Redox factor-1: an extra-nuclear role in the regulation of endothelial oxidative stress and apoptosis (original) (raw)
Introduction
Exposure of cells or tissue to oxidative stimuli such as hypoxia/reoxygenation (H/R), ischemia/reperfusion (I/R), and inflammatory cytokines induces the generation of reactive oxygen species (ROS) resulting in alterations in intracellular reduction/oxidation (redox) state. Cellular redox changes can have profound effects on the activities of transcription factors, and such regulation of transcriptional activation has emerged as one of the important mechanisms that prokaryotic and eukaryotic cells employ for the expression of specific genes important in inflammation, death and survival.1
Stimuli that result in oxidative stress employ a number of cellular ROS generating systems. Among these is the NAD(P)H oxidase regulated by the ubiquitous small GTPase rac1. Many components of the phagocyte NADPH oxidase are expressed in non-pahgocytic cells, including cardiovascular cell types.2 The functional significance of this oxidase is evident by the fact that rac1-regulates the production of ROS in a variety of cell types in response to a many oxidative stimuli such as tumor necrosis factor (TNF)3 and H/R.4
Redox factor-1 (ref-1) is a ubiquitous 37 kD bi-functional protein. Ref-1 stimulates the DNA binding activities of the AP-1 family of transcription factors by a redox-dependent mechanism.5,6 This effect is mediated through reduction of a conserved cysteine residue located at the DNA-binding domains of c-fos and c-jun.7 Through a similar reducing action, ref-1 is also capable of modulating or activating other classes of transcription factors that regulate cell growth, differentiation, survival, and death including NF-κB, p53, Egr-1, c-Myb, HLF, and Pax-8.6,8,9,10,11,12 Ref-1 also has DNA repair activity. It is an apurinic/apyrimidinic endonuclease (APE), an important enzyme in the base excision repair (BER) pathway.13 Apurinic/Apyrimidinic (AP) damage of DNA occurs with diverse oxidative stimuli, is highly mutagenic, and AP repair is indispensable for the genomic integrity and viability of cells. Although the DNA repair and transcription factor reducing properties of ref-1 are well known, other fundamental mechanisms via which it may regulate redox-sensitive transcription, and influence cell death are yet to be elucidated.
Ref-1 has been implicated in protection against cell death resulting from oxidative stimuli. Depletion of ref-1 renders cells more sensitive to hyperoxia,14 and a decrease in ref-1 expression is associated with apoptosis in ischemic neuronal tissue.15 However, the role of ref-1 and the cellular phenotypes that it regulates in cardiovascular tissue is not known. The vascular endothelium is the first line of defense against a variety of oxidative stimuli, and redox-regulated activation and death of endothelial cells have been implicated in the pathogenesis of vascular disorders. We therefore investigated the role of ref-1 in regulating the response of vascular endothelial cells to the oxidative stimulus hypoxia/reoxygenation (H/R).
Here, we report that ref-1 protects endothelial cells from apoptosis, and prevents endothelial cell activation of the inflammatory transcription factor NF-κB in response to H/R through a novel mechanism of action: suppression of intracellular H2O2 regulated by the small GTPase rac1. Although our experiments are limited to endothelial cells, the ubiquitous expression of ref-1 and rac1, and their importance in the regulation and response of cells to oxidative stimuli, suggests that the functional interaction between these two proteins described herein may also be relevant to the redox biology of other cell types.
Results
Expression of ref-1 in the cytoplasm and nucleus of endothelial cells: translocation to the nucleus with H/R
We first determined the sub-cellular expression pattern of ref-1 under basal normoxic conditions, and after hypoxia/reoxygenation (H/R) (Figure 1A,B). Under normoxic conditions, endogenous ref-1 was present in both the cytoplasm and nuclei of uninfected endothelial cells, and cells infected with the control Adβgal virus. Hypoxia/reoxygenation (H/R) of uninfected, and Adβgal-infected cells led to a significant increase in nuclear/cytoplasmic ref-1. H/R or hypoxia alone for 6 h resulted in no significant change in total expression of ref-1 (not shown).
Figure 1
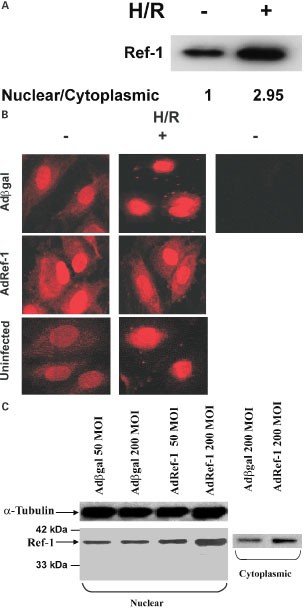
Basal and adenoviral-induced endothelial ref-1 expression and sub-cellular distribution during normoxia and with hypoxia/reoxgenation (H/R). (A) Western blot for ref-1 in nuclear fractions of endothelial cells under normoxic conditions, and after hypoxia 6 h/reoxygenation 2 h. Nuclear/cytoplasmic ratio is calculated after normalization for protein loading. (B) Immunostaining for ref-1 in uninfected, Adβgal- and Adref-1-infected cells, with and without H/R. Viruses used at 200 MOI, hypoxia 6 h, and reoxygenation 2 h.–ref-1 Ab indicates omission of primary ref-1 antibody, but inclusion of secondary antibody. (C) Western blot for ref-1 in cytoplasmic and nuclear fractions of cells infected with Adβgal and Adref-1. Protein loading of nuclear and cytoplasmic fractions is not comparable
Adenovirus-mediated cytoplasmic and nuclear overexpression of ref-1
With the goal of examining the role of ref-1 in oxidative stress and apoptosis, we used a recombinant adenovirus encoding full-length ref-1, Adref-1, to overexpress ref-1 in endothelial cells. Adenoviral vectors are capable of gene transfer efficiencies approaching 100% in HUVECs.3 Infection of HUVECs with Adref-1 resulted in significant, and dose-dependent overexpression of ref-1 compared to cells infected with Adβgal (Figure 1B,C). Importantly, infection with Adref-1 led to an increase in both cytoplasmic and nuclear expression of ref-1 (Figure 1C). Moreover, when compared to Adβgal-infected cells, Adref-1-infected cells showed significantly higher cytoplasmic ref-1 expression even after H/R (Figure 1B). This demonstrates that infection with Adref-1 is able to significantly increase both cytoplasmic and nuclear ref-1 expression during normoxic conditions and after the H/R stimulus.
