T-cell regulation: with complements from innate immunity (original) (raw)
The successful elimination of most pathogens requires crosstalk between the innate and adaptive arms of the immune system. The innate immune system recognizes pathogen-associated molecular patterns1 through pattern-recognition receptors, such as Toll-like receptors (TLRs) and nucleotide-binding oligomerization domain (NOD) receptors, and through the activation of complement by lectins (carbohydrate-binding proteins) and natural antibodies2,3. These interactions are followed by the release of cytokines, chemokines, opsonins, anaphylatoxins and defensins.
The complement system was identified more than 100 years ago as an effector arm of the antibody response that destroys bacteria by lysis4,5. The complement cascade is initiated through three pathways: the classical pathway, the lectin pathway and the alternative pathway2,3 (Fig. 1a). The classical, C1q (complement component 1q)-dependent pathway is activated by the binding of C1q to complement-fixing antibodies, which are bound to antigen on the surface of bacteria, whereas the lectin pathway is activated by proteins, such as mannan-binding lectin (MBL), in association with MBL-associated serine proteases (MASPs), attaching to carbohydrate patterns on bacteria. Activation of the alternative pathway occurs when C3b covalently binds to hydroxyl or amino groups on the surface of a target microbe. Low-level activation of the alternative pathway takes place continuously through the spontaneous hydrolysis (known as tickover) of C3 in plasma and does not involve specific recognition molecules. This pathway also amplifies the activation of C3 (amplification loop).
Figure 1: Activation and regulation of the complement system.
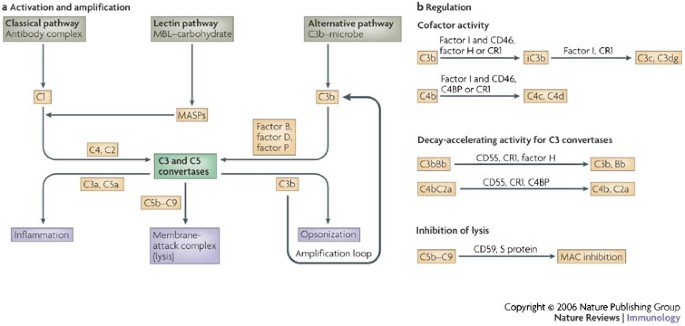
a | Complement can be activated through three pathways: the classical, lectin and alternative pathways. Complement component 1 (C1), composed of C1q , C1r and C1s, and mannose-binding lectin (MBL)-associated serine proteases (MASPs) link the complement system to classical and lectin pathways, respectively. These molecules also contain proteases that cleave C4 and C2, which then form the C3 and C5 convertases for the classical and lectin pathways. Factor B is the C2 equivalent for the alternative pathway. It is cleaved by factor D. Factor P (also known as properdin) stabilizes the alternative pathway convertases. All three pathways culminate in the formation of the C3 and C5 convertases that, in turn, generate the anaphylatoxins C3a and C5a, the membrane-attack complex (MAC; C5b–C9) and the opsonin C3b. These effectors function as chemokines, chemoattractants and activators of immunocompetent cells (C3a and C5a), mediate direct lysis of target cells (C5b–C9) or induce immune adherence and phagocytosis of the pathogen (C3b). C3b also amplifies the alternative pathway (through the amplification loop). b | Self tissue is protected from inappropriate complement deposition by fluid-phase and cell-bound regulators. C3b and C4b undergo limited proteolytic cleavage by the serine protease factor I. This enzyme requires a cofactor protein (membrane proteins CD46 and complement receptor 1 (CR1) or fluid-phase regulators factor H and C4-binding protein (C4BP)). The C3 and C5 convertases are also regulated through disassembly by regulators that have decay-accelerating activity (such as CD55, CR1, factor H and C4BP). Only the decay-accelerating activity for C3 convertases is shown. The formation of the MAC is controlled by CD59 and S protein (vitronectin).
Although triggered differently, these pathways culminate in the formation of the C3 convertases (C3bBb and C4bC2a) and C5 convertases (C3bBbC3b and C4bC2aC3b), which involves cleavage of C2 and C4 (classical and lectin pathways) or the serine proteases factor B and factor D (alternative pathway). This then results in the generation of the main effector molecules of the complement system: the opsonins C3b and C4b, the anaphylatoxins C3a and C5a, and the membrane-attack complex (MAC; membrane-bound C5b–C6–C7–C8–C9, denoted as C5b–C9) (Fig. 1a). Deposition of clusters of C3b (or C4b) on a pathogen leads either to immune adherence and subsequent ingestion by phagocytic cells (opsonization), or to lysis by engagement of the MAC, a process known as the lytic mechanism. In addition, the release of C3a and C5a mediates an acute inflammatory reaction, including the activation and directed migration of a wide range of immunocompetent cells2,3,6.
