Genome sequence of Kobresia littledalei, the first chromosome-level genome in the family Cyperaceae (original) (raw)
Sample sequencing and genome size estimation
High-quality genomic DNA for sequencing was extracted from leaf tissue of K. littledalei, which was collected in July 2018 from DangXiong in the Tibet Autonomous Region of China. The sample was in anthesis, and located at altitudes of up to 4,263 m.
The Illumina library with insert sizes of 350 bp was arranged with a Genomic DNA Sample Preparation kit from Illumina. It was then sequenced using a HiSeq 2500 platform, also from Illumina. This yielded 168.79 million reads, ∼50.64 Gb of raw sequence data, which covered ~121.95X of the genome (Table s1). Large DNA fragments longer than 10 kb were enriched and were then sequenced using a PacBio Sequel system. From this, we obtained 5,618,892 reads that had an N50 length of 17,273 base pairs and a mean of 11,099 base pairs. In total, 62.37G bases were obtained, which is ~150.20X coverage of the genome (Table s1). Leaf tissue of K. littledalei was used to construct a library for Hi-C analysis, and the NEBNext Ultra II DNA library Prep Kit from Illumina (NEB) was used to prepare the Hi-C library, which we then sequenced using the Illumina HiSeq X Ten platform. 230,316,080 paired-end reads of 150 bp were obtained from the Illumina sequencing platform for the Hi-C library, which covered ~166.40X of the genome (Table s1).
The size and heterozygosity level of the K. littledalei genome were estimated through k-mer spectrum analysis using sequences generated by Illumina DNA sequencing technology2. The depth distribution of the derived 17-mers clearly showed two separate peaks and the main volume peak of k-mer frequency was 96, based on which we estimated the heterozygosity level and repeat frequencies of the K. littledalei genome to be 1.68% and 53.93%, respectively; the genome size was estimated to be 415.24 Mb (Fig. s1, Table s2).
Assembly of the Kobresia littledalei C. B. Clarke genome
First, PacBio long reads were self-corrected to obtain pre-assembly reads. The pre-assembly reads were assembled into consensus sequences by FALCON through the “Overlap-Layout-Consensus” algorithm3. Consensus sequences were corrected using Illumina short reads to improve the precision in Pilon4. The preliminary genome assembly of K. littledalei includes 1210 contigs with N50 = 2,253,412 bp and longest scaffold = 11,050,451 bp. The genome is approximately 759 M in length and the GC content of the genome is 35.74% (Table s3).
Purge Haplotigs was used to filter redundant sequences due to heterozygosity5. The final assembled K. littledalei genome contained 212 scaffolds with an N50 length of 3,054,069 bp and a cumulative size of 373,821,983 bp. The longest scaffold reached 11,045,779 bp, and the GC content of the genome was 35.44% (Table 1)
Table 1 Statistics of assembled Kobresia littledalei C. B. Clarke assembly and annotation.
We used the procedures described by DC. Zhang et al.6 to anchor the scaffolds into pseudo-chromosomes. We first used HiCUP v0.6.17 to map and process reads obtained from the Hi-C library. Each of the reads from one pair were uniquely mapped to the assembly and kept for downstream filtration. Invalid pairs generated from fragments of the wrong size, PCR duplication, re-ligation, internal fragments, dangling ends, circularization, and contiguous sequences were removed. K. littledalei has 2n = 58 chromosomes, as determined by karyotype analyses. We corrected some small errors in the results of the FALCON assembly by clustering contigs with the contig contact frequency matrix. We obtained 523 total contigs by grouping contigs with errors into shorter contigs. Using Lachesis v1.08 (pseudo-chromosome number set as 29), we clustered 337 contigs into pseudo-chromosomes using the refined alignments. This corresponds to 96.28% of the assembly by base count and 64.44% of the assembly by sequence number.
We built an interaction matrix by HiC-Pro using clean reads from the Hi-C library to confirm the accuracy of the Hi-C scaffolding at the pseudo-chromosome level9. The genome was split into equal-sized bins of 500k, while the contact numbers were designated between each pair of bins. We confirmed the genome quality and structure using a contact map plotted with HiCPlotter10 (Fig. 2).
Fig. 2
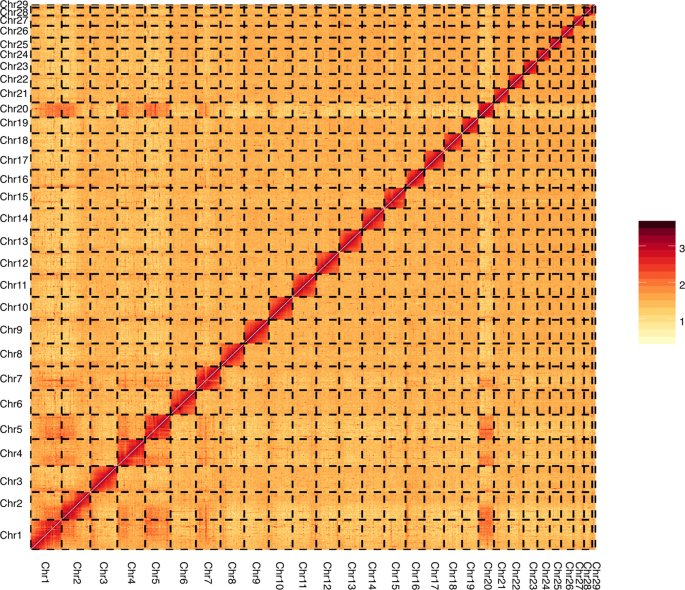
Heat map of chromatin contact matrices generated by aligning a Hi-C dataset to the Kobresia littledalei genome. The frequency of interactions was calculated using a window size of 500 K.
