Control of ceramide-induced apoptosis by IGF-1: involvement of PI-3 kinase, caspase-3 and catalase (original) (raw)
Introduction
The sphingolipid ceramide is known to be a pro-apoptotic lipid mediator, and apoptosis induced by many kinds of stresses is accompanied by an increase of intracellular ceramide.1,2 It was reported that N-hexanoylsphingosine (C6-ceramide) caused an increase of reactive oxygen species (ROS) probably by affecting electron transport in mitochondria,3 whereas N-acetylsphingosine (C2-ceramide)-induced apoptosis was suppressed by pretreatment with anti-oxidants such as N-acetylcysteine (NAC) and glutathione (GSH).4,5,6,7,8 These results suggest that ROS generation and oxidative damage are closely involved in apoptosis downstream of ceramide action. In addition, since ceramide was reported to release cytochrome c from mitochondria and activate caspase-3 along with apoptotic protease activating factor-1 (Apaf-1) and pro caspase-9, caspase-3 seems to be another candidate of downstream signaling of ceramide.9,10 Indeed, both caspase-3 activation and apoptosis induced by C2-ceramide were blocked by caspase-3 inhibitors such as DEVD-CHO and DMQD-CHO.11,12,13 Although oxidative stress and ceramide are well known to activate caspase-3, it is, at present, poorly understood how ceramide increases oxidative damage through caspase-3 activation.14,15
On the other hand, several recent studies have revealed that insulin-like growth factor 1 (IGF-1) acted against pro-apoptotic stresses such as etoposide, doxorubicin, tumor necrosis factor-α, c-myc overexpression and serum deprivation,16,17,18 and that a decrease of the IGF-1 receptor level or expression of dominant-negative IGF-1 receptor caused massive apoptosis in tumor transplantation models,19 suggesting a role of IGF-1 in apoptosis inhibition as well as cell survival. Regarding the mechanism by which IGF-1 inhibits ceramide-related apoptosis, it was shown that IGF-1 suppressed caspase-3 activation in retinal ganglion cells and cardiac muscle cells16,20 and ceramide generation probably through the inhibition of sphingomyelinase in MCF-7 cells.21,22 It was also reported that C2- or C6-ceramide inhibited Akt kinase through PI-3 kinase inhibition,23,24 which occurred in association with caveolin 1 recruitment in raft microdomains,25 suggesting that PI-3 kinase-dependent survival signal was inhibited by ceramide. However, it is still unclear whether survival signals induced by IGF-1/PI-3 kinase alter oxidative damage and caspase-3 mediated by ceramide.
Therefore, we investigated here how IGF-1/PI-3 kinase affects C2-ceramide-induced caspase-3 activation, increase of oxidative damage judged by lipid peroxidation, and apoptosis. The results showed that IGF-1 inhibits ceramide-increased apoptosis and oxidative damage by restoring the action of ROS scavenger catalase through inhibition of caspase-3 in a PI-3 kinase-dependent manner.
Results
Inhibitory action of IGF-1 on C2-ceramide-induced apoptosis and lipid peroxidation
The proportion of apoptotic cells with nuclear condensation and fragmentation, as judged by the DAPI method, increased to 60% after 24 h of 5 μM C2-ceramide treatment in serum-free medium, and pretreatment with IGF-1 inhibited the ceramide induced apoptosis in a time- and dose-dependent manner (Figure 1A,B). The proportion of apoptotic cells induced by 5 μM C2-ceramide decreased from 60 to 37% and to 22% 24 h after treatment with 100 and 1000 ng/ml IGF-1, respectively. The similar inhibition by IGF-1 of ceramide-induced apoptosis was also detected when apoptosis was examined by the extent of annexin V binding ability to the cell surface outer membrane. The mean fluorescence intensity of annexin V and the proportion of annexin V positive cells induced by 5 μM C2-ceramide decreased from 120 to 78% and from 68 to 35% by pretreatment with 100 ng/ml IGF-1, respectively (Figure 1C). In addition, increase of ceramide-induced lipid peroxidation, which was judged by detecting a decrease of fluorescence of cis-parinaric acid (CPA), was suppressed from 158 to 123% and to 110% of control level by pretreatment with 100 and 1000 ng/ml IGF-1, respectively (Figure 1D). These results suggested that IGF-1 exerted its anti-apoptotic effect against ceramide-induced apoptosis through inhibiting oxidative damage.
Figure 1
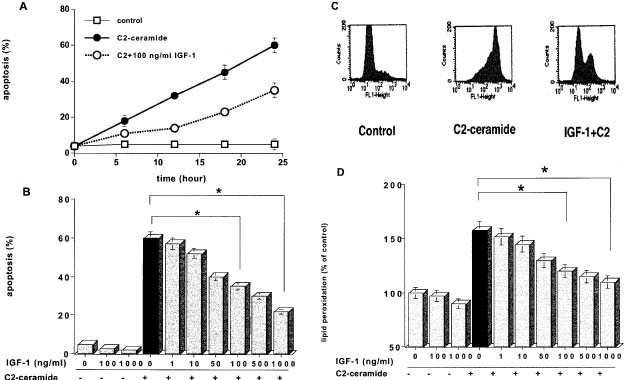
Inhibitory action of IGF-1 on ceramide-induced apoptosis and lipid peroxidation. HL-60 cells were pretreated with 100 ng/ml (A and C) or various concentrations of IGF-1 (B and D) for 30 min before treatment with 5 μM C2-ceramide in serum-free media, and harvested at various times (A) or 24 h after treatment (B, C and D). Apoptosis was judged by the DAPI staining method (A and B) or annexin V binding ability (C), and lipid peroxidation was measured by the cis-parinaric acid (CPA) fluorescence reducing method as described under ‘Materials and Methods’ (D). The results are the averages of at least three independent experiments (A, B and D) or the representative of three different experiments (C). The bars indicate 1 S.D. The significance of differences of apoptosis levels between the treatments with or without IGF-1 was determined by ANOVA test. *Indicates P<0.01 (B and D)
Effects of wortmannin on the inhibition by IGF-1 of C2-ceramide-induced apoptosis and lipid peroxidation
To investigate whether the activation of PI-3 kinase is required for mediating the inhibitory action of IGF-1, we examined the effect of a specific PI-3 kinase inhibitor, wortmannin, on the inhibition by IGF-1 of C2-ceramide-induced apoptosis and lipid peroxidation. While the per cent of apoptosis induced by 5 μM C2-ceramide decreased from 59 to 30% in the presence of 100 ng/ml IGF-1, pretreatment with 20 nM wortmannin restored the per cent of apoptosis from 30 to 64% despite the presence of IGF-1 (Figure 2A). In addition, the level of 5 μM C2-ceramide-increased lipid peroxidation was inhibited from 158 to 118% of control level in the presence of 100 ng/ml IGF-1, but restored again to 159% of control level by pretreatment with 20 nM wortmannin (Figure 2B). Thus, these results suggested that the inhibition by IGF-1 of ceramide-induced apoptosis and lipid peroxidation was dependent on PI-3 kinase activation. To confirm this notion, we next examined whether activation of PI-3 kinase by overexpressing its gene suppressed ceramide-induced apoptosis and lipid peroxidation. For this purpose, HL-60 cells were transfected with a transient expression plasmid encoding the catalytic p110 subunit of PI-3 kinase gene with a myc tag at its C-terminus, or with an expression plasmid encoding a gene point-mutated at the ATP-binding site of the p110 subunit, which expressed an enzymatically inactive PI-3 kinase protein, as the control. As shown in Figure 2C, PI-3 kinase-overexpressing (PI-3k) cells and PI-3 kinase-dead (kinase dead) cells expressed similar amount of PI-3 kinase protein, but the activity of PI-3 kinase was much higher in PI-3k cells than kinase dead cells. Ceramide-induced apoptosis and lipid peroxidation less increased in PI-3k cells than in kinase-dead cells (Figure 2D, P<0.01). Apoptosis and lipid peroxidation induced by 20 μM C2-ceramide for 24 h in 10% FCS less increased in PI-3k cells (34±3% and 136±17%, respectively) as compared to kinase-dead cells (53±4% and 172±20%, respectively). These results further confirmed that IGF-1 action against C2-ceramide-induced apoptosis and oxidative damage was mediated through PI-3 kinase.
