Regulation of both apoptosis and cell survival by the v-Src oncoprotein (original) (raw)
Introduction
A number of cell lines and primary cells of mesenchymal origin become quiescent when deprived of growth factors or matrix interactions. Even in the absence of both conditions such quiescent cells remain viable for some time.1,2,3 In contrast, the same cells transformed by either polyomavirus or RSV (Rous sarcoma virus) do not enter viable quiescence when denied both serum growth factors and matrix; they either multiply in suspension or become non-viable, a phenomenon that permitted the selective isolation of revertants of polyomavirus transformed cells and of ts (temperature sensitive) transformation mutants of RSV.2,3 We recently characterised how v-Src, the oncoprotein of RSV, prevented withdrawal from the cell cycle in the absence of growth factors4 and we now wish to understand how v-Src influences viability in these non-quiescent cells.
Several oncoproteins can induce the paradoxical outcomes of cell multiplication and senescence or apoptosis, depending on the cellular context.5 For example c-Myc, E2F-1 and v-Jun induce both cell proliferation and apoptosis6,7,8 whilst Ras, operating through the Raf pathway has similar effects.9 However, Ras stimulation of PI3-K protects against apoptosis9 and this is thought to be a reason why Ras and other anti-apoptotic oncogenes such as Bcl-2 demonstrate oncogenic cooperation with c-Myc,10,11,12 each oncogene countering the other's antiproliferative properties.
The way in which oncoproteins such as Myc and E2F-1 induce apoptosis is beginning to be characterised.13,14 v-Src transformation stimulates c-Myc expression15,16 suggesting a likely pro-apoptotic effect. However v-Src can also activate several kinases which can inhibit cell death including ERK17,18 and PI3-K19 which induces cell survival in epithelial cells following detachment via the downstream serine/threonine kinase, Akt.20 This suggests that v-Src may promote cell death or protect from cell death under different conditions.
We made use of Rat-1 cells transformed by a temperature sensitive v-Src (ts LA 29) which we used previously to characterise the mitogenic effects of v-Src.4,18,21 Here we use the reversibility of ts v-Src to examine the effect of inactivating v-Src in transformed cells grown under low serum conditions. We found that the cells exited the cell cycle and were committed to a programme of cell death. We characterised the death as apoptosis by morphological changes including membrane blebbing, DNA and nuclear fragmentation and biochemical changes including the activation of caspases22 and the activation of the stress induced kinases, JNK and p38.23,24,25 Over expression of the anti-apoptotic protein Bcl-226 prevented cell death and treatment with the caspase inhibitor Z-VAD-FMK27 and the p38 inhibitor SB20358028 simultaneously but not independently significantly reduced cell death. This suggested that the apoptotic pathway required either caspases or p38 and interacted functionally with Bcl-2.
Since v-Src transformed cells did not undergo apoptosis when v-Src was active we assessed whether apoptosis seen when v-Src was switched off was due to loss of v-Src survival signals. We investigated the effect of switching off v-Src on PI3-K and ERK activity and found that ERK and Akt (which is activated downstream of PI3-K) were rapidly inactivated. We tested whether inactivation of either kinase could induce cell death in v-Src transformed cell using LY294002 to inhibit PI3-K29 and PD98059 to inhibit ERK activating kinase (MEK1).30 We found that LY294002 was sufficient to induce apoptosis when added to v-Src transformed cells in low serum but did not induce apoptosis when added to normal Rat-1 cells grown under low serum conditions. This suggests that, in the absence of serum survival signals, v-Src promotes apoptosis but the transformed cells are protected from cell death by survival signals from v-Src mediated by PI3-K which could explain why v-Src does not require a co-operating oncoprotein. We suggest that the anti-apoptotic effects of v-Src could be as important to the transforming ability of the v-Src oncoprotein as the effects on mitogenesis.
Results
v-Src activity prevents transformed cells undergoing apoptosis in low serum
Rat-1 cells expressing the ts LA 29 v-Src mutant of RSV (ts LA 29 Rat-1) are transformed at the permissive temperature for v-Src activity (35°C) and morphologically normal at restrictive temperature (39.5°C). When such cells were placed in 0.2% serum medium (low serum) for 24 h they continue to cycle at 35°C (Figure 1A, 0 h and4). On inactivating v-Src by shift to 39.5°C we used flow cytometry to show that there was the expected rapid reduction in the number of cells in S-phase (as judged by the number of cells which incorporated BrdU during a 1 h pulse, Figure 1A, 4 and 8 h). The same phenomenon is seen initially when v-Src is inactivated in the presence of serum growth factors.4 In contrast to the cells' behaviour in high serum, however, v-Src inactivation in low serum led to many cells becoming loosely adherent (detached) within a few hours, with rounded or blebbing morphology (Figure 1B) and fragmented nuclei (Figure 1C) which could be labelled using the TUNEL (TdT-mediated dUTP nick end labelling) technique31 (Figure 1D). Almost all cells had detached in this way by 24 h after v-Src inactivation.
Figure 1
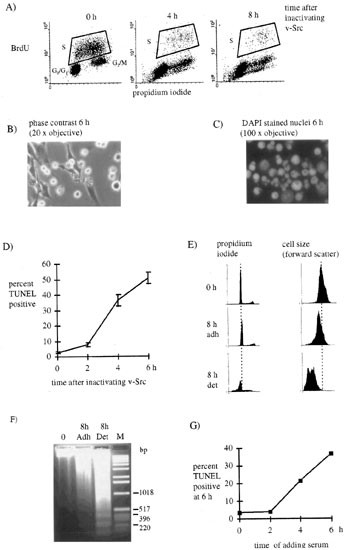
Inactivation of v-Src under low serum conditions causes cell cycle exit and apoptosis. (A) At intervals after inactivation of v-Src cells were allowed to incorporate BrdU for 1 h. They were then fixed and stained with FITC labelled antibodies against BrdU and the DNA stain, propidium iodide and analyzed by flow cytometry. The dot plots show propidium iodide staining on the X-axis and FITC labelling (BrdU) on the Y-axis. Cells which incorporated BrdU and were in S-phase are indicated. (B and C) ts-LA 29 Rat-1 cells 6 h after inactivation of v-Src. (B)photographed under phase contrast and (C) fixed and stained with DAPI. (D) Cells were harvested at intervals after v-Src inactivation, fixed and labelled by the TUNEL technique. Graph shows the means and standard errors of seven experiments. (E and F) Flow cytometric and DNA analysis of v-Src transformed cells under low serum conditions (0 h) and following inactivation of v-Src for 8 h when loosely adherent (det.) and adherent cells (adh.) were harvested separately. (E) Cells were fixed, stained with propodium iodide and analyzed by flow cytometry. Left panel shows cell number against propidium iodide staining while right panel shows cell number against cell size (forward scatter). (F) DNA extracted from the cells was analyzed by 2% agarose gel electrophoresis and stained with ethidium bromide (M – markers). (G) At intervals after inactivation of v-Src we added 5% serum. Cells were harvested 6 h after v-Src inactivation, fixed and labelled by the TUNEL technique. A representative experiment is shown
When detached and still adherent cells were harvested separately 8 h after inactivating v-Src in low serum, flow cytometry showed the detached cells to have a peak DNA content less than the G0/G1 peak (Figure 1E, left) and a smaller cell size (Figure 1E, right). The still adherent cells were smaller than cells in which v-Src was active (Figure 1E) but not as small as the detached cells. DNA isolated from detached and adherent cells separated into 200 base pair ladders, again the adherent cells had an intermediate phenotype with the laddering being more apparent in the detached cell DNA (Figure 1F). Thus inactivation of v-Src in transformed cells which lack serum survival factors led to the rapid onset of changes characteristic of apoptosis; cell shrinkage, blebbing and detachment, nuclear fragmentation, TUNEL labelling and DNA laddering. These changes could be prevented by replacing serum up to 2 h after inactivating v-Src but from 4 h after v-Src inactivation, the cells were progressively committed to a programme of cell death (Figure 1G) that eventually involved the whole population. Our results indicate that v-Src protected from cell death under low serum conditions but cells became committed to death when v-Src was switched off. Thus under low serum conditions survival signals from v-Src prevent commitment to death in v-Src transformed cells.