Overexpression of ref-1 suppresses H/R-induced apoptosis
Apoptosis plays an important part in post-hypoxic or post-ischemic cell death and tissue injury.16,17 We therefore investigated the role of ref-1 in programmed death of endothelial cells induced by H/R (Figure 2A,B). Both uninfected and Adβgal-infected cells were susceptible to H/R-induced apoptosis. This reoxygenation-stimulated apoptosis was suppressed in Adref-1-infected cells in a dose-dependent fashion.
Figure 2
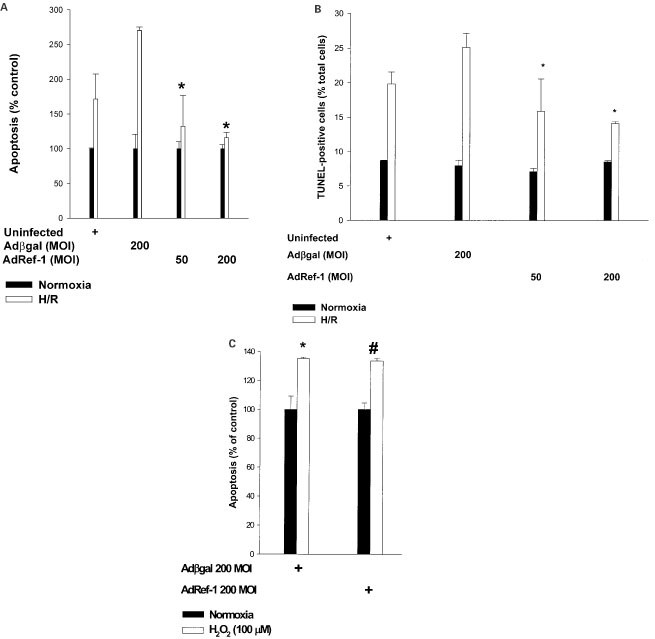
Ref-1 suppresses H/R-induced apoptosis. (A) Quantification of cell death (ELISA for cytoplasmic histone-DNA fragments) in uninfected, Adβgal-, and Adref-1-infected cells under normoxia and with H/R. Values are expressed as percent apoptosis compared to normoxic cells. Hypoxia 6 h, reoxygenation 16 h. *P<0.05 compared with Adβgal. (B) Quantification of apoptosis by TUNEL staining in uninfected, Adβgal-, and Adref-1-infected cells under normoxia and with H/R. *P<0.05 compared with Adβgal. (C) Quantification by ELISA of apoptosis induced by exogenous H2O2 in Adβgal, and Adref-1-infected. cells. Values are expressed as per cent apoptosis in the absence of H2O2. *P<0.05 compared with Adβgal without H2O2. #P=NS compared with Adβgal+H2O2. The figures are representative of two separate experiments
Oxidative stress is an important contributor to reoxygenation or reperfusion-induced apoptosis.16,18,19 Therefore, one mechanism for the above-cited effect could be a change in the cellular anti-oxidant capacity induced by ref-1. To explore this possibility we examined the susceptibility of ref-1 overexpressing cells to H2O2-induced apoptosis, and compared it to cells infected with Adβgal (Figure 2C). A modest dose (100 μM) of H2O2 was used so as not to overwhelm the cell's anti-oxidant capacities. Forced overexpression of ref-1 did not protect against H2O2-induced apoptosis when compared to Adβgal-infected cells. Thus ref-1 does not protect cells from apoptosis resulting from a direct increase in intracellular oxidative stress through addition of exogenous H2O2. This suggests that ref-1 does not affect H2O2 elimination by increasing the expression of cellular anti-oxidant defenses against H2O2 such as catalase and glutathione peroxidase, but rather protects against H/R-induced apoptosis through suppression of intracellular H2O2 production.
Ref-1 inhibits H/R and TNF-induced oxidative stress
Our next goal was to further explore the mechanism behind ref-1's ability to protect against post-hypoxic death. Therefore, we examined the role of ref-1 in regulating the increase in intracellular oxidative stress in response to H/R (Figure 3A). There was no significant difference in intracellular H2O2 levels between uninfected, Adβgal-infected, and Adref-1-infected cells under basal normoxic conditions. H/R resulted in a marked increase in intracellular H2O2 in Adβgal-infected cells. In comparison, cells infected with Adref-1 showed significant reductions in H/R-stimulated intracellular H2O2 levels. This demonstrates that overexpression of ref-1 in endothelial cells suppresses H/R-induced intracellular oxidative stress.