The coating of the pathogen with opsonic fragments derived from C3 and C4 (Ref. 6) is commonly a robust process and is minimally influenced by complement inhibitors (Table 1). However, such a powerful effector system requires tight regulation. This is achieved through plasma and membrane regulatory proteins (referred to as complement regulators) that inhibit complement activation in the fluid phase (no target) and on self (wrong target)2,3. At least three different mechanisms of regulation are known: proteolytic cleavage of deposited C3b and its cousin C4b (a process known as cofactor activity) by the serine protease factor I in conjunction with CD46 and complement receptor 1 (CR1; also known as CD35), or by factor H and C4b binding protein (C4BP); the disassembly of the C3 and C5 convertases (a process known as decay-accelerating activity) that involves CD55, CR1, factor H and C4BP; or inhibition of lysis through the blockage of MAC insertion by CD59 (Refs 6–8) (Fig. 1b).
Table 1 Complement interactions with pathogens and self
The complement system is also a main player in the handling of altered self. In response to injury, for example during apoptosis or necrosis, complement activation occurs in a more targeted and restricted manner compared with its interaction with foreign materials9,10,11,12. Here, the goal is the safe disposal of cellular debris in the absence of an immune response and with minimal collateral damage2,3,6,9.
During the past three decades, the complement system has been firmly identified as an instructor of the humoral immune response13,14,15,16,17. It serves as a natural adjuvant, lowers the threshold for B-cell activation18,19,20, facilitates the localization of antigen to follicular dendritic cells (FDCs) in lymphoid follicles21, promotes the development of optimal B-cell memory22,23 and maintains B-cell tolerance23. These functions are mostly mediated by the binding of opsonized antigens to CR1 and CR2 (also known as CD21). In view of this impressive repertoire of activities, involvement of the complement system in the regulation of T-cell responses was anticipated.
T-cell activation is induced through the presentation of antigen by mature antigen-presenting cells (APCs) in lymph nodes or secondary lymphoid structures to the T cell. The nature of the antigen, the APC and the microenvironment largely determines which type of T cell will predominate (T helper 1 (TH1) cells, TH2 cells or regulatory T cells) and whether the activated T cells will then migrate to an inflammatory site.
Recent research has shown that complement can modulate T-cell responses during the induction and effector phases, as well as in the contraction phase, of an immune response24,25,26. These effects arise through the direct modulation of the T cell itself or indirectly through the alteration of immunomodulatory cells, particularly APCs. This Review discusses the emerging roles of the complement system in the initiation, effector and termination phases of the T-cell response.
Role of complement during T-cell activation
Modulation of APC function. APCs, including dendritic cells (DCs), FDCs and macrophages, express a remarkable repertoire of complement receptors and regulators on their surfaces (Fig. 2a). Therefore, these cells are poised to recognize and interact with antigens that have been opsonized by complement27. The engagement of complement receptors and regulators on APCs modulates their maturation status and their chemokine and cytokine expression profiles, which, in turn, influences the T-cell response induced during antigen presentation16,25,26,28. So, MBL, C1q, C3b and C4b, which are tightly attached to the antigen, engage their respective receptors on APCs during antigen recognition and uptake, and then initiate changes in APC function.
Figure 2: Complement receptors and regulators on antigen-presenting cells and T cells.
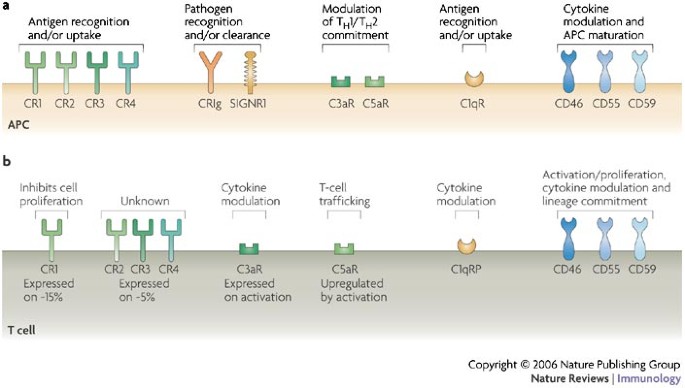
Expression profiles and immunomodulatory functions of complement-binding proteins on human antigen-presenting cells (APCs) (a) and T cells (b). C1qR, complement component C1q receptor; C3aR, complement component 3a receptor; CR, complement receptor; CRIg, complement receptor of the immunoglobulin superfamily; SIGNR1, a mouse homologue of dendritic-cell-specific ICAM3-grabbing non-integrin (DC-SIGN); TH1, T helper 1.