Figure 2
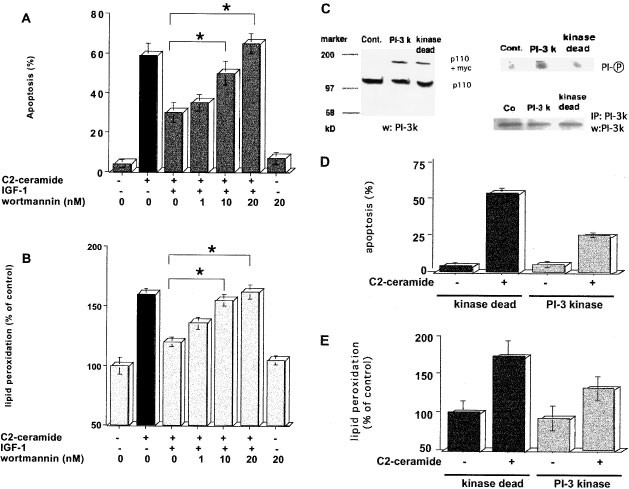
Effects of wortmannin and overexpression of PI-3 kinase on the inhibitory action of IGF-1 on ceramide-induced apoptosis and lipid peroxidation. HL-60 cells were preincubated with various concentrations of wortmannin (0, 1, 10 or 20 nM) for 10 min and 100 ng/ml IGF-1 for 30 min. Then, 5 μ C2-ceramide was added in serum-free media for 24 h. Apoptosis (A) and lipid peroxidation (B) were judged by the DAPI staining method and the reducing ability of cis-parinaric acid (CPA)-generated fluorescence, respectively, as described under Materials and Methods. HL-60 cells were transiently transfected without (control) or with plasmid containing _myc_-tagged p110 of PI-3 kinase gene (PI-3k) or _myc_-tagged p110 of PI-3 kinase-dead gene (kinase dead) by electroporation (C, left panel). After immunoprecipitation with anti-p110 of PI-3 kinase antibody PI-3 kinase activity was assessed by phosphorylating PI (C, right upper panel), and equal amount of immunoprecipitated PI-3 kinase protein was confirmed by Western blotting analysis with anti-PI-3 kinase antibody after immunoprecipitation (C, right lower panel). PI-3k and kinase-dead cells were incubated in 10% FCS-containing media for 48 h after electroporation, and then treated with 20 μM C2-ceramide for 24 h. Apoptosis (D) and lipid peroxidation (E) were judged as described in Figure 1. The results are the averages of at least three independent experiments (A, B, D and E) or the representative of three different experiments (C). The bars indicate 1 S.D. The significance of differences of apoptosis and lipid peroxidation levels between the treatments with or without wortmannin was determined by ANOVA test. *Indicates P<0.01 (A and B). Statistical significance by ANOVA test was P<0.05 for the difference of increase of apoptosis and lipid peroxidation between kinase-dead and PI-3k cells (D and E)
IGF-1 did not inhibit C2-ceramide-induced long-chain ceramide generation
When we examined the content of physiological long-chain ceramide, which was measured by a DGK assay, after treatment with C2-ceramide in HL-60 cells, we found that 5 μM C2-ceramide increased the content of intracellular long-chain ceramide in a time-dependent manner (Figure 3). As shown in Figure 1, IGF-1 at a concentration of 100 ng/ml inhibited C2-ceramide-induced apoptosis and oxidative damage, but did not decrease the content of long-chain ceramide at all, suggesting that the inhibitory action of IGF-1 against exogenous C2-ceramide was exerted downstream of intracellular ceramide action.
Figure 3
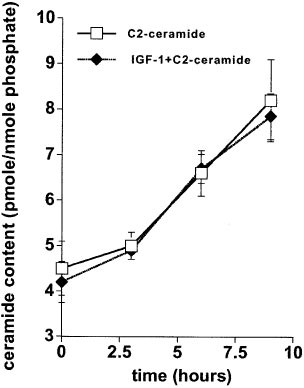
No inhibition by IGF-1 of C2-ceramide-induced long-chain ceramide generation. HL-60 cells were treated with 5 μM C2-ceramide in the presence or absence of 100 ng/ml IGF-1 for the indicated times, and then harvested for measuring ceramide levels by the DAG kinase method as described in Materials and Methods. The results are the averages from at least three independent experiments. The bars indicate 1 S.D.
IGF-1 restored C2-ceramide-depleted catalase function in a PI-3 kinase-dependent manner
We investigated the involvement of ROS scavengers such as glutathione peroxidase (GPx), glutathione (GSH) and catalase in C2-ceramide-induced apoptosis. C2-ceramide decreased catalase activity in a dose-dependent manner, but did not affect GPx activity or GSH content in HL-60 cells (data not shown). As shown in Figure 4A,B, 5 μM C2 ceramide in a serum-free condition decreased the catalase content judged by Western blotting analysis in a time-dependent manner, and the addition of 100 ng/ml IGF-1 restored the catalase depletion by C2-ceramide, and pretreatment with 20 nM wortmannin inhibited the restoration by IGF-1 of ceramide-depleted catalase content. Similarly, the activity of catalase, which was decreased from 78 to 37 U/mg protein after 24 h of C2-ceramide treatment, was restored to 60 U/mg protein by 100 ng/ml IGF-1, but this restoration of catalase activity was decreased to 35 U/mg protein by the addition of wortmannin (P<0.01). IGF-1 alone did not affect catalase activity, while 20 nM wortmannin slightly decreased its activity. In addition, the depletion of catalase content was inhibited by C2-ceramide in PI-3 kinase-overexpressing cells, but not in the kinase dead cells, while β-actin and protein level were equally detected in both cells (Figure 5A). Treatment with 20 μM C2-ceramide in 10% FCS condition catalase saw activity decreased from 88 to 55 U/mg protein in PI-3k cells and from 78 to 27 U/mg protein in the kinase-dead cells (Figure 5B, P<0.01). These results suggest that IGF-1 prevented the depletion of catalase by ceramide at both the protein level and activity levels in a PI-3 kinase-dependent manner.