Biochemical changes associated with apoptosis induced by switching off v-Src
Apoptosis is frequently associated with activation of specific proteases, caspases and with the activation of the stress activated kinases p38 and JNK. Using a biotinylated peptide, which binds to the large subunits of all active caspases tested,32 we detected active caspases in extracts from cells harvested 2.5 h after v-Src inactivation (Figure 2A). All members of the MAP kinase family including JNK and p38 are activated by dual tyrosine, serine/threonine phosphorylation in the catalytic domain by specific MAP kinase kinases.33 We used antibodies specific for the dual Thr183/Tyr185 form of JNK or Thr180/Tyr182 form of p38 to determine the amount of active p38 and JNK. We detected phosphorylated p38 and JNK 4 h after v-Src inactivation (Figure 2B). This was due to switching off v-Src and not to the change in temperature since no increase in p38 or JNK phosphorylation was seen in Rat-1 cells grown in low serum for 24 h at 35°C and shifted to 39.5°C (not shown). Several different isoforms of JNK have been reported around 54 and 46 kD which are produced from three JNK genes by alternative spicing.34 We found an isoform of JNK in apoptotic cells whose appearance correlated with a decrease in the amount of p54 JNK (Figure 2B), suggesting that it might be derived from the p54 isoform. We could also detect two JNK isoforms around 45 kD. Thus activation of caspases, JNK and p38 and the appearance of morphological changes characteristic of apoptosis (Figure 1) were all initiated between 2 and 4 h after inactivating v-Src.
Figure 2
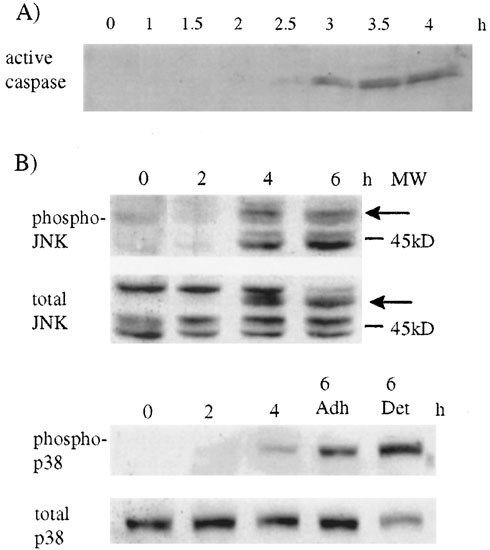
Activation of caspases, JNK and p38 during apoptosis. (A) We extracted proteins from cells harvested at intervals after v-Src inactivation and incubated with a biotinylated peptide (Z-EK(bio)D-amok) which binds to the active site of caspases. The proteins were separated by SDS–PAGE and immunoblotted with streptavidin linked to horseradish peroxidase. Active caspases were detected by ECL. (B) We extracted proteins from cells harvested at intervals after v-Src inactivation and immunoblotted using antibodies against Thr183/Tyr185 JNK or Thr180/Tyr182 p38 (upper panels) or total JNK or p38 (lower panels). Arrows indicate an isoform of JNK which appeared at 4 h
Activation of JNK and p38 can be triggered by caspases which, in some cell types, cleave and activate two upstream kinases, MEKK1 and PAK-2 (p21-activated kinase-2).35,36 Since caspases JNK and p38 were activated simultaneously (Figure 2A and B), we investigated whether caspase activity was required for the activation of JNK or p38. We added a general caspase inhibitor Z-VAD-FMK27,37 1 h before switching off v-Src and harvested the adherent and detached cells separately after 6 h. Treatment with Z-VAD-FMK prevented the cleavage of two known caspase substrates FAK (data not shown) and p27 (Figure 3A),38 indicating that caspase activity was inhibited. We found that Z-VAD-FMK slightly reduced the amount of active JNK compared to total JNK in detached cells but increased the amount of active JNK in the adherent cells (Figure 3B). More markedly, Z-VAD-FMK reduced active p38 in adherent cells and almost abolished it in detached cells (Figure 3C). This suggests that during apoptosis initiated by switching off v-Src under low serum conditions, the activation of p38 was largely dependent on the activation of caspases, while activation of JNK was not.
Figure 3
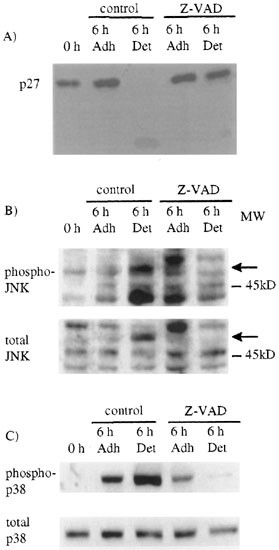
The effect of the caspase inhibitor Z-VAD-FMK on (A) p27 (B) JNK activation (C) p38 activation. (A) Proteins extracted from control or Z-VAD-FMK treated cells at 0 h and adherent and detached cells 6 h after v-Src inactivation were analyzed by immunoblotting antibodies against p27. (B and C) Proteins extracted from control or Z-VAD-FMK treated cells at 0 h and adherent and detached cells 6 h after v-Src inactivation were analyzed by immunoblotting antibodies against Thr183/Tyr185 JNK or Thr180/Tyr182 p38 (upper panels) or total JNK or p38 (lower panels) (Arrows indicate the new isoform of JNK in detached cells)
The new form of JNK (Figure 3B, arrows) was present in extracts from control detached cells but not in extracts from Z-VAD-FMK treated detached cells, which also retained the p54 isoform of JNK. The general caspase inhibitor Boc-D-FMK also inhibited the formation of the new JNK isoform, which suggests that the new isoform could have been formed by caspase cleavage of p54 JNK. The p54 isoform has a longer C-terminal tail,34 not present in p46 JNK, which could be the location of the cleavage site.
The role of caspases and p38 in apoptosis
Since we found that activation of caspases 2.5 h after switching off v-Src (Figure 2A), correlated with commitment to cell death (Figure 1G), we investigated whether inhibiting caspases would prevent cell death. We found that the caspase inhibitor Z-VAD-FMK, added at the same time as inactivating v-Src, reduced cell shrinkage in the detached cells (Figure 4A, right panels) but did not prevent cell detachment or membrane blebbing (Figure 4B), nuclear fragmentation (Figure 4C), DNA laddering (Figure 4D) or TUNEL labelling (Figure 4E). Between 4 and 6 h some of the detached control cells stopped blebbing and became rounded whereas all of the detached cells treated with Z-VAD-FMK continued blebbing (compare detached cells in Figures 1B and 4B). This suggests that caspases were not required for cell death but are required for cell shrinkage and the cessation of cell blebbing.