For example, in the absence of C1q or C3, antigen uptake is suboptimal2,3,29,30,31,32,33, resulting in reduced maturation of the APC, and leads to suboptimal T-cell activation2,3,29,30,31,32. Likewise, the anaphylatoxins C3a and C5a are liberated at sites of complement activation, where they positively modulate APC function by triggering their seven-transmembrane G-protein-coupled receptors C3a receptor (C3aR) and C5aR, respectively34 (Table 2). The biological consequences of C3aR and C5aR activation on APCs includes the migration of these cells and modulation of interleukin-12 (IL-12) production34,35,36,37,38,39. Induction of IL-12 leads to the development of a TH1 response, whereas suppression of IL-12 production favours TH2-cell development. However, both the induction and the suppression of IL-12 production has been shown to occur after C3aR and C5aR activation, depending on the disease model, the route of delivery of the antigen to be opsonized and the maturation status of the APC used34,37,38,39,40.
Table 2 Complement proteins and ligands
A related area of expanding research is the crosstalk between the complement system and TLRs27,41. In a recent study, Hawlisch et al. showed that C5a negatively regulates TLR4- and CD40-induced production of the IL-12 family of cytokines (IL-12, IL-23 and IL-27) in mouse macrophages41. Because most pathogens activate both of these innate immune surveillance systems simultaneously, an integrated analysis of their signalling pathways might shed light on the conflicting data regarding the effect of the anaphylatoxin receptors on cytokine production by APCs.
Two membrane-bound complement regulators, CD46 and CD55, also participate directly in modulating the function of APCs. The crosslinking of CD46 on human macrophages with antibodies or with pathogenic ligands (such as pili from Neisseria spp. or haemagglutinin from the measles virus) leads to calcium flux42 and the suppression of IL-12 (Ref. 43). Failure to synthesize this cytokine is one reason for the suppressed state of T-cell responses during infection with the measles virus43,44. How these regulators modulate APC function is not fully understood, but Heeger and colleagues proposed that CD55 decreases APC maturation by controlling complement deposition45. They suggest that the decay-accelerating activity of CD55 decreases the rate of formation of the C3 convertase and the rate of C3b deposition on the APC surface, thereby reducing the numbers of complement receptors engaged45. This, in turn, affects the maturation status of the APC and, consequently, T-cell priming.
Two recent studies46,47 describe previously unknown receptors for complement fragments on macrophages and indicate novel connections among complement, pathogen recognition, clearance and immune modulation. Helmy et al. identified CRIg (complement receptor of the immunoglobulin superfamily; also known as Z39Ig) as a receptor for C3b and the inactivated form of C3b (iC3b) on tissue-resident macrophages — including Kupffer cells in the liver — which mediates clearance of C3-opsonized pathogens46. Kang and colleagues observed that the lectin receptor SIGNR1 (a mouse homologue of dendritic-cell-specific ICAM3-grabbing non-integrin (DC-SIGN)) binds both C1q and the Streptococcus pneumoniae polysaccharide on splenic marginal-zone macrophages47. This binding process results in SIGNR1-dependent C3 fragment deposition on bacteria and their efficient clearance47. The generation of CRIg- and SIGNR1-deficient mice will allow analysis of their place in the regulation of adaptive immunity during infections and in autoimmunity.
Direct modulation of T-cell activation. Compared with APCs, T cells express a more limited range of complement receptors and regulators (Fig. 2b). Although the complement receptors CR1 and CR2 have a central role in the induction and regulation of B-cell responses20,23, they do not seem to have a similar place in T-cell biology. Only ∼15% of human peripheral-blood T cells express CR1 (Refs 48, 49) and ∼5% express CR2, CR3 or CR4 (Refs 48, 50). However, CR1 is upregulated on human peripheral-blood T cells on activation49, and crosslinking CR1 inhibits T-cell proliferation and IL-2 production50. No definitive function has been shown for CR2, CR3 and CR4 on CD4+ T cells48. CR3 is also expressed by differentiated CD8+ T cells51.
C1q has been proposed to bind to a number of cell-surface receptors52. The C1q receptor P (C1qRP) is expressed by most T cells, and C1q-bearing immune complexes might induce T-cell activation with concurrent secretion of interferon-γ (IFNγ) and tumour-necrosis factor (TNF)53. C5aR is also expressed by most T cells. On T-cell activation, the expression levels of C5aR increase and these cells respond to C5a with directed chemotactic migration54. These and other data on the C5a–C5aR interactions on T cells indicate a role for these proteins in T-cell trafficking34,55,56. By contrast, naive CD4+ and CD8+ T cells do not express C3aR but its expression is induced on T-cell activation57. The limited data available indicate a possible regulative effect of C3a during T-cell activation, as addition of this anaphylatoxin decreases T-cell-dependent antibody responses in a mixed lymphocyte reaction25. In addition, T cells from C3aR-deficient mice are characterized by increased cytokine production on activation40.