Figure 4
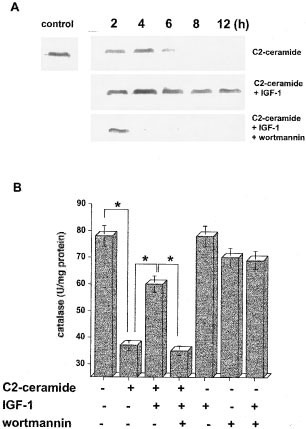
Inhibitory action of IGF-1 on ceramide-depleted catalase, and its inhibition by wortmannin. HL-60 cells were preincubated in the presence or absence of 20 nM wortmannin for 10 min and followed by treatment with 100 ng/ml IGF-1 for 30 min. Then, the cells were treated with or without 5 μM C2-ceramide and harvested various times (A; 0, 2, 4, 6, 8 and 12 h, B; 24 h) after treatment. Western blotting analysis for the amount of catalase (A) and measurement of catalase activity (B) were performed as described in ‘Materials and Methods’. The results (A) are representative of two independent experiments. The results (B) are the averages from three independent experiments, and the bars indicate 1 S.D. The significance of differences of catalase activity levels between the treatments was determined by ANOVA test. *Indicates P<0.01 (B)
Figure 5
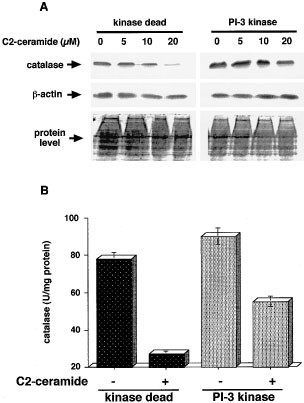
Inhibitory effect of overexpression of PI-3 kinase on ceramide-depleted catalase. PI-3 kinase-overexpressing HL-60 cells (PI-3k) and kinase-dead HL-60 cells (kinase dead) were made as described in Figure 2. PI-3k and kinase-dead cell were treated with various concentrations of C2-ceramide (A; 0, 5, 10 or 20 μM, B; 0 or 20 μM) for 24 h in 10% FCS-containing media. Western blotting analysis for catalase and β-actin protein, and Coomassie blue staining to confirm equal load of proteins (A) and measurement of catalase activity (B) were performed as described in Materials and Methods. The results (A) are the representative of two independent experiments. The results (B) are the averages from at least three independent experiments, and the bars indicate 1 S.D. Statistical significance by ANOVA test was P<0.05 for the difference of decrease of catalase activity between kinase-dead cell and PI-3k cells (B)
A catalase inhibitor, ATZ, and exogenous, purified catalase modulated the inhibition by IGF-1 of C2-ceramide-induced apoptosis and lipid peroxidation
To confirm the role of catalase in the inhibition by IGF-1 of ceramide-induced apoptosis and lipid peroxidation, we examined whether a catalase inhibitor, ATZ, and exogenous, purified catalase modulated IGF-1 action against C2-ceramide. ATZ prevented the inhibitory action of IGF-1 on 5 μM C2-ceramide-induced apoptosis while purified catalase enhanced it in a dose-dependent manner (Figure 6A). Similarly, ATZ prevented the inhibition by IGF-1 of C2-ceramide-induced lipid peroxidation while purified catalase enhanced it in a dose-dependent manner (Figure 6B). These results suggest that the extent of catalase activity was related to the inhibitory action of IGF-1 on ceramide-induced apoptosis and oxidative damage.
Figure 6
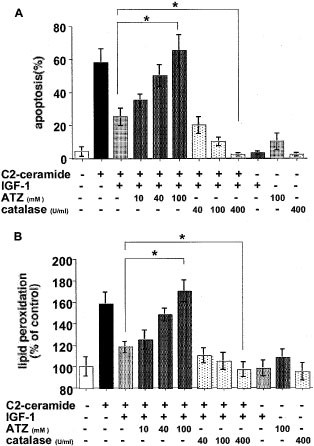
Modulation by catalase inhibitor, ATZ, and purified exogenous catalase of the inhibitory action of IGF-1 on ceramide-induced apoptosis and lipid peroxidation. HL-60 cells were treated simultaneously with C2-ceramide and IGF-1 in the presence of various concentrations of ATZ (0, 10, 40 or 100 mM) or purified catalase (0, 40, 100 or 400 U/ml). Apoptosis (A) and lipid peroxidation (B) were judged 24 h after treatment as described in Materials and Methods. The results are the averages from at least three independent experiments, and the bars indicate 1 S.D. The significance of differences of apoptosis and lipid peroxidation levels between the treatments with or without ATZ and purified catalase was determined by ANOVA test. *Indicates P<0.01
Inhibition by IGF-1 of C2-ceramide-induced decrease of mitochondrial membrane potential, and increase of cytosolic release of cytochrome c, caspase-3 cleavage and caspase-3 activity
We examined whether IGF-1 inhibited ceramide-induced apoptosis upstream of caspase-3 activation. As shown in Figure 7, an intensification of caspase-3-related signals such as mitochondrial membrane potential, cytochrome c release from mitochondria to cytosol, caspase-3 cleavage and real-timed caspase-3 activation by PhiPhiLux-using method were all detected in 5 μM C2-ceramide-induced apoptosis. These increases of caspase-3-related signals were inhibited in the presence of 100 ng/ml IGF-1, and these inhibitory action of IGF-1 was suppressed by pretreatment with 20 nM wortmannin. These results suggest that IGF-1 inhibited caspase-3 activation by augmenting mitochondrial function to restore ceramide-induced apoptosis.
Figure 7
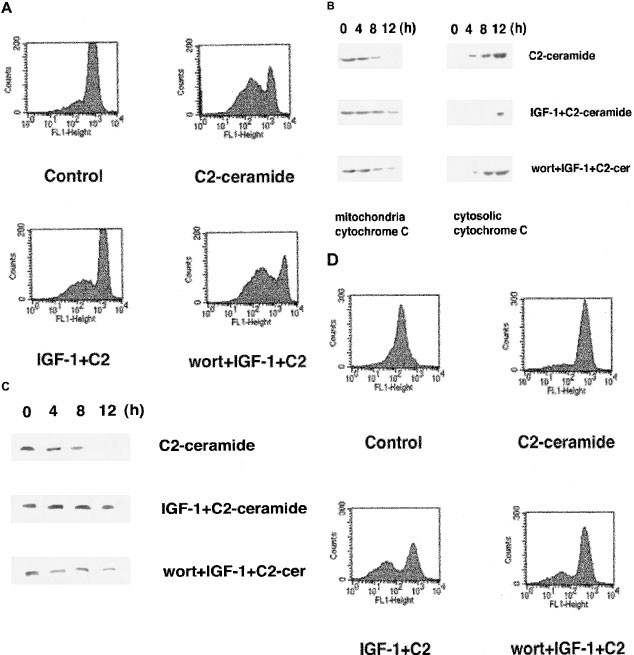
Inhibitory action of IGF-1 on ceramide-induced mitochondrial transmembrane potential, cytosolic release of cytochrome c, caspase-3 cleavage and caspase-3 activity, and its inhibition by wortmannin. HL-60 cells were preincubated in the presence or absence of 20 nM wortmannin for 10 min and followed by treatment with 100 ng/ml IGF-1 for 30 min. Then, the cells were treated with 5 μM C2-ceramide for 24 h (A and D) or the indicated times (B and C). Measurement of mitochondrial transmembrane potential using DiOC6 (A), cytosolic release of cytochrome c from mitochondria (B) and caspase-3 cleavage by Western blotting analysis (C) and real-time detection of caspase-3 activity by PhiPhilux G1D2 method (D) were performed as described in Materials and Methods. The results are representative of three independent experiments
A caspase-3 inhibitor DMQD-CHO augmented the inhibition by IGF-1 of C2-ceramide depleted catalase function
Under conditions in which IGF-1 inhibited C2-ceramide-induced apoptosis and lipid peroxidation, the activation of caspase-3 activity was examined. As shown in Table 1, when 5 μM C2-ceramide increased caspase-3 activity judged by DEVD-cleaving activity from 40 to 315 pmol/mg protein/min, 100 ng/ml IGF-1 inhibited an increase of its activity to 180 pmol/mg protein/min. A specific caspase-3 inhibitor DMQD-CHO, 100 μM of which showed a significant inhibition of ceramide-activated caspase-3 from 315 to 175 pmol/mg protein/min (P<0.01) Similarly, DMQD-CHO enhanced the inhibitory action of 100 ng/ml IGF-1 on 5 μM C2-ceramide-induced lipid peroxidation and apoptosis (from 121 to 108%, P<0.05 and from 29 to 12%, P<0.01, respectively). At the same condition DMQD-CHO also enhanced the inhibitory action of IGF-1 on C2-ceramide depleted catalase. Pretreatment with 100 μM DMQD-CHO enhanced the restoration by 100 ng/ml IGF-1 of ceramide-decreased catalase activity from 53 to 74 U/mg protein (Figure 8A, P<0.01). The enhancement of catalase at the catalase protein level was also detected by DMQD-CHO (Figure 8B). These results suggest that the regulation of catalase function by caspase-3 plays an important role in the inhibition by IGF-1 upstream of ceramide induced oxidative damage and apoptosis.