Figure 3
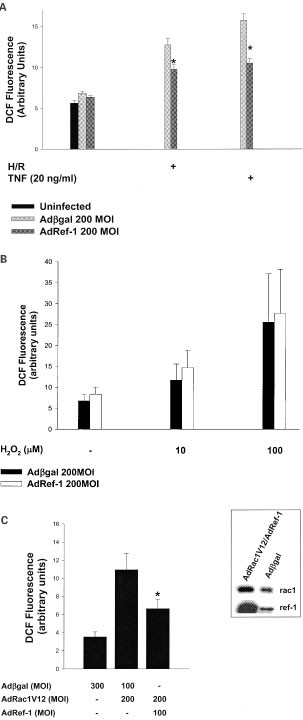
Ref-1 inhibits H/R-induced, and rac1-regulated oxidative stress. (A) Intracellular H2O2 levels in uninfected, Adβgal-, and Adref-1-infected cells, under normoxia, and with H/R and TNF. *P<0.05 compared with Adβgal. Reoxygenation was for 5 min, and TNF 15 min. (B) Intracellular H2O2 levels 5 min after addition of exogenous H2O2 in Adβgal-, and Adref-1-infected cells. (C) Intracellular H2O2 levels in cells co-infected with Adβgal-, Adrac1V12-, and Adref-1. Inset shows adenoviral-induced co-overexpression of ref-1 and rac1V12. The figures are representative of two separate experiments
Next, we determined whether ref-1's ability to suppress the rise in intracellular H2O2 was stimulus-specific. To this end, we examined the effect of ref-1 overexpression on tumor necrosis factor-α (TNF)-induced oxidative stress (Figure 3A). In Adβgal-infected cells TNF resulted in a predictable increase in intracellular H2O2 levels. In comparison, cells infected with Adref-1 demonstrated a significant reduction in TNF-induced rise in H2O2. Thus, ref-1 inhibits a mechanism that leads to an increase in oxidative stress shared by both H/R, and TNF.
Since overexpression of ref-1 did not protect against apoptosis induced by addition of exogenous H2O2, we also determined whether ref-1 had any effect on the elimination of intracellular H2O2 (Figure 3B). Addition of exogenous H2O2 resulted in a dose-dependent increase in intracellular peroxide in control Adβgal-infected cells. Overexpression of ref-1 did not suppress this rise in intracellular H2O2. This further suggests that ref-1 suppresses H/R-induced increase in intracellular H2O2 by decreasing production, and not by altering the cell's capacity to eliminate peroxide.
Both TNF and H/R activate an intracellular oxidase regulated by the ubiquitous small GTPase rac1.3,20 All the components of this multi-molecular NAD(P)H oxidase are expressed in vascular endothelial cells.2 We therefore determined the role of ref-1 in regulating the production of H2O2 by rac1 (Figure 3C). To accomplish this we co-infected cells with Adref-1 and Adrac1V12, an adenovirus encoding the activated allele of rac1. Co-infection with the two viruses resulted in co-expression of ref-1 and rac1V12. Expression of rac1V12 led to an increase in steady state intracellular H2O2 levels that was partly mitigated by overexpression of ref-1. This shows that overexpression of ref-1 suppresses rac1V12-stimulated oxidative stress.
Ref-1 overexpression suppresses H/R-induced, rac1-regulated NF-κB activation
The transcription factor nuclear factor-kappa B (NF-κB) is activated in response to oxidative stimuli in general,21 and H/R or I/R in particular.22,23,24 NF-κB activation has been implicated in a variety of inflammatory conditions including vascular inflammation seen with reperfusion injury.25 We therefore examined the effect of ref-1 overexpression on NF-κB activity (Figure 4A). H/R led to an approximately threefold induction of NF-κB transcriptional activity in both uninfected and Adβgal-infected cells. Infection with Adref-1 resulted in a significant and dose-dependent inhibition of basal, and H/R-induced NF-κB transcriptional activity.
Figure 4
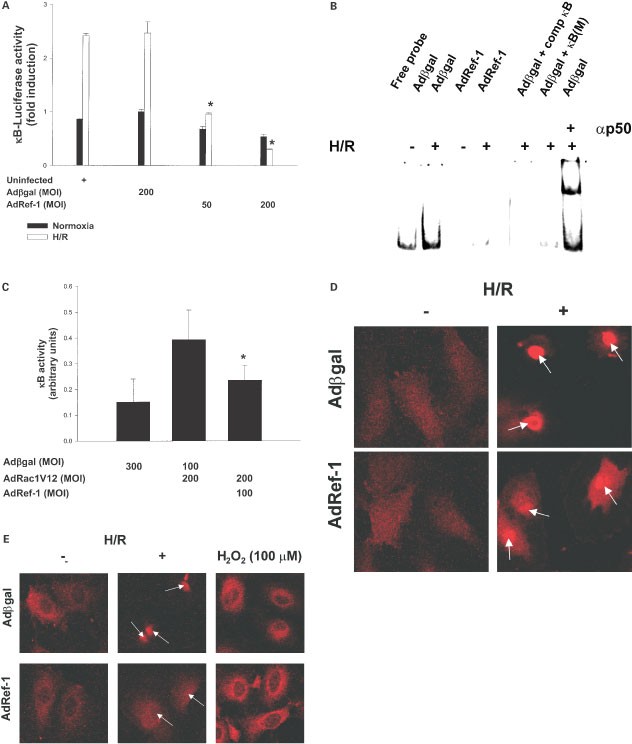
Ref-1 inhibits H/R-induced, rac1-regulated NF-κB activity by inhibiting its nuclear translocation. (A) NF-κB transcriptional activity measured by reporter assay in uninfected, Adβgal-, and Adref-1-infected cells. Values are expressed as fold induction compared with Adβgal-infected cells. *P<0.05 compared with Adβgal. The data is representative of three separate experiments. (B) EMSA for NF-κB DNA binding in Adβgal-, and Adref-1-infected cells. Where indicated, 100× excess unlabelled κB oligonucleotide (comp κB), mutant κB oligonucleotide (κB (M)), or antibody to the p50 subunit of NF-κB (αp50) were added to the binding mixture. The figure is representative of two separate experiments. (C) NF-κB reporter assay in cells co-infected with Adβgal-, Adrac1V12-, and Adref-1. *P<0.05 compared with AdRac1v12/Adβgal. Immunostaining for the p50 (D) and p65 (E) subunits of NF-κB in Adβgal-, and Adref-1-infected cells under normoxia (D,E), after H/R (D,E), and after exogenous H2O2 addition (E). Reoxgenation and H2O2 treatment were for 1 h. Arrows indicate nuclei. The figures are representative of two separate experiments
The findings from the transcriptional reporter assay were confirmed with an electrophoretic mobility shift assay (Figure 4B). H/R led to an increase in NF-κB DNA binding activity in control Adβgal-infected cells. In comparison, cells infected with Adref-1 showed significant suppression of both basal, and H/R-induced NF-κB DNA-binding activity. Supershift analysis demonstrated that the H/R-stimulated increase in DNA binding activity was primarily due to p50 subunit containing dimers. This shows that overexpression of ref-1 inhibits H/R-induced NF-κB activation in endothelial cells.