Repeat annotation
We used RepeatMasker11 to predict repeat sequences of the K. littledalei genome through homology searching of repetitive elements released by Repbase12 and ab initio identified by LTR Finder13, RepeatScout14 and RepeatModeler. We identified a total of ~202.34 M repetitive elements, which was 54.13% of the genome, after integrating ~40.04 M repetitive elements predicted by RepeatProteinMask and ~20.96 M tandem repetitive sequences predicted by TRF15 (Table s4). Among them, DNA transposons accounted for 18.53% of the genome, while long terminal repeat (LTR), long interspersed nuclear elements (LINE) and short interspersed nuclear elements (SINE) belonging to retrotransposons accounted for 27.87%, 5.42% and 0.10% of the genome respectively (Table s5).
Prediction and functional annotation of protein-coding genes
The repeat-masked K. littledalei C. B. Clarke genome was used for subsequent prediction of protein-coding genes, which integrates evidence from de novo predictions, protein homology and RNA transcripts. Augustus16, GlimmerHMM17, SNAP18, Geneid19 and Genscan20 were used for ab initio gene prediction. For homolog searches, we used proteomes of Zea mays, Brachypodium distachyon, Oryza sativa, Setaria italica, Ananas comosus and Arabidopsis thaliana. Due to the lack of RNA-seq data of K. littledalei, we used the RNA-seq data from nine species from Kobresia species. In total, 26,046 primitive gene models were predicted after integrating results of the three sources of evidence by EVM21. We then filtered and polished these gene models through expression level and evidence number, and 22,979 genes with FPKM > 1 or supported by more than two lines of evidence were retained (Table s6). For gene models only supported by one line of evidence, we searched SwissProt22, KEGG23, NCBI-nr, InterPro24 and Pfam for homologs. Gene models with homologs in any of the databases were retained, resulting in 157 genes. In total, 23,136 gene models were identified. The average length of genes and CDS are 3,545.25 bp and 1,163.41 bp, respectively, and there are 5.39 exons in each gene with length of 215.84 bp per exon (Table 1). Among them, 12,726 gene models are supported by all three lines of evidence (Fig. s2).
To assess the completeness of the gene identification, we conducted BUSCO analysis on 23,136 gene models. For 1,440 expected embryophyta genes, 86.2% complete and 3.6% fragmented gene models were identified in K. littledalei. The identified gene model was 89.8%, which is less than pearl millet (95.4%), broomcorn millet (98%), sugarcane (95.4%) and other recently published Poaceae species25,26,27. We also download the genome sequence of A. comosus and conducted BUSCO analysis; 92.6% complete and 2.7% fragmented gene models were identified28. To explore the reason, we also conducted BUSCO analysis on the transcriptomes (leaf) of Kobresia tibetica, K. royleana and K. pygmaea assembled by Trinity, and 81.4%, 74.6% and 79.3% complete gene models plus 4.1%, 5.6% and 6.0% fragmented gene models were identified, respectively (Table s7). Simultaneously, the transcriptomes of three other Cyperaceae species, Cyperus papyrus (shoot without flower), Lepidosperma gibsonii (leaves and buds) and Mapania palustris (leaf shoots) were downloaded from the 1,000 plants (1KP)29 project. The complete gene models were 57.1%, 64.7% and 39.5% and the fragmented gene models were 14.4%, 13.3% and 23.3% assessed by BUSCO analysis (Table s7).
Functional annotation of protein coding genes was obtained by mapping protein sequences to SwissProt22, KEGG23 and NCBI-nr protein databases by BLASTP to get the best hit. Simultaneously, functional annotation of protein coding genes was inferred by protein domains identified by searching the protein sequence against the InterPro24 and Pfam30 databases using InterProScan31 and HMMER32. The Gene Ontology (GO)33 terms were obtained by Blast2GO34. A total of 22,892 (98.95) out of 23,136 genes have integrated functional annotation (Table s8).
Genome structure of K. littledalei
The genus Kobresia, which includes about 70 species, is distributed predominantly in the alpine mountains of the Northern Hemisphere, and a majority of the 59 species found throughout China live on the Qinghai-Tibet Plateau. The basic chromosome numbers of species in Kobresia vary a lot (x = 16, x = 26, x = 29 and so on), which indicates that great changes have occurred in the chromosome structure of Kobresia. Moreover, it is reported that more than one-half of the tested species are polyploid in Kobresia, which is high compared with 5.7% in the closely related genus Carex35. The changes of chromosome structure and polyploidization in Kobresia likely indicate how these species adapted to the harsh environment of the Qinghai-Tibetan Plateau. The length of chromosomes of K. littledalei ranges from ~2.46 M to ~19.67 M. The centromeric regions were identified using an approach described by Robert et al.36. The base centromere repeat was 162 bp and highly abundant tandem repeats were identified on 18 chromosomes. The highly abundant tandem repeats dispersed on chromosome with high TE density like Chr1, Chr2, Chr5 and Chr16 (Fig. 3).