Table 1 Table DMQD-CHO augmented the inhibitory action of IGF-1 on ceramide-increased caspase-3 activity, lipid peroxidation and apoptosis
Figure 8
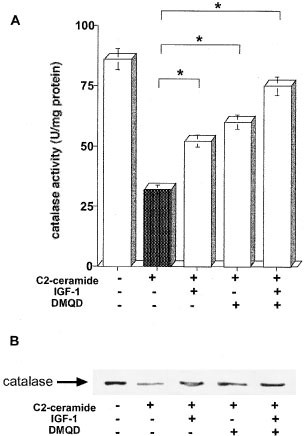
Augmentation by a caspase-3 inhibitor DMQD-CHO of the inhibitory action of IGF-1 on ceramide-depleted catalase. HL-60 cells were treated with 5 μM C2-ceramide in the presence of 100 ng/ml IGF-1, 100 μM DMQD-CHO or both 100 ng/ml IGF-1 and 100 μM DMQD-CHO for 24 h. Measurement of catalase activity (B) and Western blotting analysis for catalase (B) were performed as described in Materials and Methods. The results (A) are the averages from at least three independent experiments, and the bars indicate 1 S.D. The results (B) are the representative of two independent experiments. The significance of differences of catalase activity levels between the treatments was determined by ANOVA test. *Indicates P<0.01 (B)
Discussion
IGF-1 promotes cell survival at physiological concentrations in pro-apoptotic conditions such as interleukin-3 withdrawal, c-myc overexpression and potassium deprivation.18,26,27 Conversely, functional impairment of the IGF-1 receptor using dominant-negative IGF-1 receptor and antisense strategies induced massive apoptosis and delay of tumor growth, respectively.19,28 HL-60 cells, which we used in this study, express IGF-1 receptor, and show IGF-1-dependent cell survival in serum-free medium.29 It has been reported that many compounds, such as 4-hydroxy-2-nonenal, cupric nitrilotriacetate, TNF-α and IL-1α, induce apoptosis through increasing lipid peroxidation.15,30,31 In contrast, the suppression of lipid peroxidation inhibited TNF-α- or NO-induced apoptosis.32,33 These data suggest that oxidative damage, judged by lipid peroxidation, may be intimately involved in the inhibition of apoptosis by IGF-1. Since ceramide-induced increase of ROS generation judged by 2′,7′-dichlorofluorescein diacetate (DCFH-DA) method was significant but faint (data not shown), we decided to measure lipid peroxidation as a marker for accumulation of oxidative damage. As shown in Figure 1, IGF-1 inhibited C2-ceramide-induced HL-60 cell apoptosis through inhibiting oxidative damage in a dose- and time-dependent manner. In addition, the inhibition of lipid peroxidation and apoptosis by IGF-1 was shown to be dependent on PI-3 kinase signalling because the inhibitory action of IGF-1 was suppressed by a PI-3 kinase inhibitor wortmannin or overexpression of dead PI-3 kinase (Figure 2). These results demonstrate that the anti-apoptotic effect of IGF-1 through PI-3 kinase is closely related to the inhibition of oxidative damage.
Regarding the mechanism by which ceramide inhibits IGF-1/ PI-3 kinase-dependent cell survival, it was previously reported that ceramide inhibited the activity or nuclear translocation of PKB/Akt kinase by causing dephosphorylation of serine 473 through inhibition of PI-3 kinase-dependent kinase 2 or ceramide-activated protein phosphatase.23,24,34,35 Recently, modulation of caveolin-1 in microdomain of the membrane was also suggested to be involved in inhibition of PI-3 kinase activation by ceramide.25 In contrast, PI-3/Akt kinase inhibited stress-induced apoptosis without increasing ceramide generation.21,36 Interleukin-10 and interleukin-13 were also reported to inhibit pro-inflammatory cytokine-induced ceramide production through PI-3 kinase.22 Here, we found that the amount of long-chain ceramide generated by C2-ceramide was not inhibited at all when IGF-1 inhibited C2-ceramide-induced apoptosis (Figure 3), suggesting that IGF-1/PI-3 kinase inhibits apoptosis downstream of ceramide action rather than affecting stress-induced ceramide generation.
Insulin was shown to inhibit hydrogen peroxide-induced apoptosis through activating PI-3 kinase,37 but the present work seems to be the first report to show that IGF-1/PI-3 kinase inhibits an increase of oxidative damage downstream of ceramide action. Moreover, although it remains unproven, another important possibility suggested by the data in Figure 3 is that C2-ceramide might have an indirect effect, which is independent on a ceramide-mimicking action, on physiological ceramide generation.
It seems that oxidative damage and caspases are common pro-apoptotic signals involved in ceramide-induced apoptosis. This notion was supported by the report that apoptosis and ceramide generation were clearly inhibited by antioxidants, including GSH, NAC, catalase and GPx,6,9,38 or by caspase-3 inhibitors regardless of the kind of stress.39,40 Therefore, it would be important to investigate the mechanism by which IGF-1/PI-3 kinase inhibits ceramide-induced HL-60 cell apoptosis from the standpoint of the mechanistic connection between oxidative damage and caspase-3.
We here focused on whether IGF-1 affected the function of ROS scavenger catalase in a manner dependent on PI-3 kinase. As shown in Figures 4 and 5, catalase depletion by C2-ceramide was restored by IGF-1 or PI-3 kinase overexpression at both protein and activity levels, while this restoration was suppressed by the addition of wortmannin. Moreover, a catalase inhibitor ATZ suppressed the inhibitory action of IGF-1 on ceramide-induced oxidative damage and apoptosis in a dose-dependent manner, while purified recombinant catalase augmented the same inhibition as shown in Figure 6. These results strongly suggest that IGF-1/PI-3 kinase pathway inhibited ceramide-induced apoptosis by releasing oxidative damage through catalase function.
Ceramide was reported to activate caspase-3 via cytosolic release of cytochrome c from mitochondria and caspase-9 activation, resulting in induction of apoptosis.11,41 Although IGF-1 blocked apoptosis induction by inhibiting caspase-3 activation in retinal ganglion cells and cardiac muscle cells,16,20 the mechanistic connection between caspase-3 activation and the increase of oxidative damage remains to be elucidated. How does IGF-1 affect ceramide-induced activation of caspase-3 to increase oxidative damage? IGF-1 inhibited ceramide-induced decrease of mitochondrial membrane potential, and release of cytosolic cytochrome c, cleavage of caspase-3 and caspase-3 activation, suggesting that the inhibition of caspase-3 via mitochondrial function might be the target of IGF-1 action (Figure 7). As shown in Table 1, ceramide-induced caspase-3, oxidative damage and apoptosis were suppressed by the pretreatment with a synthetic tetrapeptide DMQD-CHO, which is a specific inhibitor of caspase-3 designed from the cleaving site of protein kinase C δ by caspase-3.13 These results indicated that caspase-3 is located upstream of the oxidative damage in C2-ceramide-induced apoptosis. In addition, the restoration by IGF-1 of ceramide-depleted catalase was enhanced at both activity and protein levels in the presence of DMQD-CHO (Figure 8). It has been reported that inhibition of hydrogen peroxide generation by catalase blocked apoptosis owing to a reduction of caspase-3 activity,42,43 and that NO stimulated hydrogen peroxide production by affecting mitochondria function, and subsequently increased caspase-3 activity.44,45 However, the results we showed here, conversely, demonstrated that caspase-3 induced oxidative damage through inhibiting catalase function. Therefore, caspase-3 and oxidative damage may consist with the signalling cycle to enhance the induction of apoptosis.