Figure 4
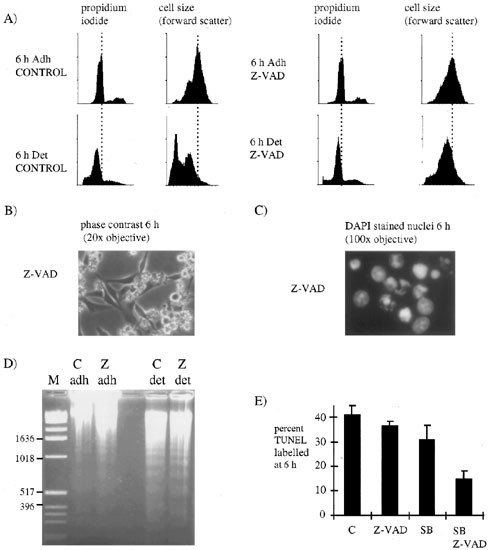
The effect of Z-VAD-FMK and SB203580 on cell death. (A) Control or Z-VAD-FMK treated adherent or detached cells were harvested 6 h after inactivating v-Src. They were fixed and stained with propodium iodide then analyzed by flow cytometry. Left panels show cell number against propidium iodide staining while right panels show cell number against cell size (forward scatter). (B) 6 h after inactivating v-Src cells treated with Z-VAD-FMK were photographed under phase contrast or (C) fixed and stained with DAPI. (D) We extracted DNA from control [C] or Z-VAD-FMK treated [Z] adherent or detached cells harvested 6 h after inactivating v-Src and analyzed it by 2% agarose gel electrophoresis and staining with ethidium bromide. (E) 6 h after inactivating v-Src cells treated with Z-VAD-FMK or SB203580 were harvested, fixed and labelled by the TUNEL technique. Results shown are the average and standard errors of four replicates from two different experiments
Previous work has shown that cell blebbing in the presence of Z-VAD-FMK requires the activity of p38.24 Cells treated with Z-VAD-FMK retained some p38 activity (Figure 3C) raising the possibility that signalling through p38 was sufficient to induce blebbing and cell death. We added a selective inhibitor of p38, SB20358028 at the same time as inactivating v-Src and investigated the effect on cell blebbing and TUNEL labelling. We found that SB203580 had no effect on the number of cells undergoing cell blebbing (data not shown) and no significant effect on the number of TUNEL positive cells (Figure 4E). However, when we added both Z-VAD-FMK and SB203580 we observed a 65% drop in the number of TUNEL positive cells (Figure 4E) and a similar fall in the number of cells undergoing cell blebbing (data not shown). Thus, in order to prevent cell death it was necessary to block both caspases and p38 suggesting that either was sufficient to induce cell death under these conditions.
v-Src mediated protection against apoptosis
Cell death following v-Src inactivation in low serum may be caused by down regulation of v-Src survival signals. Among the many down stream effects of v-Src transformation is the activation of two kinases PI3-K and ERK (MAP kinase) both of which have been shown to mediate survival signals.17,20 Therefore, we investigated the effect of switching off v-Src on the activities of PI3-K and ERK using antibodies specific for phospho Ser473 Akt and phospho p44/42 ERK. Akt phosphorylation is a good indicator of PI3-K activity since it is recruited to the membrane by the phospholipid products of PI3-K where it is activated by phosphorylation at Thr308 and Ser473.39 ERK is activated by phosphorylation on Thr202/Tyr204 by MEK1.33 We found a rapid decline in the activated Akt and ERK after switching off v-Src (Figure 5A). The upper band detected with the phospho-ERK antibody at 0 h did not appear in all experiments and its identity is not known. ERK phosphorylation returned at 4 h, simultaneous with the phosphorylation of p38 and JNK (Figure 2C) and the onset of apoptosis, whereas Akt protein was down regulated from 4 h consistent with cleavage by caspases as reported by Widmann et al.40. The decline in ERK and Akt phosphorylation was due to switching off v-Src and not to the increase in temperature from 35 to 39.5°C as this was not seen in extracts from Rat-1 cells grown in low serum for 24 h at 35°C then shifted to 39.5°C (not shown). Since both the ERK and PI3-K were rapidly inactivated when v-Src was switched off, this showed that both were controlled by v-Src activity in ts LA 29 Rat-1 cells and are candidate mediators of v-Src induced survival signals.
Figure 5
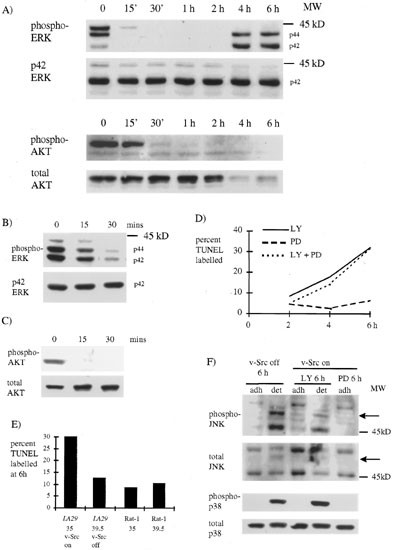
The role of ERK and PI3-K pathways in cell death. (A) We extracted proteins from the cells at intervals after v-Src inactivation and analzyed them by immunoblotting using antibodies against phospho Thr202/Tyr204 p42/44 ERK or phospho-Ser473 Akt (upper panels) or p42 ERK or total Akt (lower panels). (B and C) v-Src transformed cells were incubated in low serum at the permissive temperature for 24 h. (B) Cells were harvested at intervals after the addition of PD980959 and proteins extracted from the cells were analyzed by immunoblotting using antibodies against phospho Thr202/Tyr204 p42/44 ERK (upper) or p42 ERK (lower). (C) Cells were harvested at intervals after the addition of LY294002 and proteins extracted from the cells were analyzed by immunoblotting using antibodies against phospho-Ser473 Akt (upper) or total Akt (lower). (D) v-Src transformed cells incubated in low serum for 24 h were harvested at intervals after the addition of PD980959 (dashed line), LY294002 (unbroken line), both inhibitors (dotted line). Cells were fixed and labelled using the TUNEL technique. The graph shows a representative experiment. (E) Rat-1 cells or ts LA 29 Rat-1 cells were grown in normal growth medium at the restrictive temperature for the v-Src oncoprotein (39.5°C) for several days or kept at the permissive temperature (35°C). Cells were then transferred to low serum for 24 h, treated with LY294002 for a further 6 h, harvested, fixed and labelled by the TUNEL technique. (F) v-Src transformed cells incubated in low serum for 24 h were harvested after switching off v-Src for 6 h or after treatment with PD98059 or LY294002 for 6 h when v-Src was on. Proteins extracted from the adherent or detached cells were analyzed by immunoblotting using antibodies against Thr183/Tyr185 JNK or Thr180/Tyr182 p38 (upper panels) or total JNK or p38 (lower panels). Arrows show the new isoform of JNK seen in detached cells
To test whether either pathway relayed v-Src induced survival signals we used PD98059, a specific inhibitor of MEK1,30 to inactivate ERK or LY294002, a specific inhibitor of PI3-K.29 We cultured v-Src transformed cells for 24 h in low serum conditions then added LY294002 or PD98059 and assessed the effect on phosphorylation of Akt or ERK by immunoblotting and on cell survival by TUNEL. We found that PD98059 reduced ERK phosphorylation (Figure 5B) and LY294002 reduced Akt phosphorylation (Figure 5C). However, whereas LY294002 induced apoptosis, PD98059 did not induce apoptosis, nor did it increase the amount of apoptosis induced by LY294002 when the two inhibitors were added together (Figure 5D). Importantly, we found that the induction of apoptosis by LY294002 in low serum conditions were specific to v-Src transformed cells. ts LA 29 Rat-1 cells are morphologically normal when grown at the restrictive temperature for v-Src (39.5°C). We grew ts LA 29 Rat-1 cells in normal growth medium at 39.5°C for several days or normal Rat-1 cells at either 35 or 39.5°C. After incubation in low serum for 24 h we added LY294002 and found no increase in TUNEL labelling after 6 h (Figure 5E). This suggests that the requirement for survival signals mediated by PI3-K activity is a characteristic of v-Src transformed cells incubated under low serum conditions.