Unexpected roles are also beginning to be defined for complement regulators during T-cell activation. For example, the crosslinking of CD46 during CD4+ T-cell priming induces strong proliferation and the synthesis of large amounts of IL-10 and granzyme B24,58,59. In addition, the activation of CD46, in conjunction with phorbol 12-myristate 13-acetate, induces high IFNγ secretion by T cells60. Furthermore, a new role for another complement regulator in modulating T-cell immunity was recently described in CD55-deficient mice45,61. These animals have increased numbers of IFNγ-producing T cells after immunization with ovalbumin or the myelin oligodendrocyte glycoprotein (MOG) peptide in a model of experimental autoimmune encephalomyelitis45,61. Finally, crosslinking the MAC regulator CD59 on human T cells transduces intracellular signals and leads to increased proliferation and IL-2 production62.
Role of complement in the T-cell effector phase
Shaping the T H 1/T H 2-cell response. As discussed in the previous section, complement affects T-cell activation indirectly through modulating APC function or directly by altering T-cell priming. Through these activities, complement shapes the subsequent TH1/TH2 effector response. Support for such a crucial role in protective T-cell responses comes primarily from experiments using C3-deficient mice in influenza and lymphocytic choriomenigitis viral infection models63,64,65. In both cases, the CD4+ T-cell response to the virus was impaired in C3 −/− mice. In addition, the CD8+ T-cell response was decreased in the influenza system64. How complement is activated in these models and the mechanism(s) of C3 dependency remain unanswered. One possibility is that natural antibodies bind the virus and initiate C3 deposition through the classical pathway. In line with this hypothesis is the dependency of an optimal CD8+ T-cell response on natural IgM to activate complement after immunization against visceral leishmaniasis66. The absence of C3 probably results in reduced APC maturation and limited T-cell activation.
Another possibility — and the two mechanisms are not exclusive — is that the anaphylatoxins C3a and C5a and their respective receptors are necessary for inducing optimal adaptive immunity. Informative studies by Wetsel and colleagues showed that C3a has a central role in the priming and instruction of T-cell responses37. In these experiments, C3aR-deficient mice showed decreased TH2-cell responses, including reduced IL-4 production, and were protected against airway hyperreactivity, a TH2-dependent process, in a mouse model of asthma37. By contrast, in a mouse model of atopic dermatitis, C3aR-deficient mice showed an enhanced TH2-cell response and no disease protection40. The predominant TH2-cell response was dependent on C3aR-induced IL-4, IL-5 and IL-10 synthesis by DCs40. The reason for these contradictory results is not known.
The other anaphylatoxin, C5a, also regulates T-cell responses. Mice lacking C5aR or treated with a C5aR antagonist have a reduced ability to clear an infection with Pseudomonas aeruginosa67 and to mount virus-specific CD8+ T-cell responses68. In addition, C5-deficient animals have exacerbated TH2-cell responses in an experimental allergic asthma model41,69. Interestingly, the C5 gene was identified as a susceptibility locus for human allergen-induced asthma69. Although C5a induces TH1-cell responses in these models, a role for C5a in the induction of a TH2-cell phenotype has also been shown70. Therefore, these data suggest complex and pleomorphic effects of the anaphylatoxins on the production of the key cytokines that drive T-cell effector functions.
The explanation for the observed phenotype of _Cd55_-knockout mice used in studies by Liu61 and Heeger45 is controversial. The key finding of both studies was an enhanced TH1-cell response in CD55-deficient mice, including increased IFNγ production. However, one group attributes the observed negative regulation of T cells by CD55 to a direct effect on the T cell itself61, whereas the other favours a mechanism that involves alteration of APC function45. Future studies will clarify whether one or other, or both, of these cell types contribute to the phenotype.
In an in vivo experimental system, CD46, a structural and functional cousin of CD55, also regulated T-cell effector responses. Marie and colleagues generated mice expressing human CD46 with either of its two regularly expressed cytoplasmic tails, CYT1 or CYT2 (Ref. 71). CYT1 and CYT2 differ in size and amino-acid sequence, and both contain motifs necessary for signalling71. The researchers found that CD4+ T cells from mice expressing CD46 with the CYT1 cytoplasmic tail proliferated strongly, produced IL-10 and inhibited the contact-hypersensitivity reaction after concurrent T-cell receptor (TCR) and CD46 activation. By contrast, TCR- and CD46-activated T cells bearing CD46 with its CYT2 cytoplasmic tail showed weak proliferation and low IL-10 production but a heightened contact-hypersensitivity reaction71. This study was the first to confirm a direct in vivo T-cell regulative function for CD46 and suggests that CYT1 is the cytoplasmic domain that mediates the development of IL-10-producing regulatory T cells after CD46 crosslinking (see later section).