Activation of rac1 in response to stimuli including cytokines and I/R is essential for full induction of NF-κB transcriptional activity.16,26 We therefore also examined whether ref-1 modulates NF-κB activation specifically induced by rac1 (Figure 4C). Expression of the activated rac1V12 allele resulted in significant increase in NF-κB transcriptional activity that was partly abrogated overexpression of ref-1. This, taken together with the inhibitory effect of ref-1 on rac1-stimulated H2O2 production, further implies that ref-1 specifically inhibits a component(s) of the rac1-regulated machinery that leads to oxidative stress and NF-κB activation.
We also determined the stage in NF-κB activation affected by ref-1. Basal nuclear and cytoplasmic levels of the p50 and p65 subunits of NF-κB and their re-distribution with H/R were assessed by immunostaining (Figure 4D,E). H/R led to nuclear translocation of both p50 and p65 in control Adβgal-infected cells. In contrast, overexpression of ref-1 suppressed the H/R-stimulated nuclear translocation of p50 and p65, when compared to Adβgal-infected cells. This suggests that ref-1 inhibits the H/R-stimulated signaling mechanism(s) that leads to nuclear translocation of NF-κB.
Finally, we determined the effect of ref-1 overexpression on H2O2-stimulated NF-κB re-distribution (Figure 4E). Unlike H/R, addition of exogenous H2O2 to endothelial cells did not lead to nuclear translocation of the p65 subunit. However, H2O2 did result in strong peri-nuclear localization. In comparison with control Adβgal-infected cells, cells overexpressing ref-1 did not show any difference in this H2O2-stimulated re-distribution of NF-κB. This shows that similar to its inability to inhibit exogenous H2O2-induced apoptosis, ref-1 is also unable to affect cellular re-distribution of NF-κB in response to addition of exogenous H2O2.
Discussion
Reports to date have shown that nuclear ref-1 operates via two mechanisms. Through its N-terminus cysteine residues it modulates transcription by reduction of nuclear transcription factors, and its C-terminus functions as an AP endonuclease. In our efforts to examine the role of ref-1 in endothelial cell apoptosis we uncovered that ref-1 is also capable of inhibiting oxidative stress, most likely by modulating H2O2 production, a mechanism hitherto unappreciated. This conclusion is supported by the following findings: (1) overexpression of ref-1 resulted in suppression of H/R and TNF-stimulated, and rac1-induced rise in intracellular H2O2, and (2) overexpression of ref-1 did not affect the elimination of exogenously supplied H2O2.
The ability of ref-1 to inhibit stimulus-induced oxidative stress provides an additional mechanism for its protective effect against various pro-apoptotic oxidative stimuli. However, it is also possible that the DNA repair and transcriptional-enhancing properties of ref-1 play a role in suppressing cell death. Regardless of the precise mechanism(s) through which ref-1 inhibits apoptosis, its ability to regulate the production of cytoplasmic H2O2 can only be explained by an extra-nuclear function.
Ref-1 was first isolated from nuclear fractions of HeLa cells.27 Although ref-1 does have a nuclear localizing signal at its N-terminus, in vitro studies have also demonstrated localization to the cytoplasm.13 Moreover, numerous studies show that in vivo, ref-1 has a differential cellular and subcellular expression pattern28,29,30,31 suggesting a potential physiologic extra-nuclear role. Using an overexpression strategy we were able to uncover one such extra-nuclear function. In endothelial cells, the oxidative stimulus H/R led to a shift in the sub-cellular distribution of ref-1. This re-distribution is in accordance with another report showing that exogenous H2O2 is a potent stimulus for cytoplasmic to nuclear translocation of ref-1.32 In contrast, adenoviral-driven change in nuclear/cytoplasmic ref-1 under normoxia, and particularly after H/R, was associated with suppression of oxidative stress, and NF-κB translocation. It is worth pointing out that Adref-1 did lead to a marked increase in nuclear ref-1. However, this did not translate to an increase in H/R-induced NF-κB DNA binding or transcriptional activity. This is explained by the temporal sequence of NF-κB activation. DNA binding and transcription occur after, and are dependent upon, nuclear translocation. In addition, although this report focuses on ref-1's regulation of oxidative stress as a mechanism for it's effect on NF-κB activation, it is worth considering that cytoplasmic ref-1 may also, directly or indirectly, affect other steps in the NF-κB activation sequence such as the activity of IκB kinases, or binding affinity to IκB.
Arguably, the most intriguing finding of this study is that ref-1 suppresses rac1-induced oxidative stress and NF-κB activation. It is important to emphasize that our data show a functional interaction between ref-1 and rac1, and do not suggest a direct association between these two proteins. Nonetheless, the observation that both TNF and H/R-induced oxidative stress was inhibited by ref-1 is also consistent with such a functional interaction, since both these oxidative stimuli lead to ROS production, at least partly through a rac1-regulated NAD(P)H oxidase.3,20 Importantly, all the components of this oxidase are expressed in endothelial cells. In this regard, it is noteworthy that ref-1 physically binds with thioredoxin (TRX), another redox-regulatory protein.33 TRX is capable of binding to p40phox, a component of the phagocyte cytoplasmic NADPH oxidase, and critical thiol residues on TRX are necessary for this interaction.34 This raises the interesting possibility that cytoplasmic ref-1 via its critical cysteines, alone or in concert with TRX, may also modulate the activity, of one or more components of a rac1-regulated NAD(P)H-dependent oxidase expressed in endothelial cells. However, it would also be fair to say that the effect of ref-1 in modulating rac1-regulated H2O2 production does not exclude other mechanisms by which it may inhibit cytoplasmic oxidative stress. Notably, emerging evidence suggests that ref-1 is also localized to the mitochondria,35,36 a major site of production of H2O2 and other ROS that have been implicated in apoptosis. Furthermore, independent of oxidant production, extra-nuclear ref-1 may also participate in other important cellular functions and processes (reviewed in37,38,39,40,41,42).