If PI3-K is responsible for mediating v-Src survival signals we would expect that death induced by inhibiting PI3-K would be mediated by the same pathway as death induced by switching off v-Src. To investigate this we harvested adherent and detached cells treated with LY294002 to examine the appearance of the new isoform of JNK, which we showed was dependent on caspase activity (Figure 3A) and the activation of p38 and JNK. Cells treated with PD98059 (most of which remained attached) served as a control. Treatment of serum deprived v-Src transformed cells with LY294002 induced p38 and JNK phosphorylation and the new isoform of JNK in the detached cells, whereas PD98059 had no effect (Figure 5F). In contrast to results shown in Figures 2B and 3B, we did not see any active p38 in the adherent cells in Figure 5F, suggesting that cells with active p38 were weakly attached and could be harvested with detached cells. These results indicate that apoptosis induced by inhibiting PI3-K under low serum conditions is accompanied by similar biochemical events to that induced by switching off v-Src, supporting the role for PI3-K in mediating v-Src survival signals.
Cell death is inhibited by the anti-apoptotic protein Bcl-2
Over expression of Bcl-2 or Bcl-XL can inhibit some but not all types of apoptosis.41,42 Therefore we tested whether over expression of Bcl-2 would inhibit cell death induced by switching off v-Src. We transfected ts LA 29 Rat-1 cells with a mammalian expression vector for mouse Bcl-2 and isolated clones which over express Bcl-2 to differing extents (Figure 6A). We found that Bcl-2 inhibited cell death induced by v-Src inactivation or by LY294002 in a dose-dependent manner (TUNEL labelling shown in Figure 6B). However, Bcl-2 did not block the fall in Akt or ERK phosphorylation after v-Src was switched off (Figure 6C). These results show that Bcl-2 blocks cell death due to switching off v-Src or inhibiting PI3-K without blocking inactivation of Akt.
Figure 6
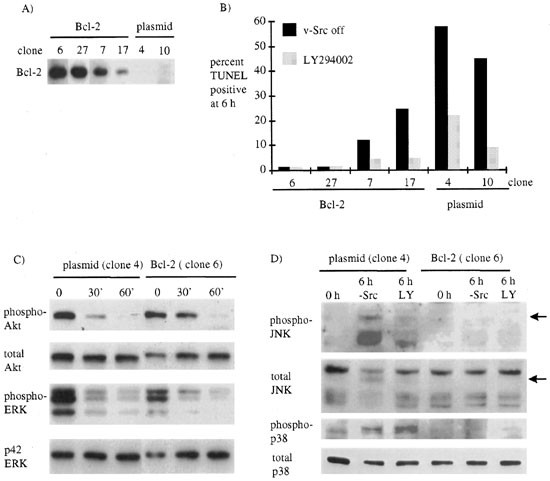
Inhibition of cell death by Bcl-2. (A) We extracted proteins from cell lines engineered to over express Bcl-2 or containing plasmid DNA and analyzed the level of Bcl-2 expression by immunoblotting with antibodies specific for Bcl-2. (B) v-Src transformed Bcl-2 expressing or control cells were incubated in low serum for 24 h. LY294002 was added or v-Src switched off and the cells were harvested 6 h later, fixed and stained using the TUNEL assay. The results shown are representative of several different experiments. (C) v-Src transformed Bcl-2 expressing and control cells were incubated in low serum for 24 h then harvested cells at intervals after switching off v-Src. Proteins were extracted and analyzed by immunoblotting using antibodies against phospho-Ser473 Akt or phospho Thr202/Tyr204 p42/44 ERK (upper panels) or Akt or p42 ERK (lower panels). (D) The v-Src transformed Bcl-2 expressing and control cells were incubated in low serum for 24 h then harvested 6 h after switching off v-Src (-Src) or adding LY294002 (LY). Proteins were extracted and analyzed by immunoblotting using antibodies against Thr183/Tyr185 JNK or Thr180/Tyr182 p38 (upper panels) or total JNK and p38 (lower panels). Arrows indicate the new isoform of JNK
The Bcl-2 family of anti-apoptotic proteins inhibit apoptosis by preventing activation of caspases, JNK and p38.26,41,43,44,45 Our results suggest that Bcl-2 blocked cell death downstream of Akt, thus to define more precisely where Bcl-2 acted we determined if it could inhibit phosphorylation of JNK and p38 or the caspase dependent mobility shift in JNK. JNK and p38 did not become phosphorylated in clones expressing high levels of Bcl-2 whereas they were phosphorylated in clones containing the plasmid alone (Figure 6D). Similarly, the new JNK isoform (indicted by arrows) was not seen in clones with high Bcl-2 levels (Figure 6D). These results suggest that Bcl-2 blocks cell death upstream of the activation of caspases, JNK and p38 and downstream of PI3-K and Akt.
Discussion
The apoptotic activities resulting from the activation of mitogenic oncoproteins may protect normal cells from unscheduled proliferation, and this is one postulated reason for co-operation between mitogenic and anti-apoptotic oncogenes.10,11,12 Unlike other oncoproteins, the deregulated tyrosine kinase v-Src does not require a co-operating partner to induce full neoplastic transformation, however, as a potent mitogen, v-Src might also be expected to promote apoptosis. If so, then v-Src itself must induce a countervailing anti-apoptotic process if it is to succeed in inducing neoplasia on its own. We show here that v-Src does indeed induce both pro-apoptotic and anti-apoptotic effects in Rat-1 cells cultured with insufficient serum survival factors.
The potentially deleterious effect of v-Src on cell viability was implied in very early studies, which showed that, in low serum, v-Src expressing cells either multiplied or died, but could not become quiescent.3 We demonstrate here that v-Src transformed cells in low serum are primed to die by apoptosis when v-Src is switched off (Figure 1) or PI3-K is inhibited (Figure 5D). Apoptosis was specific to v-Src transformed cells in low serum, since inhibition of PI3-K in low serum did not lead to apoptosis in normal Rat-1 cells or ts LA 29 Rat-1 cells in which v-Src had been inactive for several days (Figure 5E). Since ts LA 29 Rat-1 cells display this apoptosis upon switching off v-Src in low serum, either the oncoprotein or serum must provide an overriding survival signal (Figure 1G). The speed and magnitude of the apoptotic response in this conditional system may make it useful for studying details of apoptotic commitment and execution. We, however, have concentrated on the dual roles of v-Src in this phenomenon and their consequences for Src biology.
Possible mechanisms of priming apoptosis by v-Src under low serum conditions
A proapoptotic effect of v-Src in low serum has not been previously described and, although we do not know how it is mediated, the properties of v-Src offer several interrelated possibilities.
Firstly, v-Src upregulates c-Myc, which has been shown both to promote cell proliferation and to prime Rat-1 cells for apoptosis under low serum conditions (comprehensively reviewed in46). Secondly, there is evidence that activated Ras promotes apoptosis through Raf signalling9 and v-Src also activates this pathway.18 Thirdly, v-Src disrupts cell cycle controls4,18,21 and the ability to induce apoptosis under low serum conditions, common to mitotic oncoproteins such as E2F-1,6,47 v-Jun8 and c-Myc,7 could be a consequence of deregulated cell cycle progression. Variations of the dual signal model for Myc function postulate that the oncogene coordinately stimulates distinct pathways for apoptosis and proliferation.46 v-Src may also show this functional duality since addition of the MEK1 inhibitor, PD98059, throughout the 24 h incubation in low serum arrested v-Src transformed Rat-1 in G0/G1 but did not prevent apoptosis when v-Src was switched off (data not shown).
