Diagnostic investigations in individuals with mental retardation: a systematic literature review of their usefulness (original) (raw)
Introduction
Background
Mental retardation (MR) is a frequently occurring disorder with a major impact on the life of the affected person, the family, and society. Establishing an aetiologic diagnosis is usually a challenge for every specialist involved, as the spectrum of possible underlying disorders is enormous and the range of available additional investigations extensive. Still the sheer knowing of the cause, the recurrence risk, the short-term and long-term prognosis, treatment options, availability of special services, contacts with other parents of children, and other issues are of great importance to parents, and often also forms the first step towards acceptance of the disability. Furthermore, the costs of a complete diagnostic work-up in a child with MR are considerable, and can be a major burden to many health care systems. This obliges clinicians to reconsider the usefulness of every diagnostic investigation.
The ability to determine a cause of MR is based largely on the use of specific diagnostic tools. In a given diagnostic setting, the physician depends on their availability and guidelines for application. Such guidelines should be established in an evidence-based manner, that is, based on information from original empirical studies on quality, yield, and usefulness of the diagnostic investigations. Until now, available guidelines have been based foremost on expert opinion,1 with one recent exception, which became available after the present study was completed.2
Aims of the review
We initiated a systematic search for and analysis of all papers published in peer-reviewed journals in seven different languages during the last 35 years, reporting the application of one or more of the following major diagnostic investigations: dysmorphologic examination, neurologic examination, neuroimaging, cytogenetic investigations (routine karyotyping and subtelomeric FISH analysis), fragile X screening, metabolic investigations. An additional goal was to investigate whether the yield differed depending on (1) setting (institution, outpatient clinic, school, population survey), (2) severity of MR, and (3) gender.
We have chosen these six investigations because (1) these are the most frequently applied, (2) these investigations have been applied in numerous studies in various populations and in various settings, and (3) each of the investigations may yield information sufficient for establishing an aetiologic diagnosis. This is rarely the case in other investigations such as ophthalmologic or electrophysiologic investigations, which we have excluded from this review.
Presentation of results
The different parts of this systematic literature review use the same methodology, independent of the diagnostic technique under study. This includes definitions, search strategy, selection criteria, the yield of the search strategy, selection procedure of studies using the Quorum flow diagram,3 data extraction and analyses, study quality assessment, and statistical analyses.
When reading and interpreting this review, it is important to note that inclusion or exclusion was dependent first and foremost on the availability in the paper of quantitative data on the accuracy and yield of diagnostic techniques in patient groups with MR. We are aware of the fact that potentially valuable information on other aspects of MR aetiology and management is not included in the present review due to our focused study aims. Furthermore, the choice of diagnostic techniques does not indicate that other investigations in persons with MR are useless. An example may be that in our opinion every retarded child needs to be regularly checked for visual and auditive abilities, as disturbances may have a significant impact on the total functioning of a child.
Methods
This systematic review was designed according to the Cochrane Reviewers Handbook version 4. 1.4.4 All consecutive steps and phases of the review are depicted in Figure 1.
Figure 1
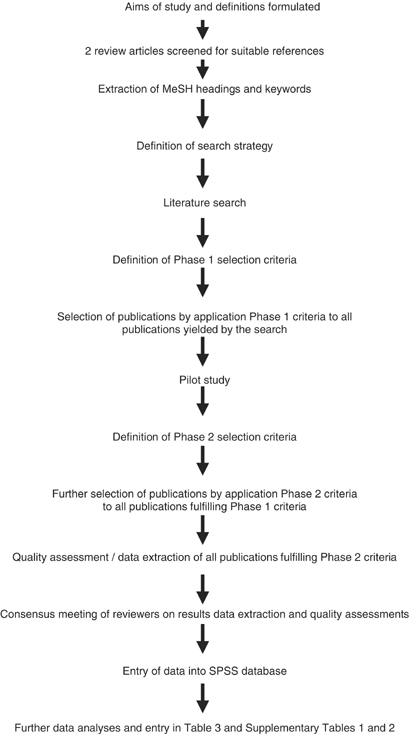
Flow chart of consecutive methodologic steps of systematic review. All steps were performed by two independent reviewers.
Definitions
Prior to designing the search strategy, the following definitions were formulated:
MR: The definition of MR of the American Association on Mental Retardation was used:5 MR refers to substantial limitations in present functioning. It is characterised by significantly subaverage intellectual functioning, existing concurrently with related limitations in two or more of the following applicable adaptive skills: communication, self-care, home living, social skills, community use, self-direction, health and safety, functional academics, leisure, and work. MR manifests before the age of 18.
Severity of MR: This was categorised according to the World Health Organization classification6 and DSM-IV criteria:7 profound (IQ=0–20); severe (IQ=21–35); moderate (IQ=36–50); mild (IQ=51–70); borderline (IQ=71–85).