Ref-1 activates p53 to bind to DNA,10 and promotes the _trans_-activating and pro-apoptotic functions of p53.43 At first, this seems to contradict our observation that it has a protective role in H/R-induced apoptosis. However, most in vitro and in vivo studies have shown that hypoxia, and H/R-induced apoptosis occur independent of p53 status.17,44 Moreover, prior studies examining the role of ref-1 in apoptosis have not distinguished between cytoplasmic and nuclear expression. Thus the manner in which ref-1 modulates apoptotic death may depend on its cellular compartmentalization and be stimulus-specific.
A recent report examined the role of ref-1 in endothelial cell NF-κB activity and apoptosis.45 This report showed that total cellular ref-1 decreased after severe (18 h) of hypoxia, and that overexpression of ref-1 (using a liposome-based transfection protocol) resulted in an increase in NF-κB activation, and suppression of and TNF/hypoxia-induced apoptosis. However, the subcellular distribution of exogenous and endogenous ref-1, with or without oxidative stimuli (hypoxia or TNF), was not examined by the authors. Nor did the authors attempt to determine whether the degree of ref-1 down-regulation or overexpression correlates with the extent of oxidative stress resulting from TNF or hypoxia. Our novel observations that forced adenoviral-mediated overexpression of ref-1 leads to (1) suppression of oxidative stress, and (2) inhibition of NF-κB activation add new insights into the role of ref-1 in endothelial cell biology, and are in contradiction with the findings of the above-mentioned report. The different conclusions of the two studies may be a result of differences in methodological approaches to overexpression coupled with differences in the degree of scrutiny regarding the sub-cellular compartmentalization of ref-1 in endothelial cells.
In summary, this report shows that ref-1 has an important function in suppressing intracellular oxidative stress and apoptosis in endothelial cells, through modulation of a rac1-regulated oxidase, implicating an extra-nuclear function of ref-1 (Figure 5). Similar observations in an animal model of hepatic ischemia/reperfusion injury (data not shown) suggest that this extra-nuclear function may not be limited to endothelial cells. Moreover, taken in conjunction with other reports, our data suggests a paradigm in which ref-1 plays dual and opposing roles in the regulation of NF-κB activation. Cytoplasmic ref-1, through the feedback mechanism resulting in decreased ROS (H2O2) production, inhibits rac1-stimulated, redox-regulated nuclear translocation of NF-κB. Once in the nucleus, by maintaining NF-κB in the reduced state, and thereby enhancing its ability to bind DNA, ref-1 promotes κB-driven transcription. Therefore levels of nuclear versus cytoplasmic ref-1, and the ability of the cell to regulate these levels in response to oxidative stimuli, may well determine its net effect on NF-κB activity. Importantly, a similar dual function of TRX in NF-κB and heat shock factor activation has been shown.46,47 Thus, such spatial and temporal regulation of transcription factor activity by redox proteins may represent a common theme in the cellular response to oxidative stimuli.
Figure 5
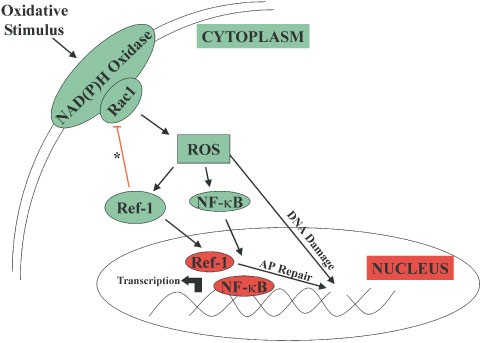
Nuclear and Cytoplasmic Roles of ref-1. Summary diagram illustrating the cytolasmic and nuclear effects of ref-1. *Signifies the role of ref-1 in suppressing rac1-regulated ROS production demonstrated in this study. Figure does not include other potential mechanisms by which ref-1 may suppress NF-κB activation, nor does it include other potential extra-nuclear actions of ref-1. AP: Apurinic/Apyrimidinic
Materials and Methods
Cell culture
Human umbilical vein endothelial cells (HUVECs) were purchased from Clonetics (San Diego, CA, USA) and were grown and maintained in endothelial growth medium. Cells were passaged by standard trypsinization, and passages 2–7 were used in all experiments.
Hypoxia/reoxygenation and quantification of apoptosis
Hypoxia (6 h) and reoxygenation were attained as previously described48 in a hypoxia chamber (Billups-Rothenberg, Inc., Del Mar, CA, USA). Apoptosis was quantified according to a previously described method3 using an ELISA that detects cytoplasmic histone-DNA fragments (Cell Death Detection ELISA, Boehringer Mannheim).