Ceramide has been shown to inhibit anti-apoptotic signals such as Bcl-2 family, heat shock protein-70, diacylglycerol/protein kinase C system, PI-3 kinase and Akt kinase.2,23,24,34,46 In contrast, we demonstrated here that the activation of IGF-1/PI-3 kinase inhibited ceramide-induced oxidative damage by preventing catalase function through caspase-3 inhibition, and consequently protected the cells from apoptotic death. The present findings suggest that the coordinated cross-talk between IGF-1/PI-3 kinase dependent survival signals and ceramide-induced pro-apoptotic signals such as oxidative damage and caspase-3 is important in determining the fate of the cells.
Materials and Methods
Materials
Human leukemia HL-60 cells were a kind gift from Dr. M Saito (National Cancer Institute, Tokyo, Japan). [γ-32P]-ATP (6000 Ci/mmol) was purchased from Amersham Pharmacia Biotech (Buckinghamshire, UK). Recombinant human insulin-like growth factor-1 was obtained from Fujisawa Pharmaceutical Co.,Ltd (Osaka, Japan). N-acetylsphingosine (C2-ceramide) was obtained from Matreya, Inc (PA, USA). Anti-PI-3 kinase p85, anti-PI-3 kinase p110, anti-caspase-3, anti-β-actin, anti cytochrome c and anti-catalase antibodies were purchased from Santa Cruz Biotechnology, Inc. (California, USA). Peroxidase- conjugated anti-rabbit immunoglobulin was purchased from TAGO, Inc. (Burlingame, USA). Cis-parinaric acid (PCA) was obtained from Pierce (Rockford, USA). PI-3 kinase expression vectors were a kind gift from Dr. A Klippel.47 Other chemicals, unless mentioned otherwise, were purchased from Sigma Co. Ltd. (St. Louis, USA).
Cell culture
Human myelogenous leukemia HL-60 cells were grown in RPMI 1640 medium (Nissui, Tokyo, Japan) supplemented with 10% fetal calf serum (FCS, JRH Biosciences) and kanamycin sulfate (80 ng/ml), and incubated at 37°C in a humidified atmosphere containing 5% CO2/95% air. The cells were counted using a hemocytometer and the viability was always greater than 95% in all experiments as assayed by 0.025% Trypan blue dye exclusion. Before the experiments, the cells were washed with phosphate-buffered saline (PBS) and incubated overnight in serum-free RPMI 1640 medium at a concentration of 2×105 cells/ml, and then treated as described in the text. The experiments using transfectants were performed in 10% FCS-containing media.
Measurement of number of apoptotic cells
The cells were treated with C2-ceramide at various conditions, harvested and then fixed in 1% glutaraldehyde for 30 min at room temperature, and stained with DAPI (4′,6-diamidino-2-phenylindole) as described before.48 At least 200 cells were counted under a fluorescent microscope in each determination, and the cells with nuclear condensation and fragmentation were judged as apoptotic cells. The results expressed as a percentage of the total cells that were positive for apoptotic feature. Control indicates no treatment.
Detection of apoptosis by FACS
To assess cells undergoing apoptosis the annexin V staining kit (BD PharMingen) was used. After treatment the cells were harvested in cold PBS, washed, and stained with 5 μl of Annexin V-FITC in 100 μl of 1× binding buffer (10 mM HEPES (pH 7.4), 140 mM NaCl, and 25 μM CaCl2) for 15 min at room temperature in the dark, diluted in 400 μl 1× binding buffer, and immediately subjected to FACS analysis (10 000 events/sample).
Measurement of lipid peroxidation and catalase activity
Cis_-parinaric acid (CPA) is a fluorescent poly-unsaturated fatty acid used as a probe to directly monitor lipid peroxidation. After treatment, the cells were washed with PBS twice and resuspended in 3 ml RPMI 1640 media without FCS, and then incubated with 5 μM CPA at 37°C in 5% CO2/95% air for 30 min. Fluorescence measurement was carried out using a spectrophotometer F-3000 (Hitachi) using 318 nm excitation and 420 nm emission filters. Catalase activity was assayed by measuring the kinetics of the loss of H2O2 as described previously.49 The cells at a concentration of 1×106/ml were washed with PBS twice and resuspended in a solution containing 0.3% Triton-X, 50 mM Tris-HCl (pH 7.5), 0.2 mM phenylmethyl-sulfonyl fluoride and 0.5% acetyltrimethylammonium bromide. This cell suspension was subjected to freeze–thawing and sonication with a model UR-20P (Tomy Seiko, Japan). The sonicate was centrifuged at 12 000×_g for 15 min. The supernatant was used as the cell lysate. The cell lysate was supplemented with phosphate buffer and H2O2 and loss of H2O2 was monitored at 240 nm using spectrophotometer.
Ceramide quantitation
After extracting the lipids according to the Bligh and Dyer method, ceramide levels in the cells were measured enzymatically by using E-coli diacylglycerol kinase (DGK), which converts ceramide and diacylglycerol to ceramide-1 phosphate and phosphatidic acid, respectively, and the amounts of ceramide were corrected by phospholipids phosphate as described elsewhere.50
Western blot analysis
The samples (50 μg) were denatured by boiling in Laemmli's sample buffer for 5 min, subjected to SDS-polyacrylamide gel electrophoresis using a 7.5% running gel, and electroblotted to an Immobilon-P Transfer Membrane (Millipore) as described.51 Non-specific binding was blocked by incubation of the membrane with PBS containing 5% skim milk and 0.1% Tween 20 for more than 1 h. Then the membrane was washed in PBS containing 0.1% Tween 20 (PBS-T) once for 15 min and once for 5 min, and incubated with a 1 : 200 dilution of anti-PI-3 kinase p85, and anti-PI-3 kinase p110 and anti-cytochrome c and anti-catalase or anti-caspase-3 antibody in PBS-T for 1 h. The membrane was washed in PBS-T once for 15 min and once for 5 min, and incubated with a 1 : 4000 dilution of peroxidase-conjugated anti-rabbit immunoglobulin in PBS-T for 1 h. After washing the membrane three times for 5 min in PBS-T, detection was performed using ECL Western blotting detection reagents (Amersham) according to the manufacturer's protocol.
Preparation of mitochondria and cytosol
Cells (1×107) were washed with PBS, resuspended in five volumes of CFS buffer (220 mM mannitol, 68 mM sucrose, 2 mM NaCl, 2.5 mM KH2PO4, 0.5 mM EGTA, 2 mM MgCl2, 5 mM pyruvate, 0.1 mM PMSF, 10 mM HEPES-NaOH, pH 7.4), and swollen on ice for 20 min. Cells were disrupted by 10–15 aspirations through a 22-gauge needle and centrifuged at 750×g for 5 min at 4°C to remove nuclei. The supernatant was centrifuged again (10 000×g, 10 min, 4°C) to recover mitochondria. The 10 000×g supernatant was ultracentrifuged at 100 000×g for 30 min at 4°C, and the remaining supernatant was used as the cytosol fraction. Both samples were used immediately or stored at −80°C until immunodetection of cytochrome c.