Finally, v-Src phosphorylation leads to disruption and turnover of the components of cellular adhesions and linking cytoskeletal elements,48 a process that became more pronounced during the 24 h incubation in low serum employed in our experiments (data not shown). The consequent attenuation of integrin signalling may be a signal for apoptosis, analogous to detachment induced cell death (‘anoikis’) in epithelial cells.49 Serum-deprived mesenchymal cells can survive in the absence of integrin signalling1,2,3 and ts LA29 Rat-1 cells at the restrictive temperature and normal Rat-1, which both have a normal morphology, resist apoptosis following PI3-K inhibition (Figure 5E). This implies that, in mesenchymal cells, either integrin mediated or other signals are sufficient to induce survival. These signals can substitute for one another since we showed previously that surrogate integrin signalling can protect serum-deprived fibroblasts from Myc-induced apoptosis.50 In serum-deprived ts LA29 Rat-1 cells at the permissive temperature inactivation of v-Src or inhibition of PI3-K may remove another survival signal from cells whose integrin survival signalling has been disrupted by morphological transformation, resulting in the onset of cell death.
v-Src induced survival signals
We showed that the v-Src survival signal is mediated, at least in part, by PI3-K, since the PI3-K inhibitor, LY294002, induced significant apoptosis in v-Src transformed cells grown under low serum conditions (Figure 5D). v-Src can bind and activate PI3-K19 and, when we switched off v-Src, we observed inactivation of PI3-K, as evidenced by reduced Akt phosphorylation (Figure 5A). Furthermore, LY294002 inhibited Akt phosphorylation, showing that this inhibitor did, indeed, act on the PI3-K pathway (Figure 5C). Taken together, these results show that v-Src survival signals mediated by PI3-K protect cells from v-Src-primed apoptosis. Previous reports showed that v-Src survival signals mediated by PI3-K protected epithelial cells from anoikis51 and Rat-1 cells from UV induced apoptosis,52 suggesting that this is a general mechanism whereby v-Src can provide a survival signal to protect against different apoptotic stimuli.
The effectors of v-Src primed apoptosis
Survival signals mediated by Akt have been shown to impinge on the apoptotic machinery in a number of ways including phosphorylation of Bad (Bcl-XL/Bcl-2 associated death factor)53 and pro-caspase 9.54 Phosphorylation prevents pro-caspase 9 activation while phosphorylated Bad is held within the cytosol in a complex with 14-3-3 protein.53,55 When Akt is inactivated Bad is dephosphorylated and moves to the mitochondrial membranes where it may titrate out the anti-apoptotic Bcl-2 family members. This may allow pro-apoptotic Bcl-2 family members to homodimerize and interact with pores in the mitochondrial membranes, resulting in release AIF56 and cytochrome c26 which in turn activates caspase 9. Thus dephosphorylation of Bad and/or caspase 9 provides a possible mechanism of both caspase activation and its inhibition by Bcl-2 when v-Src or PI3-K was switched off. Consistent with this we have found that ts LA29 Rat-1 cells express Bad protein (D Johnson and C Dive unpublished).
Additional mechanisms are required to explain why apoptosis induced by switching off v-Src or inhibiting PI3-K was simultaneous with the activation of both caspases and stress activated kinases, JNK and p38 (Figure 2). Either caspases or p38 were sufficient to induce cell death, although not necessarily all the stigmata of apoptosis, but inhibiting both enhanced cell survival (Figure 4). In addition the activation of p38 was partly caspase dependent (Figure 3B) which may be due to the cleavage and activation of an upstream kinase analogous to cleavage of PAK 236 or MEKK1.35 Therefore switching off v-Src or inhibiting PI3-K engaged at least two independent pathways. These are similar to those reported downstream of Fas45,47 since ligation of Fas or other death receptors activated the p38 and JNK pathways58,59,60,61 and independently stimulated the activation of caspase 8.57 Ras induced PI3-K activity down regulated Fas expression.62 If v-Src induced PI3-K activity has a similar effect, then switching off v-Src or inhibiting PI3-K would increase Fas expression, which may activate the Fas pathway. Bcl-2 can inhibit Fas induced cell death in some cells and can inhibit activation of JNK45 and caspase 841 at the cell membrane. We found that Bcl-2 inhibited the activation of JNK, p38 and caspases when v-Src or PI3-K were switched off in low serum. If inhibiting PI3-K in ts LA 29 Rat-1 cells in low serum induced Fas expression then Bcl-2 may prevent cell death by blocking JNK, p38 and caspase activation downstream of Fas.
Conclusions
v-Src can both prime for apoptosis and protect against it. Apoptosis priming is not unique to ts LA29 Rat-1, since we observed it also in v-Src transformed chick embryo fibroblasts but not in transformed NIH3T3 cells (data not shown). Pro- and anti-apoptotic functions are also shown by activated Ras9 v-Src and Ras share, at least in part, the same anti-apoptotic pathway. However, Ras, unlike v-Src, requires cooperating oncogenes for primary cell transformation, in which its pro-apoptotic function prevails over the anti-apoptotic effect. With v-Src, the priming for apoptosis is only apparent when serum survival factors are limiting, conditions under which v-Src transformed cells are finely balanced between proliferation and death. These phenomena may, nonetheless, have implications for a role for Src in neoplasia. Many tumours, particularly those of the colon, have high levels of c-Src expression63,64 and a few metastatic colon tumours have mutated c-Src which could be important in tumour progression.65 We showed previously how v-Src induces transformation via effects on cell growth and behaviour.4,18,21,66 Here we suggest that the ability to induce cell survival could be an equally important transforming activity of v-Src and could be another mechanism whereby deregulated c-Src contributes to tumour formation in vivo. Manipulating pathways involved in the alternative death priming effects of v-Src could have therapeutic potential, therefore it would be interesting to characterise these pathways in v-Src transformed Rat-1 cells.
Materials and Methods
Cell culture and induction of cell death
Rat-1 cells and ts LA 29 Rat-1 cells were maintained as in Johnson et al.4 BrdU (bromodeoxyuridine) incorporation and labelling was as in Johnson et al.4 For the induction of cell death, cells were seeded at 1.5×106 per 90 mm plate, the following day the medium was replaced with medium containing 0.2% serum. After 24 h they were either shifted to restrictive temperature (39.5°C) to inactivate v-Src or various inhibitors were added.
To harvest adherent and detached cells we seeded 6×106 cells on a 150 mm plate. The following day the medium was replaced with medium containing 0.2% serum. After 24 h they were shifted to 39.5°C for 6–8 h. The detached cells were harvested by washing carefully and the adherent cells were either harvested by trypsinization or lysed on the plate.
BrdU immunostaining, propidium iodide staining and flow cytometry
As in Johnson et al.4
Generation of cell lines which over express Bcl-2
pSFFV-Bcl-2 expression plasmid was from Stanley J Korsmeyer, Washington University, St Louis, Missouri, USA. ts LA 29 Rat-1 cells were transfected with pSFFV-Bcl-2 or pSFFV using DOTAP (Boehringer Mannheim). Single G418 resistant colonies were isolated, grown up and tested for expression of Bcl-2 by immunoblotting. All colonies isolated from cells transfected with pSFFV-Bcl-2 expressed Bcl-2 but we could not detect Bcl-2 in colonies from cells transfected with pSFFV.
Western blot analysis
As in Johnson et al.4 except phospho-specific antibodies were used as recommended by the manufacturers.
Antibodies
Phospho-specific antibodies to Akt, JNK and p38 were from New England Biolabs (#9271S, #9251L, #9211S), phospho-ERK was from Promega (#V8031). Total JNK antibody, raised against full-length recombinant JNK was a gift from Billy Clark, Beatson Institute, Glasgow and the p42 ERK antibody, raised against a peptide from the C-terminus of p42 ERK (EETARFQPGYRS),67 was a gift from Anne Wyke, Beatson Institute, Glasgow. Bcl-2 antibody was from Pharmingen (#15021A), p27 antibody was kind gift of Steve Coats, Amgen, CA, USA.