Fig. 3

Features of the Kobresia littledalei genome.
Evolutionary and comparative genomic analysis
To explore the evolutionary relationship of K. littledalei, we used OrthoFinder34 to cluster its genes with those from eight other commelinid monocots: B. distachyon, O. sativa, Z mays, Sorghum bicolor, A. comosus, Elaeis guineensis, Musa acuminata, Phyllostachys heterocycla and one dicot A. thaliana. From these ten species, we identified 826 one-to-one single-copy genes that were used to construct a maximum likelihood (ML) tree to show the evolutionary relationships using RaxML with the GTRGAMMA model37. Divergence times were estimated using the ‘mcmctree’ program incorporated in the PAML package38. According to the phylogenetic tree, K. littledalei separated from Poaceae about 97.6 million years ago after separating from Bromeliaceae about 114.3 million years ago (Fig. 4a).
Fig. 4
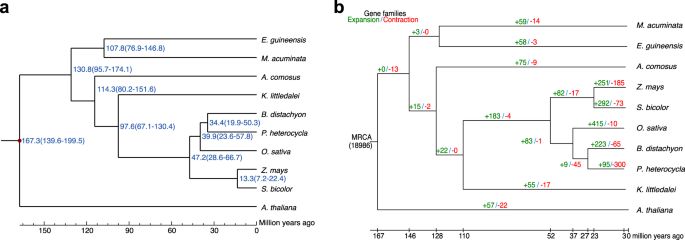
The phylogenetic relationships and divergence times of commelinid plants, and contraction and expansion of gene families. (a) The phylogenetic relationships and divergence times of commelinid plants. Phylogenetic reconstructions using concatenation of 1,077 genes and the maximum likelihood (ML) method with A. thaliana as the distant outgroup. Divergence times were estimated using the ‘mcmctree’ program incorporated in the PAML package. (b) Contraction and expansion of gene families. Numbers in green represent expanded families on this clade, and numbers in red represent contracted families on this clade.
To clarify the genome duplication history of K. littledalei, we screened the paralogs within syntenic blocks of K. littledalei by McScan39 and calculated the distribution of the rate of transversions on fourfold degenerate synonymous sites (4DTv). There is one peak with values of 4DTv at 0.63–0.68, which indicated that one whole genome duplication (WGD) event occurred before the rho WGD event that occurred ~70 MYA in the grass lineage40. To investigate the genome duplication history in Poales, we also screened the orthologs with syntenic blocks between K. littledalei and A. comosus, O. sativa and S. bicolor separately. Simultaneously, we calculated the 4DTv of the paralogs in A. comosus, O. sativa and S. bicolor, which showed an obvious WGD with a 4DTv value of 0.4. The 4DTv peaks between K. littledalei and A. comosus, O. sativa and S. bicolor are between 0.6–0.8, near the WGD of K. littledalei but earlier than the WGD of A. comosus, O. sativa and S. bicolor. Taken together, Cyperaceae separated from Bromeliaceae and Poaceae during the time of the WGD of K. littledalei and they were subjected to WGD independently after differentiation (Fig. 5).
Fig. 5
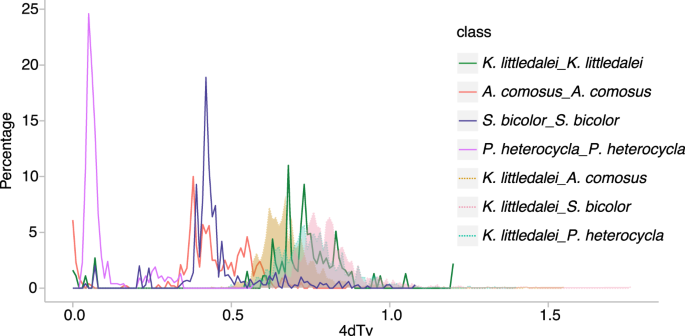
Whole genome duplication events in Kobresia littledalei and other Poaceae plants.
Comparing gene families among seven monocots, including K. littledalei, O. sativa, S. bicolor, A. comosus, E. guineensis, M. acuminata and P. heterocycla, we identified 23,136 K. littledalei genes in 12,006 families, with 8,645 gene families shared among them and 117 gene families unique in K. littledalei (Fig. s3). The expansion and contraction of the gene families, which can implicate the evolutionary dynamics of genes, were indicated by gene copy number in each family. In total, 17 gene families were contracted, and 55 gene families were expanded in K. littledalei (Fig. 4b). The expanded gene families included F-box containing protein, agamous-like MADS-box protein, and B3 domain containing protein (Table s9).