Measurement of mitochondrial membrane potential
Changes of mitochondrial membrane potential were studied with the use of DiOC6. DiOC6 is incorporated into mitochondria in strict nonlinear dependence to mitochondrial membrane potential and emits exclusively within the spectrum of green light. After treatment, 1×106 cells were treated continuously with 40 nM DiOC6 in the culture medium for 15 min at 37°C. After treatment with DiOC6, the cells were washed twice with PBS, resuspended in 1 ml of PBS, and analyzed with a flow cytometer (FACScan; Becton Dickinson, San Jose, CA, USA). The data were recorded with FL1 photomultiplier, and 10 000 cells per sample were acquired in histograms using the data analysis program CELL Quest.
Measurement of DEVDase activity
The cells after each treatment were homogenized in lysis buffer containing 10 mM HEPES/KOH (pH 7.4), 2 mM EDTA, 0.1% CHAPS, 5 mM dithiothreitol (DTT), 1 mM PMSF, 100 μM pepstatin, 0.15 U/ml aprotinin, and 50 μg/ml leupeptin, and centrifuged at 10 000×g for 10 min. The supernatant was collected as an enzyme source and added to the reaction mixture (10% sucrose, 10 mM HEPES/KOH (pH 7.4), 5 mM DTT, 0.1% CHAPS, and 10 μM DEVD-MCA) which was followed by incubation at 25°C for 60 min. Fluorescence was measured by a microplate reader (MTP-100F, Corona Electric, Japan) using 360 nm excitation and 450 nm emission filters. Concentrations of 7-amino-4-methyl-coumarin (AMC) liberated as a result of cleavage were calculated comparing with standard AMC solutions.
Detection of caspase-3 activity by FACS
The caspase-3 activity was evaluated as the protease activity of caspase-3 by using the PhiPhiLux G1D2 kit (MBL, Nagoya, Japan). A substrate of PhiPhiLux G1D2, which can penetrate into the cell nucleus, is converted to the fluorescent form when it is cleaved by the protease activity of caspase-3. The assessment was performed according to the manufacturer's recommendations. Briefly, HL-60 cells were incubated with 75 μl of PhiPhiLux G1D2 for 1 h at 37°C in 5% CO2, and caspase-3 activity was analyzed by fluorescence-activated cell sorting caliber flow cytometry.
Expression constructs and transfection
The expression plasmid constructs used were the constructs for constitutively activated PI-3 kinase (the catalytic p110 subunit of PI-3 kinase, Myc-tagged at the C-terminus in pEF-Bos vector) and kinase dead PI-3 kinase (the catalytic p110 subunit, point-mutated at the ATP- binding site, Myc-tagged in pEF-Bos vector) which have been previously described.47 HL-60 cells were transiently transfected by the electroporation method. Fifty micrograms of expression plasmid cDNA were mixed with per 5×107 cells and the mixture was exposed to a high-voltage pulse (300 V, 960 μF) by using a Bio-Rad electroporation apparatus with a capacitor extender. The transfected cells were cultured in RPMI 1640 media supplemented with 20% fetal calf serum and kanamycin sulfate (80 ng/ml), and incubated at 37°C in a humidified atmosphere containing 5% CO2/95% air. Prior to experiments 48 h after transfection, the medium was replaced with 10% fetal calf serum, and then the cells were treated as described in the text.
Measurement of PI-3 kinase enzymatic activity
After treatment the cells (1×107) were lysed in 700 μl of lysis buffer (50 mM Tris-HCl (pH 7.6), 0.5% TritonX-100, 300 mM NaCl, 5 mM EDTA, 10 μg/ml leupeptin, 10 μg/ml aprotinin, 1 mM PMSF, 1 mM sodium orthovanadate), and then the lysates were subjected to immunoprecipitation with anti-PI 3 kinase (p85α) antibody. Immunoprecipitants were washed once with cold PBS, twice with washing buffer A (0.5 M LiCl, 0.1 M Tris-HCl (pH 7.4)), and once with washing buffer B (0.1 M Tris-HCl (pH 7.4), 0.1 M NaCl, 1 mM EDTA), and then assayed for PI-3 kinase activity. Note: all wash solutions contained 0.2 mM sodium orthovanadate. For protein immunoblot analysis using anti-p85 antibody, one-third of this immunoprecipitant mixture was electrophoresed on an SDS-polyacrylamide gel and the separated proteins were blotted onto an Immobilon-P Transfer Membrane (Millipore) as described.47 Immunoprecipitants were resuspended in 50 μl of PI-3 kinase reaction buffer (30 mM HEPES (pH 7.4), 30 mM MgCl2, 200 μM adenosine, 40 μM ATP), and PI (final concentration 0.2 mg/ml) was preincubated at 4°C in this buffer for 20 min. The kinase reaction was started by addition of 20 μCi [γ-32P]-ATP, and after incubation at 30°C or 15 min, the reaction was terminated by addition of 100 μl of 1N HCl. The lipid-containing organic phase was applied to oxalate-coated TLC plates and developed in chloroform/methanol/water/ammonium hydroxide (60/47/11.3/2) and lipid species were visualized by autoradiography.
Statistical examination
Statistical significances in this work were calculated by Student's t-test and ANOVA test.
Abbreviations
IGF-1:
insulin-like growth factor
Pl-3:
phosphatidylinositol-3
C2-ceramide:
N-acetylsphingosine
ROS:
reactive oxygen species
NAC:
N-acetylcysteine
C6-ceramide:
N-hexanoylsphingosine
GSH:
glutathione
APAF-1:
apoptotic protease activating factor-1
References
- Obeid LM, Hannun YA . 1995 Ceramide: a stress signal and mediator of growth suppression and apoptosis J. Cell Biochem. 58: 191–198
Article CAS Google Scholar - Okazaki T, Kondo T, Kitano T, Tashima M . 1998 Diversity and complexity of ceramide signalling in apoptosis Cell Signal. 10: 685–692
Article CAS Google Scholar - Quillet-Mary A, Jaffrezou JP, Mansat V, Bordier C, Naval J, Laurent G . 1997 Implication of mitochondrial hydrogen peroxide generation in ceramide- induced apoptosis J. Biol. Chem. 272: 21388–21395
Article CAS Google Scholar - Degli Esposti M, McLennan H . 1998 Mitochondria and cells produce reactive oxygen species in virtual anaerobiosis: relevance to ceramide-induced apoptosis FEBS Lett. 430: 338–342
Article CAS Google Scholar - Garcia-Ruiz C, Colell A, Mari M, Morales A, Fernandez-Checa JC . 1997 Direct effect of ceramide on the mitochondrial electron transport chain leads to generation of reactive oxygen species. Role of mitochondrial glutathione J. Biol. Chem. 272: 11369–11377
Article CAS Google Scholar - Kiryu-Seo S, Sasaki M, Yokohama H, Nakagomi S, Hirayama T, Aoki S, Wada K, Kiyama H . 2000 Damage-induced neuronal endopeptidase (DINE) is a unique metallopeptidase expressed in response to neuronal damage and activates superoxide scavengers Proc. Natl. Acad. Sci. USA 97: 4345–4350
Article CAS Google Scholar - Davis MA, Flaws JA, Young M, Collins K, Colburn NH . 2000 Effect of ceramide on intracellular glutathione determines apoptotic or necrotic cell death of JB6 tumor cells Toxicol. Sci. 53: 48–55