Inhibitors
Inhibitors were purchased from CalBiochem. Stocks were made in DMSO (dimethylsulphoxide) as follows: Z-VAD-FMK 10 mM stock, used at 100 μM, SB203580 66 mM stock, used at 50 μM, LY294002 80 mM stock, used at 50 μM, PD98059 10 mM stock used at 50 μM.
DAPI (4′,6-Diamidino-2-pheylindole) staining
Cells were grown in chamber slides, 1×104 cells per chamber in 0.4 ml culture medium. They were washed twice in PBS (phosphate buffered saline) then fixed in 3.7% formaldehyde in PBS for 10 min. They were washed in PBS and permeabilised in 0.5% Triton-X-100, 1% BSA in PBS for 30 min. They were washed once in PBS then mounted in Vectashield (Vector Labs) containing 0.1 μg/ml DAPI.
DNA laddering
Cells were harvested by trypsinization, washed in PBS then suspended in TNE (10 mM Tris-Cl pH 8.2, 400 mM NaCl, 2 mM EDTA (ethylenediaminetetraacetic acid)) at a concentration of 5×106 per ml. Cells were lysed by addition of 0.5% SDS and proteins digested by adding 0.25 mg/ml proteinase K and incubating at 37°C overnight. The lysates were extracted with water saturated phenol then chloroform and precipitated with 1.5 volumes of 100% ethanol and spun for 5 min in a microfuge. After a 70% ethanol wash the pellet was dissolved in 100 μl 10 mM Tris pH 8.0, 1 mM EDTA and 0.1 mg/ml DNase free RNase A was added. Tubes were incubated at 37°C for 30 min then 5–10 μg was run on a 2% agarose gel.
TUNEL31
Cells were harvested and washed once in PBS. They were fixed for 15 min on ice in 1% formaldehyde in PBS then centrifuged at 1000 r.p.m. for 5 min. They were resuspended in 100 μl PBS and 1 ml ice cold 70% ethanol was added. They were stored overnight at 4°C and the next day they were rehydrated in PBS on ice for 30 min. Aliquots of 106 cells were spun and resuspended in 0.2 M potassium cacodylate, 2.5 mM Tris-HCl pH 6.6, 2.5 mM CoCl2, 0.25 mg/ml, BSA, 5 units of TdT (terminal deoxynucleotidyl transferase) and 0.5 nmoles of biotinylated dUTP per tube. They were incubated at 37°C for 30 min, washed in 1 ml of PBS then spun and resuspended in 100 μl 4×SSC (1×SSC=0.15 M NaCl, 0.015 M NaCitrate), 0.1% Triton X-100, 5% low fat dried milk and 5 μg/ml avidin-FITC (fluorescein isothiocyanate). They were incubated for 30 min at room temperature in the dark then washed by addition of 1 ml PBS containing 0.1% Triton X-100, spun and resuspended in 500 μl of PBs containing 10 μg/ml propidium iodide. They were analyzed by flow cytometry and the percentage TUNEL positive cells determined by FITC fluorescence.
Detection of active caspases32
Cells were lysed by freezing on dry ice and thawing at 37°C three times then incubated with Z-EK(bio)D-amok (N-(Nα-benzyloxycarbonylglutamyl-Nε-biotinyllysyl)aspartic acid[(2,6-dimethylbenzoyl)oxy]methyl ketone) (1 μM added from a 25 μM stock in DMSO) at 37°C for 15 min. They were diluted with 2×SDS sample buffer, boiled for 3 min and sonicated briefly. They were subject to SDS–PAGE (polyacrylamide gel electrophoresis) on 16% gels, transferred to nitrocellulose, probed with peroxidase-labeled streptavidin and visualised using ECL (enhanced chemiluminescence).
Abbreviations
BAD:
Bcl-XL/Bcl-2 associated death factor
BrdU:
5-Bromo-2′-deoxyuridine
DAPI:
(4′,6-Diamidino-2-pheylindole)
DMSO:
dimethylsulphoxide
DMEM:
Dulbecco's modified Eagle's medium
ECL:
enhanced chemiluminescence
EDTA:
ethylenediaminetetraacetic acid
ERK:
extracellular signal-regulated kinase
FITC:
fluorescein isothiocyanate
FMK:
fluoromethylketone
GSK-3:
glycogen synthase kinase-3
JNK:
Jun N-terminal kinase
MAP:
mitogen-activated protein
MEK:
MAP kinase kinase/ERK kinase
MEKK:
MEK kinase
PAGE:
polyacrylamide gel electrophoresis
PAK:
p21-activated kinase
PBS:
phosphate buffered saline
PI3-K:
phosphatidylinositol 3-kinase
RSV:
Rous sarcoma virus
TdT:
terminal deoxynucleotidyl transferase
ts :
temperature sensitive
TUNEL:
TdT-mediated dUTP nick end labelling
Z-:
benzyloxycarbonyl
Z-EK(bio)D-aomk:
N-(Nα-benzyloxycarbonylglutamyl-Nε-biotinyllysyl)aspartic acid[(2,6-dimethylbenzoyl)oxy]methyl ketone
References
- Stoker M, O'Neill C, Berryman S and Waxman V . (1968) Anchorage and growth regulation in normal and virus-transformed cells. Int. J. Cancer 3: 683–693
Article CAS Google Scholar - Wyke JA . (1973) The selective isolation of temperature-sensitive mutants of Rous sarcoma virus. Virology 52: 587–590
Article CAS Google Scholar - Wyke J . (1971) A method of isolating cells incapable of multiplication in suspension culture. Exp. Cell Res. 66: 203–208
Article CAS Google Scholar - Johnson D, Frame MC and Wyke JA . (1998) Expression of the v-Src oncoprotein in fibroblasts disrupts normal regulation of the CDK inhibitor p27 and inhibits quiescence. Oncogene 16: 2017–2028
Article CAS Google Scholar - Hueber AO and Evan GI . (1998) Traps to catch unwary oncogenes. Trends Genet. 14: 364–367
Article CAS Google Scholar - Shan B and Lee WH . (1994) Deregulated expression of E2F-1 induces S-phase entry and leads to apoptosis. Mol. Cell Biol. 14: 8166–8173.