A similar regulative role in virus-induced T-cell responses was discovered for CD59. Mice deficient for CD59 showed enhanced virus-specific CD4+ T-cell responses after immunization with recombinant vaccinia virus72. Notably, the observed effect in this study was shown to be independent of complement activation and does not therefore involve regulation of MAC formation. Such downregulation of T-cell activity by CD59 in mice contrasts with findings in human T cells, in which CD59, when crosslinked with antibodies, functions as a co-stimulatory molecule by inducing T-cell proliferation and IL-2 production independent of MAC formation62. The above data are consistent with the emerging understanding that crosslinking or activating complement receptors and/or regulators on T cells induces signalling events that influence proliferation and cytokine production by these cells.
However, these studies also raise an important issue: namely that there are substantial differences between mouse and human (and even other primate) complement receptors and regulators. Although CR1 and CR2 are two separate proteins in humans, mice express them as one protein derived from a single gene in a more limited array of cell types7,8. Moreover, human CR1 is a 250-kDa protein with three C3b and C4b binding sites, whereas mouse CR1 is a 60-kDa amino-terminal attachment to CR2 with one binding site for C3b and C4b7,8. In addition, the complement regulatory proteins CD55 and CD59 are present in mice as two different protein forms, each with distinct expression profiles, whereas in humans CD55 and CD59 are only expressed in one form7,8. Furthermore, CD46 is not expressed by somatic cells in the mouse73,74, and another protein, complement-receptor-related protein (CRRY), apparently replaces the role of CD46 as a cofactor in the cleavage of C3b and C4b (Ref. 75). CRRY has co-stimulatory properties for mouse CD4+ T cells76 and crosslinking of this protein on the surface of these cells induces proliferation and IL-4 production76.
Overall, the complement-activation system does not seem to be as robust in mice as it is in humans. For example, the ability of mouse complement to lyse sheep red blood cells is one-tenth to one-hundredth as efficient as human complement. Most of the studies regarding the role of complement in T-cell function — specifically in vivo work — have been carried out in mouse models. On account of these issues, the results obtained should be transferred to the human system with caution. Information on the affect of complement on signalling in effector T cells is limited. The complement-mediated signalling pathways activated during the APC–T-cell interactions are largely unknown and probably complex. Available data indicate, however, that these responses might not follow a pre-set, fixed pathway but are likely to be customized to pathogen classes and to vary considerably from one tissue type to another.
Role of complement in the contraction phase
An exciting development in the field of T-cell immunity is the idea that the complement system affects the termination of the T-cell response. Two main mechanisms for the termination of the T-cell response by complement are emerging: the modulation of apoptosis and induction of regulatory T cells.
Modulation of apoptosis. During the contraction phase of the effector T-cell response most of the cells undergo apoptosis, leaving only a small number of viable T cells to constitute the memory pool. Efficient uptake and safe disposal of these apoptotic cells are necessary to avoid inflammation and autoimmune disease. C1q binds to late apoptotic cells and initiates the activation of the classical pathway, which leads to their opsonization by C3b (Refs 9, 10). C1q therefore aids in the uptake of these cells by macrophages. In addition, CD46 has an unanticipated function in recognizing apoptotic T cells77. Elward et al. observed that apoptotic T cells rapidly lose CD46 from their surfaces77. CD46 was shown to cluster in the apoptotic blebs and then shed in microparticles. The authors proposed that expressing CD46 on the surface of a T cell prevents the cell from being phagocytosed and that losing CD46 on the induction of apoptosis removes this protective signal and allows engulfment by phagocytes. In addition, losing this major complement regulatory molecule allows for greater C3b deposition, which, in turn, increases phagocytic uptake of the apoptotic cells78.
An additional role for complement, beyond providing assistance in the safe removal of apoptotic cells, has been suggested. In this work, CD55 and CD59 were shown to directly modulate CD95 (also known as FAS)-mediated apoptosis of human CD4+ T cells79. Localization of CD28, CD55 and CD59 into lipid rafts was observed on T-cell activation; CD28 was concentrated into microdomains, which are enriched with ganglioside GM1, within the lipid rafts, whereas CD55 and CD59 were excluded from these microdomains79. Whereas co-ligation of CD28 with CD95 amplified the CD95 signalling pathway, co-ligation of CD95 with CD55 or CD59 inhibited the apoptotic signal79. Although the biological significance of this finding is not clear, it will be of interest to investigate whether these events are linked to the inhibitory role of CD55 in TCR-mediated T-cell activation45,61.