Article CAS Google Scholar - Mansat de Mas V, Bezombes C, Quillet Mary A, Bettaieb A, D'Orgeix AD, Laurent G, Jaffrezou JP . 1999 Implication of radical oxygen species in ceramide generation, c Jun N-terminal kinase activation and apoptosis induced by daunorubicin Mol. Pharmacol. 56: 867–874
Article CAS Google Scholar - Liu B, Hannun YA . 1997 Inhibition of the neutral magnesium-dependent sphingomyelinase by glutathione J. Biol. Chem. 272: 16281–16287
Article CAS Google Scholar - Zou H, Henzel WJ, Liu X, Lutschg A, Wang X . 1997 Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3 [see comments] Cell 90: 405–413
Article CAS Google Scholar - Anjum R, Ali AM, Begum Z, Vanaja J, Khar A . 1998 Selective involvement of caspase-3 in ceramide induced apoptosis in AK-5 tumor cells FEBS Lett. 439: 81–84
Article CAS Google Scholar - Kondo T, Matsuda T, Kitano T, Takahashi A, Tashima M, Ishikura H, Umehara H, Domae N, Uchiyama T, Okazaki T . 2000 Role of c-jun expression increased by heat shock- and ceramide-activated caspase-3 in HL-60 cell apoptosis. Possible involvement of ceramide in heat shock-induced apoptosis J. Biol. Chem. 275: 7668–7676
Article CAS Google Scholar - Hirata H, Takahashi A, Kobayashi S, Yonehara S, Sawai H, Okazaki T, Yamamoto K, Sasada M . 1998 Caspases are activated in a branched protease cascade and control distinct downstream processes in Fas-induced apoptosis J. Exp. Med. 187: 587–600
Article CAS Google Scholar - Matsura T, Kai M, Fujii Y, Ito H, Yamada K . 1999 Hydrogen peroxide-induced apoptosis in HL-60 cells requires caspase-3 activation Free Radic. Res. 30: 73–83
Article CAS Google Scholar - Ma Y, Ogino T, Kawabata T, Li J, Eguchi K, Okada S . 1999 Cupric nitrilotriacetate-induced apoptosis in HL-60 cells association with lipid peroxidation, release of cytochrome C from mitochondria, and activation of caspase-3 Free Radic. Biol. Med. 27: 227–233
Article CAS Google Scholar - Wu W, Lee WL, Wu YY, Chen D, Liu T, Jang A, Sharma PM, Wang PH . 2000 Expression of constitutively active phosphatidylinositol 3 kinase inhibits activation of caspase 3 and apoptosis of cardiac muscle cells J. Biol. Chem. 275: 40113–40119
Article CAS Google Scholar - Mooney A, Jobson T, Bacon R, Kitamura M, Savill J . 1997 Cytokines promote glomerular mesangial cell survival in vitro by stimulus-dependent inhibition of apoptosis J. Immunol. 159: 3949–3960
CAS PubMed Google Scholar - Parrizas M, Saltiel AR, LeRoith D . 1997 Insulin-like growth factor 1 inhibits apoptosis using the phosphatidylinositol 3′-kinase and mitogen-activated protein kinase pathways J. Biol. Chem. 272: 154–161
Article CAS Google Scholar - D'Ambrosio C, Ferber A, Resnicoff M, Baserga R . 1996 A soluble insulin-like growth factor I receptor that induces apoptosis of tumor cells in vivo and inhibits tumorigenesis Cancer Res. 56: 4013–4020
CAS PubMed Google Scholar - Kermer P, Klocker N, Labes M, Bahr M . 2000 Insulin-like growth factor-I protects axotomized rat retinal ganglion cells from secondary death via PI3-K-dependent Akt phosphorylation and inhibition of caspase-3 In vivo J. Neurosci. 20: 2–8
Article CAS Google Scholar - Burow ME, Weldon CB, Collins Burow BM, Ramsey N, McKee A, Klippel A, McLachlan JA, Clejan S, Beckman BS . 2000 Cross-talk between phosphatidylinositol 3-kinase and sphingomyelinase pathways as a mechanism for cell survival/death decisions J. Biol. Chem. 275: 9628–9635
Article CAS Google Scholar - Pahan K, Khan M, Singh I . 2000 Interleukin-10 and interleukin-13 inhibit proinflammatory cytokine-induced ceramide production through the activation of phosphatidylinositol 3-kinase J. Neurochem. 75: 576–582
Article CAS Google Scholar - Zhou H, Summers SA, Birnbaum MJ, Pittman RN . 1998 Inhibition of Akt kinase by cell-permeable ceramide and its implications for ceramide-induced apoptosis J. Biol. Chem. 273: 16568–16575
Article CAS Google Scholar - Schubert KM, Scheid MP, Duronio V . 2000 Ceramide inhibits protein kinase B/Akt by promoting dephosphorylation of serine 473 J. Biol. Chem. 275: 13330–13335
Article CAS Google Scholar - Zundel W, Swiersz LM, Giaccia A . 2000 Caveolin 1-mediated regulation of receptor tyrosine kinase-associated phosphatidylinositol 3-kinase activity by ceramide Mol. Cell. Biol. 20: 1507–1514
Article CAS Google Scholar - Juin P, Hueber AO, Littlewood T, Evan G . 1999 c-Myc-induced sensitization to apoptosis is mediated through cytochrome c release Genes Dev. 13: 1367–1381
Article CAS Google Scholar - Leverrier Y, Thomas J, Mathieu AL, Low W, Blanquier B, Marvel J . 1999 Role of PI3-kinase in Bcl-X induction and apoptosis inhibition mediated by IL-3 or IGF-1 in Baf 3 cells Cell Death Differ. 6: 290–296
Article CAS Google Scholar - Pass HI, Mew DJ, Carbone M, Matthews WA, Donington JS, Baserga R, Walker CL, Resnicoff M, Steinberg SM . 1996 Inhibition of hamster mesothelioma tumorigenesis by an antisense expression plasmid to the insulin-like growth factor-1 receptor Cancer Res. 56: 4044–4048
CAS PubMed Google Scholar - Estrov Z, Meir R, Barak Y, Zaizov R, Zadik Z . 1991 Human growth hormone and insulin-like growth factor-1 enhance the proliferation of human leukemic blasts J. Clin. Oncol. 9: 394–399
Article CAS Google Scholar - Kalinich JF, Ramakrishnan R, McClain DE, Ramakrishnan N . 2000 4-Hydroxynonenal, an end-product of lipid peroxidation, induces apoptosis in human leukemic T- and B-cell lines Free Radic. Res. 33: 349–358
Article CAS Google Scholar - Bohler T, Waiser J, Hepburn H, Gaedeke J, Lehmann C, Hambach P, Budde K, Neumayer HH . 2000 TNF-alpha and IL-1alpha induce apoptosis in subconfluent rat mesangial cells. Evidence for the involvement of hydrogen peroxide and lipid peroxidation as second messengers Cytokine. 12: 986–991
Article CAS Google Scholar - Manna SK, Mukhopadhyay A, Aggarwal BB . 2000 Resveratrol suppresses TNF induced activation of nuclear transcription factors NF-kappa B, activator protein-1, and apoptosis: potential role of reactive oxygen intermediates and lipid peroxidation J. Immunol. 164: 6509–6519
Article CAS Google Scholar - Wei T, Chen C, Hou J, Zhao B, Xin W, Mori A . 1999 The antioxidant EPC-K1 attenuates NO-induced mitochondrial dysfunction, lipid peroxidation and apoptosis in cerebellar granule cells Toxicology 134: 117–126
Article CAS Google Scholar - Schmitz-Peiffer C, Craig DL, Biden TJ . 1999 Ceramide generation is sufficient to account for the inhibition of the insulin-stimulated PKB pathway in C2C12 skeletal muscle cells pretreated with palmitate J. Biol. Chem. 274: 24202–24210
Article CAS Google Scholar - Salinas M, Lopez-Valdaliso R, Martin D, Alvarez A, Cuadrado A . 2000 Inhibition of PKB/Akt1 by C2-ceramide involves activation of ceramide-activated protein phosphatase in PC12 cells Mol. Cell. Neurosci. 15: 156–169