Article CAS Google Scholar - Evan GI, Wyllie AH, Gilbert CS, Littlewood TD, Land H, Brooks M, Waters CM, Penn LZ and Hancock DC . (1992) Induction of apoptosis in fibroblasts by c-myc protein. Cell 69: 119–128
Article CAS Google Scholar - Clark W and Gillespie DA . (1997) Transformation by v-Jun prevents cell cycle exit and promotes apoptosis in the absence of serum growth factors. Cell Growth Differ. 8: 371–380
CAS PubMed Google Scholar - Kauffman-Zeh A, Rodriguez-Viciana P, Ulrich E, Gilbert C, Coffer P, Downward J and Evan G . (1997) Suppression of c-Myc-induced apoptosis by Ras signalling through PI(3)K and PKB. Nature 385: 544–548
Article Google Scholar - Fanidi A, Harrington EA and Evan GI . (1992) Cooperative interaction between c-Myc and Bcl-2 proto-oncogenes. Nature 359: 554–556
Article CAS Google Scholar - Bissonnette RP, Echeverri F, Mahboubi A and Green DR . (1992) Apoptotic cell death induced by c-Myc is inhibited by Bcl-2. Nature 359: 552–524
Article CAS Google Scholar - Strasser A, Harris AW, Bath ML and Cory S . (1990) Novel primitive lymphoid tumours induced in transgenic mice by cooperation between Myc and Bcl-2. Nature 348: 331–333
Article CAS Google Scholar - Rohn JL, Hueber AO, McCarthy NJ, Lyon D, Navarro P, Burgering BM and Evan GI . (1998) The opposing roles of the Akt and c-Myc signalling pathways in survival from CD95-mediated apoptosis. Oncogene 17: 2811–2818
Article CAS Google Scholar - Palmero I, Pantoja C and Serrano M . (1998) p19ARF links the tumour suppressor p53 to Ras. Nature 395: 125–126
Article CAS Google Scholar - Sovova V, Friis R, Fidlerova H and Hlozanek I . (1993) c-Myc gene activation as a permanent trait of RSV-infected quail cells. Int. J. Cancer 53: 983–987
Article CAS Google Scholar - Aziz N, Cherwinski H and McMahon M . (1999) Complementation of defective colony-stimulating factor 1 receptor signaling and mitogenesis by Raf and v-Src. Mol. Cell. Biol. 19: 1101–1115
Article CAS Google Scholar - Yeh JH, Hsu SC, Han SH and Lai MZ . (1998) Mitogen-activated protein kinase kinase antagonized fas-associated death domain protein-mediated apoptosis by induced FLICE-inhibitory protein expression. J. Exp. Med. 188: 1795–1802
Article CAS Google Scholar - Wyke AW, Frame MC, Gillespie DA, Chudleigh A and Wyke JA . (1995) Mitogenesis by v-Src: fluctuations throughout G1 of classical immediate early AP-1 and mitogen-activated protein kinase responses that parallel the need for the oncoprotein. Cell Growth Differ. 6: 1225–1234
CAS PubMed Google Scholar - Cantley LC, Auger KR, Carpenter C, Duckworth B, Graziani A, Kapeller R and Soltoff S . (1991) Oncogenes and signal transduction. Cell 64: 281–302
Article CAS Google Scholar - Downward J . (1998) Mechanisms and consequences of activation of protein kinase B/Akt. Curr. Opin. Cell Biol. 10: 262–267
Article CAS Google Scholar - Wyke AW, Cushley W and Wyke JA . (1993) Mitogenesis by v-Src: a need for active oncoprotein both in leaving G0 and in completing G1 phases of the cell cycle. Cell Growth Differ. 4: 671–678
CAS PubMed Google Scholar - Villa P, Kaufmann SH and Earnshaw WC . (1997) Caspases and caspase inhibitors. Trends Biochem. Sci. 22: 388–393
Article CAS Google Scholar - Xia Z, Dickens M, Raingeaud J, Davis RJ and Greenberg ME . (1995) Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science 270: 1326–1331
Article CAS Google Scholar - Huot J, Houle F, Rousseau S, Deschesnes RG, Shah GM and Landry J . (1998) SAPK2/p38-dependent F-actin reorganization regulates early membrane blebbing during stress-induced apoptosis. J. Cell Biol. 143: 1361–1373
Article CAS Google Scholar - Basu S and Kolesnick R . (1998) Stress signals for apoptosis: ceramide and c-Jun kinase. Oncogene 17: 3277–3285
Article Google Scholar - Reed JC . (1998) Bcl-2 family proteins. Oncogene 17: 3225–3236
Article Google Scholar - Slee EA, Zhu H, Chow SC, MacFarlane M, Nicholson DW and Cohen GM . (1996) Benzyloxycarbonyl-Val-Ala-Asp (OMe) fluoromethylketone (Z-VAD.FMK) inhibits apoptosis by blocking the processing of CPP32. Biochem. J. 315: 21–24
Article CAS Google Scholar - Lee JC, Laydon JT, McDonnell PC, Gallagher TF, Kumar S, Green D, McNulty D, Blumenthal MJ, Heys JR, Landvatter SW, et al. (1994) A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature 372: 739–746
Article CAS Google Scholar - Vlahos CJ, Matter WF, Hui KY and Brown RF . (1994) A specific inhibitor of phosphatidylinositol 3-kinase,2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002). J. Biol. Chem. 269: 5241–5248
CAS PubMed Google Scholar - Alessi DR, Cuenda A, Cohen P, Dudley DT and Saltiel AR . (1995) PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J. Biol. Chem. 270: 27489–27494
Article CAS Google Scholar - Gorczyca W, Gong J and Darzynkiewicz Z . (1993) Detection of DNA strand breaks in individual apoptotic cells by the in situ terminal deoxynucleotidyl transferase and nick translation assays. Cancer Res. 53: 1945–1951
CAS PubMed Google Scholar - Martins LM, Mesner PW, Kottke TJ, Basi GS, Sinha S, Tung JS, Svingen PA, Madden BJ, Takahashi A, McCormick DJ, Earnshaw WC and Kaufmann SH . (1997) Comparison of caspase activation and subcellular localization in HL-60 and K562 cells undergoing etoposide-induced apoptosis. Blood 90: 4283–4296
CAS Google Scholar - Kyriakis JM and Avruch J . (1996) Protein kinase cascades activated by stress and inflammatory cytokines. Bioessays 18: 567–577
Article CAS Google Scholar - Gupta S, Barrett T, Whitmarsh AJ, Cavanagh J, Sluss HK, Derijard B and Davis RJ . (1996) Selective interaction of JNK protein kinase isoforms with transcription factors. EMBO J. 15: 2760–2770
Article CAS Google Scholar - Cardone MH, Salvesen GS, Widmann C, Johnson G and Frisch SM . (1997) The regulation of anoikis: MEKK-1 activation requires cleavage by caspases. Cell 90: 315–323
Article CAS Google Scholar - Rudel T and Bokoch GM . (1997) Membrane and morphological changes in apoptotic cells regulated by caspase-mediated activation of PAK2. Science 276: 1571–1574
Article CAS Google Scholar - Garcia-Calvo M, Peterson EP, Leiting B, Ruel R, Nicholson DW and Thornberry NA . (1998) Inhibition of human caspases by peptide-based and macromolecular inhibitors. J. Biol. Chem. 273: 32608–32613
Article CAS Google Scholar - Levkau B, Koyama H, Raines EW, Clurman BE, Herren B, Orth K, Roberts JM and Ross R . (1998) Cleavage of p21Cip1/Waf1 and p27Kip1 mediates apoptosis in endothelial cells through activation of Cdk2: role of a caspase cascade. Mol. Cell. 1: 553–563
Article CAS Google Scholar - Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P and Hemmings BA . (1996) Mechanism of activation of protein kinaseB by insulin and IGF-1. EMBO J. 15: 6541–6551
Article CAS Google Scholar - Widmann C, Gibson S and Johnson GL . (1998) Caspase-dependent cleavage of signaling proteins during apoptosis. A turn-off machine for anti-apoptotic signals. J. Biol. Chem. 273: 7141–7147
Article CAS Google Scholar - Kawahara A, Kobayashi T and Nagata S . (1998) Inhibition of Fas-induced apoptosis by Bcl-2. Oncogene 17: 2549–2554
Article CAS Google Scholar - Scaffidi C, Fulda S, Srinivasan A, Friesen C, Li F, Tomaselli KJ, Debatin KM, Krammer PH and Peter ME . (1998) Two CD95 (APO-1/Fas) signaling pathways. EMBO J. 17: 1675–1687