Induction of regulatory T cells. Downregulation of the effector T-cell response through the development of a lineage of T cells with suppressive properties is a recently described role for complement in the contraction of an immune response. The concurrent crosslinking of the TCR and CD46 on naive peripheral-blood CD4+ T cells with specific antibodies, C3b dimers or a pathogenic ligand (for example, the streptococcal M protein, which interacts with CD46 on human CD4+ T cells) induces the development of T cells with regulatory properties24,59,80,81,82,83 (Fig. 3).
Figure 3: Characteristics of complement-induced regulatory T cells.
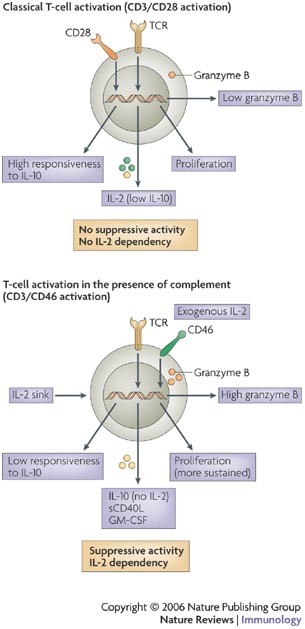
Functional differences between CD3- and CD28-stimulated compared with CD3- and CD46-stimulated naive CD4+ T cells are summarized. The activation of T cells in the presence of complement ligands induces cells with a cytokine- and granzyme-profile that is distinct from those activated through the classical CD28 co-stimulatory pathway. Importantly, CD46 activation does not lead to interleukin-2 (IL-2) production24. GM-CSF, granulocyte/macrophage colony-stimulating factor; sCD40L, secretory CD40 ligand; TCR, T-cell receptor.
Active suppression of autoreactive T cells by regulatory T cells has emerged as an essential process to prevent autoimmunity and mediate peripheral tolerance. In addition, the suppression of resident effector CD4+ TH1 and TH2 cells directed against enteric commensals and other foreign but benign factors at the host–environment interface is a crucial factor in mucosal immune homeostasis84,85. Regulatory T cells are currently divided into two main subsets. Natural CD4+CD25+ T regulatory (TReg) cells originate in the thymus, are specific for self-antigens and act predominantly through a contact-dependent mechanism that probably involves granzyme A in humans59,80,81,82. However, in mice it is thought that natural TReg cells might use granzyme B for their suppressive activity86. These natural TReg cells constitutively express the transcription factor forkhead box P3 (FOXP3) and the IL-2 receptor α-chain (CD25), and require exogenous IL-2 for their function and expansion81,82,87. Inducible T regulatory 1 (TR1) and TH3 cells are generated in the periphery against both self and foreign antigens81,82. They also require exogenous IL-2 and mediate their suppressive effect primarily by the secretion of IL-10 (TR1 cells) or transforming growth factor-β (TGFβ; TH3 cells)84,88. Inducible regulatory T cells might or might not express FOXP3 and show variable expression of CD25 (Refs 59, 81).
Crosslinking CD46 during TCR activation leads to the development of T cells with the ability to regulate the activation of classically activated (CD3 and CD28) bystander T cells, similar to inducible T regulatory cells24. The CD46-mediated signalling pathways leading to this phenotype have not been defined. However, these cells express normal levels of many activation markers, including CD25, CD69, CD62 ligand (CD62L) and CD152, and acquire a normal memory phenotype (expression of CD45RO)24. These CD46-induced regulatory T cells proliferate strongly and suppress the activation of bystander T cells through the secretion of the immunosuppressive cytokine IL-10 (Refs 24, 74). They require no pre-existing basal expression of FOXP3 (Ref. 59) but their induction, function and expansion is, as it is for other inducible regulatory T cells, highly dependent on an exogenous source of IL-2 (Ref. 24) (Fig. 3). CD46-induced regulatory T cells also synthesize granzyme B and perforin, and show contact-dependent cytotoxicity towards autologous immunocompetent T cells, including activated CD4+ and CD8+ T cells59,80. Therefore, regulatory T cells generated through CD3 and CD46 activation have three distinct mechanisms for effector T-cell suppression: secretion of IL-10, synthesis of granzyme B and competition for IL-2 as a growth factor.
T cells with these properties are predicted to aid in the control and contraction of an effector T-cell response. After the clonal expansion of effector T cells, sufficient amounts of IL-2 and CD46 ligand (C3b- or C4b-opsonized immune complexes) would become available to induce IL-10-secreting, granzyme-B-expressing regulatory T cells. We propose that these regulatory T cells then downregulate effector T-cell function to prevent immunopathology caused by an overexuberant immune response (Fig. 4). In this manner, complement-induced regulatory T cells might participate in terminating an effector immune response to an invading pathogen and in preventing chronic inflammation.