Article CAS Google Scholar - Goswami R, Kilkus J, Dawson SA, Dawson G . 1999 Overexpression of Akt (protein kinase B) confers protection against apoptosis and prevents formation of ceramide in response to pro-apoptotic stimuli J. Neurosci. Res. 57: 884–893
Article CAS Google Scholar - Aikawa R, Nawano M, Gu Y, Katagiri H, Asano T, Zhu W, Nagai R, Komuro I . 2000 Insulin prevents cardiomyocytes from oxidative stress-induced apoptosis through activation of PI3 kinase/Akt Circulation 102: 2873–2879
Article CAS Google Scholar - Liu B, Andrieu-Abadie N, Levade T, Zhang P, Obeid LM, Hannun YA . 1998 Glutathione regulation of neutral sphingomyelinase in tumor necrosis factor-alpha induced cell death J. Biol. Chem. 273: 11313–11320
Article CAS Google Scholar - Janicke RU, Sprengart ML, Wati MR, Porter AG . 1998 Caspase-3 is required for DNA fragmentation and morphological changes associated with apoptosis J. Biol. Chem. 273: 9357–9360
Article CAS Google Scholar - Ohta T, Kinoshita T, Naito M, Nozaki T, Masutani M, Tsuruo T, Miyajima A . 1997 Requirement of the caspase-3/CPP32 protease cascade for apoptotic death following cytokine deprivation in hematopoietic cells J. Biol. Chem. 272: 23111–23116
Article CAS Google Scholar - Yoshimura S, Banno Y, Nakashima S, Takenaka K, Sakai H, Nishimura Y, Sakai N, Shimizu S, Eguchi Y, Tsujimoto Y, Nozawa Y . 1998 Ceramide formation leads to caspase-3 activation during hypoxic PC12 cell death. Inhibitory effects of Bcl-2 on ceramide formation and caspase-3 activation J. Biol. Chem. 273: 6921–6927
Article CAS Google Scholar - Chen YC, Lin-Shiau SY, Lin JK . 1998 Involvement of reactive oxygen species and caspase 3 activation in arsenite-induced apoptosis J. Cell. Physiol. 177: 324–333
Article CAS Google Scholar - Sengupta J, Sinha P, Mukhopadhyay C, Ray PK . 1999 Molecular modeling and experimental approaches toward designing a minimalist protein having Fc-binding activity of Staphylococcal protein A Biochem. Biophys. Res. Commun. 256: 6–12
Article CAS Google Scholar - Rossig L, Fichtlscherer B, Breitschopf K, Haendeler J, Zeiher AM, Mulsch A, Dimmeler S . 1999 Nitric oxide inhibits caspase-3 by S-nitrosation in vivo J. Biol. Chem. 274: 6823–6826
Article CAS Google Scholar - Yabuki M, Kariya S, Ishisaka R, Yasuda T, Yoshioka T, Horton AA, Utsumi K . 1999 Resistance to nitric oxide-mediated apoptosis in HL-60 variant cells is associated with increased activities of Cu,Zn-superoxide dismutase and catalase Free Radic. Biol. Med. 26: 325–332
Article CAS Google Scholar - Esposti MD, Hatzinisiriou I, McLennan H, Ralph S . 1999 Bcl-2 and mitochondrial oxygen radicals. New approaches with reactive oxygen species-sensitive probes J. Biol. Chem. 274: 29831–29837
Article CAS Google Scholar - Klippel A, Reinhard C, Kavanaugh WM, Apell G, Escobedo MA, Williams LT . 1996 Membrane localization of phosphatidylinositol 3-kinase is sufficient to activate multiple signal-transducing kinase pathways Mol. Cell. Biol. 16: 4117–4127
Article CAS Google Scholar - Kondo T, Matsuda T, Tashima M, Umehara H, Domae N, Yokoyama K, Uchiyama T, Okazaki T . 2000 Suppression of heat shock protein-70 by ceramide in heat shock induced HL-60 cell apoptosis J. Biol. Chem. 275: 8872–8879
Article CAS Google Scholar - Beers RF, Sizer IW . 1952 A spectrophotometric method for measuring the break down of hydrogen peroxide by catalase J. Biol. Chem. 195: 133–140
CAS Google Scholar - Okazaki T, Bielawska A, Bell RM, Hannun YA . 1990 Role of ceramide as a lipid mediator of 1 alpha,25-dihydroxyvitamin D3-induced HL-60 cell differentiation J. Biol. Chem. 265: 15823–15831
CAS Google Scholar - Sawai H, Okazaki T, Yamamoto H, Okano H, Takeda Y, Tashima M, Sawada H, Okuma M, Ishikura H, Umehara H et al. 1995 Requirement of AP-1 for ceramide induced apoptosis in human leukemia HL-60 cells J. Biol. Chem. 270: 27326–27331
Article CAS Google Scholar - Demura Y, Ameshima S, Ishizaki T, Miyamori I . 1999 [Glutathione peroxidase] Nippon Rinsho. 57: 448–450
PubMed Google Scholar - Xia YM, Hill KE, Burk RF . 1989 Biochemical studies of a selenium-deficient population in China: measurement of selenium, glutathione peroxidase and other oxidant defense indices in blood J. Nutr. 119: 1318–1326
Article CAS Google Scholar - Takahashi A, Alnemri ES, Lazebnik YA, Fernandes-Alnemri T, Litwack G, Moir RD, Goldman RD, Poirier GG, Kaufmann SH, Earnshaw WC . 1996 Cleavage of lamin A by Mch2 alpha but not CPP32: multiple interleukin 1 beta-converting enzyme-related proteases with distinct substrate recognition properties are active in apoptosis Proc. Natl. Acad. Sci. USA 93: 8395–8400
Article CAS Google Scholar
Acknowledgements
We thank Dr. A. Klippel for the kind gift of expression plasmids of PI-3 kinase. This work was supported by the grants of Japanese Ministry of Science, Education and Culture and Ono Pharmaceutical Co., Ltd. (to T Okazali).
Author information
Authors and Affiliations
- Department of Hematology and Oncology, Graduate School of Medicine, Kyoto University, 54 Syogoin-Kawaramachi, Sakyo-ku, Kyoto, 606-8507, Japan
T Kondo, T Kitano, K Iwai, M Watanabe, Y Taguchi, T Yabu, T Uchiyama & T Okazaki - Laboratory of Membrane Biochemistry and Biophysics, Graduate School of Biostudies, Kyoto University, Yoshida, Sakyo-ku, Kyoto, 606-8501, Japan
Y Taguchi - Department of Medicine, Osaka Dental University, 1-5-17 Otemae, Cyuoh-ku, Osaka, 540, Japan
H Umehara & N Domae
Authors
- T Kondo
You can also search for this author inPubMed Google Scholar - T Kitano
You can also search for this author inPubMed Google Scholar - K Iwai
You can also search for this author inPubMed Google Scholar - M Watanabe
You can also search for this author inPubMed Google Scholar - Y Taguchi
You can also search for this author inPubMed Google Scholar - T Yabu
You can also search for this author inPubMed Google Scholar - H Umehara
You can also search for this author inPubMed Google Scholar - N Domae
You can also search for this author inPubMed Google Scholar - T Uchiyama
You can also search for this author inPubMed Google Scholar - T Okazaki
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toT Okazaki.
Additional information
Edited by M Miura
Rights and permissions
About this article
Cite this article
Kondo, T., Kitano, T., Iwai, K. et al. Control of ceramide-induced apoptosis by IGF-1: involvement of PI-3 kinase, caspase-3 and catalase.Cell Death Differ 9, 682–692 (2002). https://doi.org/10.1038/sj.cdd.4401019
- Received: 03 July 2001
- Revised: 07 December 2001
- Accepted: 07 January 2002
- Published: 24 May 2002
- Issue Date: 01 June 2002
- DOI: https://doi.org/10.1038/sj.cdd.4401019