Adenoviruses
Recombinant replication-deficient adenoviruses were used in all experiments. Adref-1 encoding the ref-1 cDNA was constructed by homologous recombination in HEK293 cells. Adβgal, encoding the inert E. Coli lacZ gene, and Adrac1V12, encoding the constitutively active rac1 GTPase, have been described previously.3 Adβgal-infected cells, or uninfected cells were used as controls in all experiments. Recombinant adenoviruses were propagated, purified, and tittered, and HUVECs were infected at the specified multiplicity of infection (MOI) as previously described.3
Western blotting and immunostaining
Nuclear and cytoplasmic protein was extracted and fractionated as previously described.49 Western blots were performed with an antibody against ref-1 (SC-334, Santa Cruz Biotechnology), or rac1 (23A8, UBI) and bound immunocomplexes were visualized by enhanced chemiluminescence (Amersham). In some experiments, mouse monoclonal antibody against α-tubulin (DM1A, Sigma) was used to confirm equal protein loading. Immunostaining employed primary antibodies against ref-1, p50 (SC-7178, Santa Cruz Biotechnology), or p65 (SC-372, Santa Cruz Biotechnology), and secondary fluorescein-tagged antibody that were used according to the manufacturer's recommendations. Images were captured on a Zeiss confocal laser-scanning microscope.
Measurement of intracellular reactive oxygen species
Intracellular H2O2 was detected with the peroxide-sensitive fluorophore 2′,7′-dichlorodihydrofluorescein diacetate (DCF-DA) as previously described.3 The fluorescence was measured after 5 min of incubation with DCF using excitation and emission wavelengths of 488 and 515 nm, respectively. Absolute fluorescence of 20–25 random cells was quantified with MetaMorph™ software. Values are mean±S.D.
NF-κB reporter assay
HUVECs were co-transfected with 1.5 μg of a luciferase or CAT reporter plasmid containing three κB binding sites, and 0.75 ug of constitutive CAT or luciferase expression plasmid, using LipofectAMINETM reagent (Life Technologies) according to the manufacturer s instructions. After 16 h of reoxygenation luciferase and CAT activities in cell lysates were determined and normalized against each other to account for variations in transfection efficiency. All conditions were performed in triplicate and values are mean±S.D.
Electrophoretic mobility shift assay (EMSA)
EMSA for NF-κB DNA-binding activity 2 h after reoxygenation were carried out as previously described3 using a κB consensus sequence oligonucleotide (5′-AGT TGA GGG GAC TTT CCC AGGC-3′). In supershift experiments, antibody to p50 subunit was added to nuclear extracts for 30 min on ice prior to addition of labeled oligonucleotide.
Statistical analysis
Data are expressed as mean±S.D. Differences in measured variables between experimental and control groups were determined using a two-tailed paired _t_-test. P<0.05 was considered to be statistically significant.
Abbreviations
HUVECs:
human umbilical vein endothelial cells
H/R:
hypoxia/reoxygenation
NF-KB:
nuclear factor-kappa B
ROS:
reactive oxygen species
ref-1:
redox factor-1
redox:
reduction-oxidation
References
- Nakamura H, Nakamura K, Yodoi J . 1997 Redox regulation of cellular activation Annu. Rev. Immunol. 15: 351–369
Article CAS Google Scholar - Gorlach A, Brandes RP, Nguyen K, Amidi M, Dehghani F, Busse R . 2000 A gp91phox containing NADPH oxidase selectively expressed in endothelial cells is a major source of oxygen radical generation in the arterial wall [see comments] Circ. Res. 87: 26–32
Article CAS Google Scholar - Deshpande SS, Angkeow P, Huang J, Ozaki M, Irani K . 2000 Rac1 inhibits TNF-alpha-induced endothelial cell apoptosis: dual regulation by reactive oxygen species FASEB J. 14: 1705–1714
Article CAS Google Scholar - Kim KS, Takeda K, Sethi R, Pracyk JB, Tanaka K, Zhou YF, Yu ZX, Ferrans VJ, Bruder JT, Kovesdi I, Irani K, Goldschmidt-Clermont P, Finkel T . 1998 Protection from reoxygenation injury by inhibition of rac1 J. Clin. Invest. 101: 1821–1826
Article CAS Google Scholar - Abate C, Patel L, Rauscher FJD, Curran T . 1990 Redox regulation of fos and jun DNA-binding activity in vitro Science 249: 1157–1161
Article CAS Google Scholar - Xanthoudakis S, Miao G, Wang F, Pan YC, Curran T . 1992 Redox activation of Fos-Jun DNA binding activity is mediated by a DNA repair enzyme EMBO J. 11: 3323–3335
Article CAS Google Scholar - Walker LJ, Robson CN, Black E, Gillespie D, Hickson ID . 1993 Identification of residues in the human DNA repair enzyme HAP1 (Ref-1) that are essential for redox regulation of Jun DNA binding Mol. Cell. Biol. 13: 5370–5376
Article CAS Google Scholar - Huang RP, Adamson ED . 1993 Characterization of the DNA-binding properties of the early growth response-1 (Egr-1) transcription factor: evidence for modulation by a redox mechanism Dna And Cell Biology 12: 265–273
Article CAS Google Scholar - Huang LE, Arany Z, Livingston DM, Bunn HF . 1996 Activation of hypoxia-inducible transcription factor depends primarily upon redox-sensitive stabilization of its alpha subunit J. Biol. Chem. 271: 32253–32259
Article CAS Google Scholar - Jayaraman L, Murthy KG, Zhu C, Curran T, Xanthoudakis S, Prives C . 1997 Identification of redox/repair protein Ref-1 as a potent activator of p53 Genes Dev. 11: 558–570