Article CAS Google Scholar - Zamzami N, Brenner C, Marzo I, Susin SA and Kroemer G . (1998) Subcellular and submitochondrial mode of action of Bcl-2-like oncoproteins. Oncogene 16: 2265–2282
Article CAS Google Scholar - Frisch SM, Vuori K, Kelaita D and Sicks S . (1996) A role for Jun-N-terminal kinase in anoikis; suppression by Bcl-2 and crmA. J. Cell Biol. 135: 1377–1382
Article CAS Google Scholar - Yang X, Khosravi-Far R, Chang HY and Baltimore D . (1997) Daxx, a novel Fas-binding protein that activates JNK and apoptosis. Cell 89: 1067–1076
Article CAS Google Scholar - Prendergast GC . (1999) Mechanisms of apoptosis by c-Myc. Oncogene 18: 2967–2987
Article CAS Google Scholar - Qin XQ, Livingston DM, Kaelin Jr WG and Adams PD . (1994) Deregulated transcription factor E2F-1 expression leads to S-phase entry and p53-mediated apoptosis. Proc. Natl. Acad. Sci. USA 91: 10918–10922
Article CAS Google Scholar - Fincham VJ, Wyke JA and Frame MC . (1995) v-Src-induced degradation of focal adhesion kinase during morphological transformation of chicken embryo fibroblasts. Oncogene 10: 2247–2252
CAS PubMed Google Scholar - Frisch SM and Francis H . (1994) Disruption of epithelial cell-matrix interactions induces apoptosis. J. Cell Biol. 124: 619–626
Article CAS Google Scholar - Crouch DH, Fincham VJ and Frame MC . (1996) Targeted proteolysis of the focal adhesion kinase pp125 FAK during c-MYC-induced apoptosis is suppressed by integrin signalling. Oncogene 12: 2689–2696
CAS PubMed Google Scholar - Khwaja A, Rodriguez-Viciana P, Wennstrom S, Warne PH and Downward J . (1997) Matrix adhesion and Ras transformation both activate a phosphoinositide 3-OH kinase and protein kinaseB/Akt cellular survival pathway. EMBO J. 16: 2783–2793
Article CAS Google Scholar - Kulik G, Klippel A and Weber MJ . (1997) Antiapoptotic signalling by the insulin-like growth factorI receptor, phosphatidylinositol 3-kinase, and Akt. Mol. Cell. Biol. 17: 1595–1606
Article CAS Google Scholar - Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y and Greenberg ME . (1997) Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 91: 231–241
Article CAS Google Scholar - Cardone MH, Roy N, Stennicke HR, Salvesen GS, Franke TF, Stanbridge E, Frisch S and Reed JC . (1998) Regulation of cell death protease caspase-9 by phosphorylation. Science 282: 1318–1321
Article CAS Google Scholar - Zha J, Harada H, Yang E, Jockel J and Korsmeyer SJ . (1996) Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-X(L). Cell 87: 619–628
Article CAS Google Scholar - Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, Mangion J, Jacotot E, Costantini P, Loeffler M, Larochette N, Goodlett DR, Aebersold R, Siderovski DP, Penninger JM and Kroemer G . (1999) Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 397: 441–446
Article CAS Google Scholar - Muzio M, Chinnaiyan AM, Kischkel FC, O'Rourke K, Shevchenko A, Ni J, Scaffidi C, Bretz JD, Zhang M, Gentz R, Mann M, Krammer PH, Peter ME and Dixit VM . (1996) FLICE, a novel FADD-homologous ICE/CED-3-like protease, is recruited to the CD95 (Fas/APO-1) death-inducing signaling complex. Cell 85: 817–827
Article CAS Google Scholar - Nishitoh H, Saitoh M, Mochida Y, Takeda K, Nakano H, Rothe M, Miyazono K and Ichijo H . (1998) ASK1 is essential for JNK/SAPK activation by TRAF2. Mol. Cell 2: 389–395
Article CAS Google Scholar - Chang HY, Nishitoh H, Yang X, Ichijo H and Baltimore D . (1998) Activation of apoptosis signal-regulating kinase1 (ASK1) by the adapter protein Daxx. Science 281: 1860–1863
Article CAS Google Scholar - Yuasa T, Ohno S, Kehrl JH and Kyriakis JM . (1998) Tumor necrosis factor signaling to stress-activated protein kinase (SAPK)/Jun NH2-terminal kinase (JNK) and p38. Germinal center kinase couples TRAF2 to mitogen-activated protein kinase/ERK kinase kinase1 and SAPK while receptor interacting protein associates with a mitogen-activated protein kinase kinase kinase upstream of MKK6 and p38. J. Biol. Chem. 273: 22681–22692
Article CAS Google Scholar - Muhlenbeck F, Haas E, Schwenzer R, Schubert G, Grell M, Smith C, Scheurich P and Wajant H . (1998) TRAIL/Apo2L activates c-Jun NH2-terminal kinase (JNK) via caspase-dependent and caspase-independent pathways. J. Biol. Chem. 273: 33091–33098
Article CAS Google Scholar - Peli J, Schroter M, Rudaz C, Hahne M, Meyer C, Reichmann E and Tschopp J . (1999) Oncogenic Ras inhibits Fas ligand-mediated apoptosis by downregulating the expression of Fas. EMBO J. 18: 1824–1831
Article CAS Google Scholar - Cartwright CA, Meisler AI and Eckhart W . (1990) Activation of the pp60c-src protein kinase is an early event in colonic carcinogenesis. Proc. Natl. Acad. Sci USA 87: 558–562
Article CAS Google Scholar - Cartwright CA, Kamps MP, Meisler AI, Pipas JM and Eckhart W . (1989) pp60c-Src activation in human colon carcinoma. J. Clin. Invest. 83: 2025–2033
Article CAS Google Scholar - Irby RB, Mao W, Coppola D, Kang J, Loubeau JM, Trudeau W, Karl R, Fujita DJ, Jove R and Yeatman TJ . (1999) Activating SRC mutation in a subset of advanced human colon cancers. Nat. Genet. 21: 187–190
Article CAS Google Scholar - Fincham VJ and Frame MC . (1998) The catalytic activity of Src is dispensable for translocation to focal adhesions but controls the turnover of these structures during cell motility. EMBO J. 17: 81–92
Article CAS Google Scholar - Leevers SJ and Marshall CJ . (1992) Activation of extracellular signal-regulated kinase, ERK2, by p21 ras oncoprotein. EMBO J. 11: 569–574
Article CAS Google Scholar
Acknowledgements
We are grateful to Stanley Korsmeyer, Washington University, MO, USA for the Bcl-2 expression plasmid, C Dive, Manchester University, UK for help with Bad westerns, Steve Coats, Amgen, CA for p27 antibodies and Dave Gillespie for many helpful discussions. This work was supported by The Cancer Research Campaign, project grant number SP 2281/0101, The Association for International Cancer Research project grant number 98284, The Sylvia Aitkin Trust, and Cancer Research Campaign studentship to M Agochiya.
Author information
Authors and Affiliations
- The Beatson Institute for Cancer Research, Glasgow, G61 1BD, UK
D Johnson, M Agochiya, M Frame & J Wyke - Institute of Cell and Molecular Biology, University of Edinburgh, Edinburgh, UK
K Samejima & W Earnshaw
Authors
- D Johnson
You can also search for this author inPubMed Google Scholar - M Agochiya
You can also search for this author inPubMed Google Scholar - K Samejima
You can also search for this author inPubMed Google Scholar - W Earnshaw
You can also search for this author inPubMed Google Scholar - M Frame
You can also search for this author inPubMed Google Scholar - J Wyke
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toD Johnson.
Additional information
Edited by G Evan
Rights and permissions
About this article
Cite this article
Johnson, D., Agochiya, M., Samejima, K. et al. Regulation of both apoptosis and cell survival by the v-Src oncoprotein.Cell Death Differ 7, 685–696 (2000). https://doi.org/10.1038/sj.cdd.4400700
- Received: 21 September 1999
- Revised: 29 February 2000
- Accepted: 23 March 2000
- Published: 27 July 2000
- Issue Date: 01 August 2000
- DOI: https://doi.org/10.1038/sj.cdd.4400700