Figure 4: Complement in the T-cell response continuum.
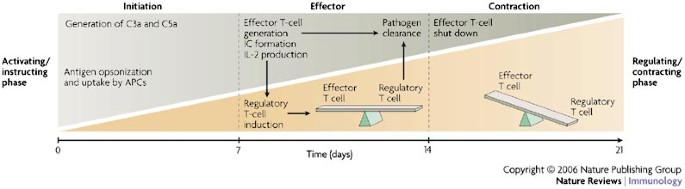
A model for how complement regulates the three phases of a T-cell immune response. During the initiation phase, the activation of complement (the generation of the opsonins complement component 3a (C3a) and C5a) facilitates antigen recognition and the maturation of antigen-presenting cells (APCs). Antigen-experienced mature APCs induce effector T-cell responses to produce interleukin-2 (IL-2), which is necessary for the induction of complement (CD46)-induced regulatory T cells. Ligands for CD46, such as opsonized immune complexes (ICs), are also generated at this time. Clonal expansion of antigen-specific effector T cells leads to the clearance of the pathogen, and the balance between effector T cells and CD46-induced regulatory T cells prevents excessive immunopathology. With continuous expansion of CD46-induced regulatory T cells (which proliferate more strongly than effector T cells24), IL-10 secretion and the number of granzyme B-expressing cells might become sufficient59 to shut down the successful effector T-cell response. The inactivation of effector T cells leads to the simultaneous contraction of the regulatory T-cell pool — as these cells are dependent on IL-2 produced by effector T cells — and the generation of an effector T-cell and regulatory T-cell memory pool24. The timescale chosen for this model is based on an acute viral or bacterial infection.
Generally, adaptive IL-10-producing regulatory T-cell populations do not allow for DC maturation on antigen encounter, because IL-10 not only inhibits T-cell activation but also DC activation89. By contrast, CD46-induced regulatory T cells permit DC activation90. This distinguishing feature is achieved through their simultaneous secretion of granulocyte/macrophage colony-stimulating factor (GM-CSF) and soluble CD40, in addition to IL-10 (Ref. 90). Soluble CD40 and GM-CSF neutralize the suppressive effect of IL-10 on DCs, but not on T cells, and thereby allow the differentiation of DCs. So, the cytokine profile of CD46-induced regulatory T cells suppresses T-cell responses through IL-10 but allows for DC maturation in the presence of this cytokine. Such 'DC-sparing' complement-induced regulatory T cells could be desirable at the host–environment interfaces. In the airway, skin and gastrointestinal tract, the cytokine profile of these regulatory T cells might ensure unresponsiveness to commensals and innocuous antigens (by suppressing an unwanted T-cell response through IL-10) while maintaining reactivity to invading pathogens (by allowing DC maturation on encounter with pathogenic antigens).
Intestinal bacteria are a constant source of antigen that provides inflammatory signals for cells of the innate immune system and a nidus for complement activation. A portion of T cells that encounter their cognate antigen at this location might be induced towards a regulatory T-cell phenotype through CD46 stimulation. Such a regulatory T-cell pool could control effector T-cell-mediated inflammation through local IL-10 secretion85. Indeed, both effector and regulatory T cells specific for the same _Escherichia coli_-derived antigen are present in the intestine91 and mice that are deficient for the Il10 or Il2 gene succumb to colitis92,93. However, allowing DCs to function normally at this location also guarantees protection against invading pathogens. That the human lamina propria contains T cells with a cytokine expression profile that is characteristic of CD46-induced regulatory T cells supports the hypothesis that complement-induced regulatory T cells are involved in mucosal immunity90.
The immunomodulatory properties of CD46 on T cells and macrophages probably account for the use of CD46 as a receptor by numerous human pathogens (for example, strains of the measles virus, herpes simplex virus 6 and certain adenoviruses, as well as Streptococci pyogenes and pathogenic Neisseria spp.)44,73,83. We have speculated that pathogens that bind to CD46 might abuse this complement receptor to induce a local suppressive milieu74,83. This concept of abusing regulatory T-cell functions has been shown for several other pathogenic microorganisms94,95.