Article CAS Google Scholar - Mitomo K, Nakayama K, Fujimoto K, Sun X, Seki S, Yamamoto K . 1994 Two different cellular redox systems regulate the DNA-binding activity of the p50 subunit of NF-kappa B in vitro Gene 145: 197–203
Article CAS Google Scholar - Tell G, Pellizzari L, Cimarosti D, Pucillo C, Damante G . 1998 Ref-1 controls pax-8 DNA-binding activity Biochem. Biophys. Res. Commun. 252: 178–183
Article CAS Google Scholar - Demple B, Herman T, Chen DS . 1991 Cloning and expression of APE, the cDNA encoding the major human apurinic endonuclease: definition of a family of DNA repair enzymes Proc. Natl. Acad. Sci. USA 88: 11450–11454
Article CAS Google Scholar - Walker LJ, Craig RB, Harris AL, Hickson ID . 1994 A role for the human DNA repair enzyme HAP1 in cellular protection against DNA damaging agents and hypoxic stress Nucleic Acids Res. 22: 4884–4889
Article CAS Google Scholar - Walton M, Lawlor P, Sirimanne E, Williams C, Gluckman P, Dragunow M . 1997 Loss of Ref-1 protein expression precedes DNA fragmentation in apoptotic neurons Brain Res. Mol. Brain Res. 44: 167–170
Article CAS Google Scholar - Ozaki M, Deshpande SS, Angkeow P, Bellan J, Lowenstein CJ, Dinauer MC, Goldschmidt-Clermont PJ, Irani K . 2000 Inhibition of the Rac1 GTPase protects against nonlethal ischemia/reperfusion-induced necrosis and apoptosis in vivo FASEB J. 14: 418–429
Article CAS Google Scholar - Webster KA, Discher DJ, Kaiser S, Hernandez O, Sato B, Bishopric NH . 1999 Hypoxia-activated apoptosis of cardiac myocytes requires reoxygenation or a pH shift and is independent of p53 J. Clin. Invest. 104: 239–252
Article CAS Google Scholar - Cho KS, Lee EH, Choi JS, Joo CK . 1999 Reactive oxygen species-induced apoptosis and necrosis in bovine corneal endothelial cells Invest Ophthalmol Vis. Sci. 40: 911–919
CAS PubMed Google Scholar - Zulueta JJ, Sawhney R, Yu FS, Cote CC, Hassoun PM . 1997 Intracellular generation of reactive oxygen species in endothelial cells exposed to anoxia-reoxygenation Am. J. Physiol. 272: L897–L902
CAS PubMed Google Scholar - Pracyk JB, Tanaka K, Hegland DD, Kim KS, Sethi R, Rovira II, Blazina DR, Lee L, Bruder JT, Kovesdi I, Goldschmidt-Clermont PJ, Irani K, Finkel T . 1998 A requirement for the rac1 GTPase in the signal transduction pathway leading to cardiac myocyte hypertrophy J. Clin. Invest. 102: 929–937
Article CAS Google Scholar - Muller JM, Rupec RA, Baeuerle PA . 1997 Study of gene regulation by NF-kappa B and AP-1 in response to reactive oxygen intermediates Methods 11: 301–312
Article CAS Google Scholar - Zwacka RM, Zhang Y, Zhou W, Halldorson J, Engelhardt JF . 1998 Ischemia/reperfusion injury in the liver of BALB/c mice activates AP-1 and nuclear factor kappaB independently of IkappaB degradation Hepatology 28: 1022–1030
Article CAS Google Scholar - Zwacka RM, Zhou W, Zhang Y, Darby CJ, Dudus L, Halldorson J, Oberley L, Engelhardt JF . 1998 Redox gene therapy for ischemia/reperfusion injury of the liver reduces AP1 and NF-kappaB activation Nat. Med. 4: 698–704
Article CAS Google Scholar - Imbert V, Rupec RA, Livolsi A, Pahl HL, Traenckner EB, Mueller-Dieckmann C, Farahifar D, Rossi B, Auberger P, Baeuerle PA, Peyron JF . 1996 Tyrosine phosphorylation of I kappa B-alpha activates NF-kappa B without proteolytic degradation of I kappa B-alpha Cell 86: 787–798
Article CAS Google Scholar - Schneider A, Martin-Villalba A, Weih F, Vogel J, Wirth T, Schwaninger M . 1999 NF-kappaB is activated and promotes cell death in focal cerebral ischemia Nat. Med. 5: 554–559
Article CAS Google Scholar - Sulciner DJ, Irani K, Yu ZX, Ferrans VJ, Goldschmidt-Clermont P, Finkel T . 1996 rac1 regulates a cytokine-stimulated, redox-dependent pathway necessary for NF-kappaB activation Mol. Cell Biol. 16: 7115–7121
Article CAS Google Scholar - Xanthoudakis S, Curran T . 1992 Identification and characterization of Ref-1, a nuclear protein that facilitates AP-1 DNA-binding activity EMBO J. 11: 653–665
Article CAS Google Scholar - Rivkees SA, Kelley MR . 1994 Expression of a multifunctional DNA repair enzyme gene, apurinic/apyrimidinic endonuclease (APE; Ref-1) in the suprachiasmatic, supraoptic and paraventricular nuclei Brain Res. 666: 137–142
Article CAS Google Scholar - Wilson TM, Rivkees SA, Deutsch WA, Kelley MR . 1996 Differential expression of the apurinic/apyrimidinic endonuclease (APE/ref-1) multifunctional DNA base excision repair gene during fetal development and in adult rat brain and testis Mutat Res. 362: 237–248
Article Google Scholar - Kakolyris S, Kaklamanis L, Engels K, Fox SB, Taylor M, Hickson ID, Gatter KC, Harris AL . 1998 Expression and subcellular localization of human AP endonuclease 1 (HAP1/Ref-1) protein: a basis for its role in human disease Histopathology 33: 561–569
Article CAS Google Scholar - Duguid JR, Eble JN, Wilson TM, Kelley MR . 1995 Differential cellular and subcellular expression of the human multifunctional apurinic/apyrimidinic endonuclease (APE/ref-1) DNA repair enzyme Cancer Res. 55: 6097–6102
