quantities of signaling molecules that can be released from synaptic sites are greatly diluted in relation to the entire volume of the dendritic arborization or nucleus. A precise machinery is therefore required to deliver the signal to nuclear target sites, but unfortunately very little is known on just how protein messenger can translocate several hundred micrometers, avoid degradation, retain signal and do this in a reliable manner.
_N_-methyl-D-aspartate receptors (NMDAR) make an essential contribution to activity-dependent gene expression in learning and memory. However, NMDAR are present at both synaptic and extrasynaptic sites, and the subcellular localization of each receptor profoundly and differentially affects the nuclear response to its activation. Activation of synaptic NMDAR induces the expression of cell survival and plasticity genes, while their extrasynaptic counterparts primarily drive the expression of cell death genes, linking the pathway to disease (Hardingham and Bading, 2010). An unresolved issue is how can the distant nucleus discriminate between synaptic and extrasynaptic NMDAR-induced signals?
Jacob is a synapto–nuclear messenger, and previous work has shown that extrasynaptic NMDAR activation induces nuclear translocation of Jacob, which results in sustained dephosphorylation and transcriptional inactivation of the transcription factor CREB, a loss of synaptic contacts, a retraction of dendrites and eventually cell death (Dieterich et al, 2008). However, Jacob also transits to the nucleus of CA1 neurons, following induction of Schaffer collateral-dependent long-term potentiation (LTP) but not long-term depression (LTD), and hence acts as a messenger for both synaptic and extrasynaptic NMDAR pathways (Behnisch et al, 2011). How does the protein get to the nucleus and what is the molecular basis for these different functions after nuclear import of Jacob? Neuronal importins are present in axons, dendrites, and synapses and they can associate with a dynein motor for active retrograde transport along microtubuli to the nucleus. Jacob utilizes this transport system after activation of both type of receptors and in a recent study we found that Jacob, following its nuclear import, can encode the synaptic and extrasynaptic origin of NMDAR signals (Karpova et al, 2013). ERK1/2-kinase binding and ERK-dependent phosphorylation of the serine180 residue in Jacob encodes synaptic but not extrasynaptic NMDAR activation. A stable trimeric complex with proteolytically cleaved fragments of the neurofilament _α_-internexin is formed, which protects Jacob and active ERK against phosphatase activity during retrograde transport. In the nucleus, this signalosome-like complex enhances ‘plasticity-related’ and ‘CREB-dependent’ gene expression as well as synaptic strength. It appears that Jacob operates as a mobile hub that docks NMDA receptor-derived signalosomes to nuclear target sites and it will be interesting to clarify the molecular identity of these complexes, because they are attractive targets for pharmacological interventions in activity-dependent gene transcription.
FUNDING AND DISCLOSURE
The authors declare no conflict of interest.
Figure 1
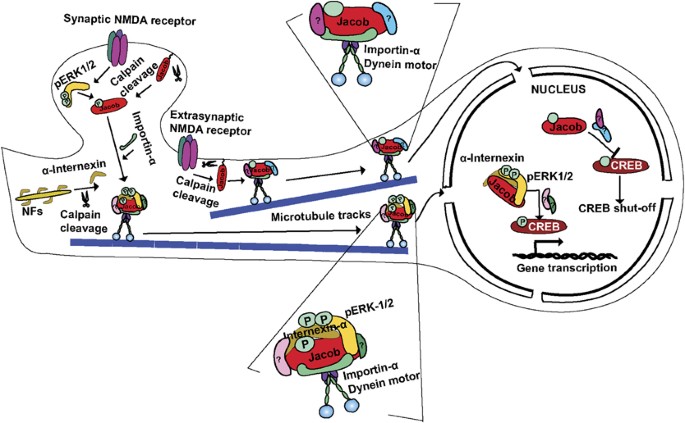
Schematic illustrating the encoding of the origin of synaptic and extrasynaptic NMDAR signals by Jacob. Neuronal activity leads to Calpain-mediated cleavage of _α_-internexin and the myristoylated N terminus of Jacob. High but not low frequency stimulation of synaptic NMDAR results in ERK activation, ERK binding, and phosphorylation of the serine180 in Jacob. This is then followed by recruitment of C-terminal fragments of _α_-internexin or a tighter assembly of an already preformed complex, which is then efficiently protected against phosphatase activity analogous to a mechanism demonstrated by Perlson et al (2005) for the retrograde transport of pERK after axon injury. In the nucleus, this complex can dock to CREB and potentially other sites and will enhance plasticity and cell survival promoting gene expression. Following the extrasynaptic NMDAR activation, ERK is not activated and remains outside of the nucleus, while non-phosphorylated Jacob will dock yet unknown protein components to CREB that eventually induce CREB shut-off and cell death.
References
- Behnisch T, Yuanxiang P, Bethge P, Parvez S, Chen Y, Yu J et al (2011). Nuclear translocation of Jacob in hippocampal neurons after stimuli inducing long-term potentiation but not long-term depression. PLoS One 6: e17276.
Article CAS Google Scholar - Dieterich DC, Karpova A, Mikhaylova M, Zdobnova I, König I, Landwehr M et al (2008). Caldendrin—Jacob: a protein liaison that couples NMDA receptor signalling to the nucleus. PLoS Biol 6: e34.
Article Google Scholar - Hardingham GE, Bading H (2010). Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat Rev Neurosci 11: 682–696.
Article CAS Google Scholar - Jordan BA, Kreutz MR (2009). Nucleocytoplasmic protein shuttling: The direct route in synapse-to-nucleus signaling. Trends Neurosci 32: 392–401.
Article CAS Google Scholar - Karpova A, Mikhaylova M, Bera S, Bär J, Reddy PP, Behnisch T et al (2013). Encoding and transducing the synaptic or extrasynaptic origin of NMDA receptor signals to the nucleus. Cell 152: 1119–1133.
Article CAS Google Scholar - Perlson E, Hanz S, Ben-Yaakov K, Segal-Ruder Y, Seger R, Fainzilber M (2005). Vimentin-dependent spatial translocation of an activated MAP kinase in injured nerve. Neuron 45: 715–726.
Article CAS Google Scholar
Acknowledgements
This study was supported by the DFG (SFB 779/TPB8), DIP grant, CBBS Magdeburg, MC-ITN NPlast, and the Leibniz Foundation (Pakt für Forschung).
Author information
Authors and Affiliations
- RG Neuroplasticity, Leibniz-Institute for Neurobiology, Magdeburg, Germany
Sujoy Bera & Michael R Kreutz
Authors
- Sujoy Bera
You can also search for this author inPubMed Google Scholar - Michael R Kreutz
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toMichael R Kreutz.
PowerPoint slides
Rights and permissions
About this article
Cite this article
Bera, S., Kreutz, M. When Synaptic Proteins Meet the Genome: Transcriptional Regulation in Cell Death and Plasticity by the Synapto–Nuclear Messenger Jacob.Neuropsychopharmacol 39, 245–246 (2014). https://doi.org/10.1038/npp.2013.204
- Published: 09 December 2013
- Issue Date: January 2014
- DOI: https://doi.org/10.1038/npp.2013.204