The results of studies of CD46-induced regulatory T cells that can regulate a bystander T-cell response make an appealing case for a role of complement in the contraction phase of an immune response24,74. However, this concept awaits verification in an in vivo system. In addition, the circumstances that lead to the induction of these cells in vivo are poorly understood. Although C3b dimers induce regulatory T-cell development through their interactions with CD46, the observation that a CD46–human IgG4 fusion protein binds to human cell lines as well as to polymorphonuclear leukocytes77 makes the idea of an unidentified new ligand or receptor for CD46 — perhaps expressed by specialized (possibly tolerogenic) APCs — intriguing. Furthermore, the recent identification of novel complement receptors (CRIg and SIGNR1) on APCs46,47 shows that there is still much to be discovered about the complement system.
Complement-induced signalling in T cells
Complement activation fragments such as C3b, C4b, C3a and C5a interact with complement receptors and regulators to modulate T-cell lineage commitment and cytokine, granzyme and perforin production. However, relatively little is known about the signalling pathways involved in these effects compared with, for example, CR2-induced signalling in B cells20,23. Although the activation of CD46 induces intracellular signalling events in several cell types25,26,73,96,97, these pathways are largely undefined. The situation is complicated by the fact that CD46 expresses two distinct cytoplasmic domains (CYT1 and CYT2), both of which have signalling capacities73. CD46 activation in macrophages induces calcium flux from intracellular stores and the recruitment of SH2-domain-containing protein tyrosine phosphatase 1 (SHP1) to the CD46 cytoplasmic domain(s)98. On epithelial cells, the phosphorylation of the intracellular domains of CD46, as well as association of CYT1 with discs large 4 (DLG4), a protein involved in the formation of cell-adherent junctions, has been shown42,99. Activation of CD46 on T cells induces the phosphorylation of the cytoplasmic tail domains of both CD46 and the scaffolding molecule LAT, as well as the activation of the guanine-nucleotide-exchange factor VAV, the GTPase RAC and the extracellular-signal-regulated kinase 1 (ERK1)/ERK2 mitogen-activated protein kinase pathway58,100.
A direct signalling role in T cells has also been described for the two glycosylphosphatidylinositol-anchored complement regulators, CD55 and CD59 (Refs 8, 25, 26, 62, 101). Crosslinking of these molecules enhances T-cell proliferation and, in the case of CD59, IL-2 production62. These biological outcomes involve SRC-family kinase LCK-mediated activation and phosphorylation events102. The signalling pathways for C3aR and C5aR have been studied in human peripheral-blood mononuclear and neutrophil populations34. These complement receptors signal through G proteins34 and downstream events include the activation of important signalling molecules such as phosphatidylinositol 3-kinase (PI3K), protein kinase C (PKC), ERK and signal transducers and activators of transcription 3 (STAT3) (Ref. 34). The processes that occur between the initial events that are mediated by the activation of complement receptors and regulators and the induction of proliferation, cytokines and granzymes remain to be revealed. The current understanding of the signalling pathways induced by complement receptors and regulators in T cells have been recently reviewed by Hawlisch34, Morgan25,26 and Russell96.
Complement and T-cell-mediated pathologies
Because an important role for complement in regulating T-cell-mediated immunity is emerging, it is not surprising that the complement system has been studied in connection with transplantation rejection103,104. The main obstacle to successful allograft transplantation is rejection of the donor graft, a principally T-cell-mediated process104,105. Complement is likely to have a more central role in this process than has previously been suspected. Mice deficient in different complement components (mainly C3 or C4) or animals treated with complement inhibitors show prolonged survival of skin allografts and primarily vascularized grafts104. This is related to a reduction in ischaemia-reperfusion injury and vessel wall inflammation and the decreased recruitment of effector cells104. The mechanisms underlying complement-mediated rejection are complex and include the induction of alloantibody responses and effector T-cell activation and infiltration. Local synthesis of complement components by epithelial and vascular tissue in the graft might also contribute to this response103. Therefore, to improve the success rate of organ and tissue transplantation, a better understanding of these processes is needed, and therapeutic manipulation of complement in parallel with the more standard immunosuppressive regimens should be considered. It is likely that complement deficiencies or dysfunctions are also associated with human T-cell-mediated diseases. Indeed, mice deficient for the complement regulator CD55 develop more severe experimental autoimmune encephalomyelitis than wild-type mice in this disease model45 and CD46-transgenic mice show abnormal T-cell responses in the contact-hypersensitivity reaction71.
Future considerations
Interaction between the complement system and T cells is a new area of research. Additional complement ligands and receptors on T cells await discovery. Analysis of their actions and of those reviewed in this update should clarify conflicting opinions in the literature (for example, the roles of the anaphylatoxins in cytokine production, the mechanism(s) of T-cell modulation by CD55 and to what extent we should or can apply results obtained in mouse models to humans) and could lead to new models of how T-cell responses are affected by complement. An improved understanding of the parameters that ultimately shape the interplay between T cells and complement will become the basis for developing new therapies that centre on manipulating complement to control T-cell responses.