CAS PubMed Google Scholar - Tell G, Zecca A, Pellizzari L, Spessotto P, Colombatti A, Kelley MR, Damante G, Pucillo C . 2000 An ‘environment to nucleus’ signaling system operates in B lymphocytes: redox status modulates BSAP/Pax-5 activation through Ref-1 nuclear translocation Nucleic Acids Res. 28: 1099–1105
Article CAS Google Scholar - Hirota K, Matsui M, Iwata S, Nishiyama A, Mori K, Yodoi J . 1997 AP-1 transcriptional activity is regulated by a direct association between thioredoxin and Ref-1 Proc. Natl. Acad. Sci. USA 94: 3633–3638
Article CAS Google Scholar - Nishiyama A, Ohno T, Iwata S, Matsui M, Hirota K, Masutani H, Nakamura H, Yodoi J . 1999 Demonstration of the interaction of thioredoxin with p40phox, a phagocyte oxidase component, using a yeast two-hybrid system Immunol. Lett. 68: 155–159
Article CAS Google Scholar - Tell G, Crivellato E, Pines A, Paron I, Pucillo C, Manzini G, Bandiera A, Kelley MR, Di Loreto C, Damante G . 2001 Mitochondrial localization of APE/Ref-1 in thyroid cells Mutat. Res. 485: 143–152
Article CAS Google Scholar - Fung H, Kow YW, Van Houten B, Taatjes DJ, Hatahet Z, Janssen YM, Vacek P, Faux SP, Mossman BT . 1998 Asbestos increases mammalian AP-endonuclease gene expression, protein levels, and enzyme activity in mesothelial cells Cancer Res. 58: 189–194
CAS PubMed Google Scholar - Wilson III DM, Barsky D . 2001 The major human abasic endonuclease: formation, consequences and repair of abasic lesions in DNA Mutat Res. 485: 283–307
Article CAS Google Scholar - Chiarini LB, Linden R . 2000 Tissue biology of apoptosis. Ref-1 and cell differentiation in the developing retina Ann. NY Acad. Sci. 926: 64–78
Article CAS Google Scholar - Fritz G . 2000 Human APE/Ref-1 protein Int. J. Biochem. Cell Biol. 32: 925–929
Article CAS Google Scholar - Evans AR, Limp-Foster M, Kelley MR . 2000 Going APE over ref-1 Mutat. Res. 461: 83–108
Article CAS Google Scholar - Arner ES, Holmgren A . 2000 Physiological functions of thioredoxin and thioredoxin reductase Eur. J. Biochem. 267: 6102–6109
Article CAS Google Scholar - Mol CD, Hosfield DJ, Tainer JA . 2000 Abasic site recognition by two apurinic/apyrimidinic endonuclease families in DNA base excision repair: the 3′ ends justify the means Mutat. Res. 460: 211–229
Article CAS Google Scholar - Gaiddon C, Moorthy NC, Prives C . 1999 Ref-1 regulates the transactivation and pro-apoptotic functions of p53 in vivo EMBO J. 18: 5609–5621
Article CAS Google Scholar - Bialik S, Geenen DL, Sasson IE, Cheng R, Horner JW, Evans SM, Lord EM, Koch CJ, Kitsis RN . 1997 Myocyte apoptosis during acute myocardial infarction in the mouse localizes to hypoxic regions but occurs independently of p53 J. Clin. Invest. 100: 1363–1372
Article CAS Google Scholar - Hall JL, Wang X, Van A, Zhao Y, Gibbons GH . 2001 Overexpression of Ref-1 inhibits hypoxia and tumor necrosis factor-induced endothelial cell apoptosis through nuclear factor-kappab-independent and -dependent pathways Circ. Res. 88: 1247–1253
Article CAS Google Scholar - Hirota K, Murata M, Sachi Y, Nakamura H, Takeuchi J, Mori K, Yodoi J . 1999 Distinct roles of thioredoxin in the cytoplasm and in the nucleus. A two-step mechanism of redox regulation of transcription factor NF- kappaB J. Biol. Chem. 274: 27891–27897
Article CAS Google Scholar - Jacquier-Sarlin MR, Polla BS . 1996 Dual regulation of heat-shock transcription factor (HSF) activation and DNA-binding activity by H2O2: role of thioredoxin Biochem. J. 318: 187–193
Article CAS Google Scholar - Ozaki M, Deshpande SS, Angkeow P, Suzuki S, Irani K . 2000 Rac1 regulates stress-induced, redox-dependent heat shock factor activation J. Biol. Chem. 275: 35377–35383
Article CAS Google Scholar - Andrews NC, Faller DV . 1991 A rapid micropreparation technique for extraction of DNA-binding proteins from limiting numbers of mammalian cells Nucleic Acids Res. 19: 2499
Article CAS Google Scholar
Acknowledgements
This work was supported by a NIH grant P01 HL65608 (K Irani), the W.W. Smith Charitable Trust (K Irani), the American Heart Association (K Irani), the Bernard Foundation, the Abraham and Virginia Weiss Endowment, and a grant from the Cardio Fellows Foundation (P Angkeow). The authors wish to thank T Finkel, and T Curran for materials and advice.
Author information
Author notes
- P Angkeow and S S Deshpande: These authors contributed equally to this work
Authors and Affiliations
- Division of Cardiology, Department of Medicine, The Johns Hopkins University School of Medicine, Baltimore, MD, USA
P Angkeow, S S Deshpande, B Qi, Y-X Liu, Y C Park, B H Jeon & K Irani - Department of Experimental Surgery and Bioengineering, National Children's Hospital, Tokyo, Japan
M Ozaki
Authors
- P Angkeow
- S S Deshpande
- B Qi
- Y-X Liu
- Y C Park
- B H Jeon
- M Ozaki
- K Irani
Corresponding author
Correspondence toK Irani.
Additional information
Edited by B Osborne
Rights and permissions
About this article
Cite this article
Angkeow, P., Deshpande, S., Qi, B. et al. Redox factor-1: an extra-nuclear role in the regulation of endothelial oxidative stress and apoptosis.Cell Death Differ 9, 717–725 (2002). https://doi.org/10.1038/sj.cdd.4401025
- Received: 06 August 2001
- Revised: 05 January 2002
- Accepted: 12 January 2002
- Published: 12 June 2002
- Issue Date: 01 July 2002
- DOI: https://doi.org/10.1038/sj.cdd.4401025