Investigations: Diagnostic investigations used to discern the aetiology of MR were: (1) dysmorphologic exam: physical examination focused on the detection of dysmorphic features, minor anomalies, and malformations; (2) neurologic exam: physical exam focused on detection of neurologic abnormalities; (3) metabolic studies: standard 24 h urinary screenings of amino acids, organic acids, oligosaccharides, acid mucopolysaccharides, and uric acid; (4) cytogenetics: high-resolution G-banded karyogram screening for numerical and structural chromosome anomalies (minimal banding quality 350–400 bands),8 and FISH analysis screening for subtelomeric rearrangements;9 (5) fragile X screening: cytogenetic screening on chromosomes prepared using medium 199 for fragile sites in region Xq27.3,10 or molecular screening of the FMR-1 gene for CGG expansions;11 (6) Neuroradiologic studies: screening for intracranial abnormalities by magnetic resonance imaging (MRI) scan, computer tomography (CT) scan, and echo cerebrum.
Aetiologic diagnosis: A disorder was considered an aetiologic diagnosis if there was sufficient literature evidence external to this review to make a causal relationship of the disorder with MR likely, and if it met the Schaefer–Bodensteiner standard (‘a specific diagnosis that can be translated into useful clinical information for the family, including providing information about prognosis, recurrence risk, and preferred modes of available therapy’).12
Search strategy
The search strategy was based on two recent reviews of the diagnostic process in individuals with MR.1,13 Two investigators (CDMvK, RCMH) independently screened the bibliographies of these two reports for references of articles possibly suitable for this review. These articles were then retrieved and their MeSH headings and textual keywords were subsequently used to set up the search strategy by two independent reviewers (AGEL, CDMvK). Publications were retrieved by a computerised search (using OVID) of MEDLINE (1966–June 2002), EMBASE (1983–June, 2002), Cochrane Database of Systematic Reviews and Controlled Clinical Trials (issue of the first quarter, 2002), Best Evidence Database (1991–June, 2002), using the following keywords: mental retardation, learning disorders, developmental disabilities, mass screening, cohort studies, case–control, retrospective studies, prospective studies. For the specific diagnostic investigations, the following keywords were used: neurologic abnormalities, chromosome abnormalities, metabolic diseases, tomography, mutations. For dysmorphologic examination, two different search strategies were performed: one, using as keywords ‘syndromes’ and ‘multiple congenital anomalies’, and a second using as keywords the terms of the 17 most frequent syndromes:14 Down syndrome, trisomy chromosome 8, trisomy chromosome 13, trisomy chromosome 18, deletion chromosome 18p, Angelman syndrome, Bardet–Biedl syndrome, Cohen syndrome, Cornelia de Lange syndrome, Cri-du-Chat syndrome, fetal alcohol syndrome, fragile X syndrome, Prader–Willi syndrome, Smith–Lemli–Opitz syndrome, Sotos syndrome, Williams syndrome, Wolf–Hirschhorn syndrome.
The references of all identified relevant studies were hand searched for additional potentially relevant publications (CDMvK).
Selection criteria
The selection was performed by two independent reviewers (CDMvK, RCMH) in two consecutive phases. The selection criteria listed in Table 2 were applied to the titles and abstracts of publications. After a pilot study, more strict criteria were formulated and were applied during a second phase to articles fulfilling the first-phase criteria. Reasons for exclusion of articles during phases 1 and 2 are listed in Table 3. Only for population surveys, less strict criteria regarding the description of the severity of MR and of MR assessment methods were applied, as the large numbers of patients in the study groups hampered an exact description of all these items. For inclusion in the review, a reasonable certainty was needed that all included patients were indeed mentally delayed, next to the general criteria.
Table 1 Overview of the databases and MeSH headings used in the computerised literature searches, and the strategy and yield of the each search
Table 2 Criteria applied for the selection of articles for this review
Table 3 Number and reasons for exclusion from the systematic review of publications on diagnostic investigations in patients with MR
Studies describing comprehensive diagnostic evaluations of patients potentially have great value, but also have the drawback that the description of the number of patients in whom a specific investigation technique is performed is often lacking. Although it seemed often likely that each technique was performed in all patients, it cannot be derived from most publications with certainty. This prohibits accurate calculation of frequency of anomalies found with each of the diagnostic techniques. Therefore, such comprehensive studies were not included in the present review, unless reliable figures regarding the number of patients who underwent the individual studies were available.
Only FISH studies screening for subtelomeric rearrangements were included. Information yielded by studies applying FISH to screen for specific interstitial microdeletions, such as a multi-FISH study for five different microdeletion syndromes,15 are not reported here. The various microdeletion syndromes each provide a distinct phenotype and this phenotype will urge the specific FISH study. These FISH studies will not be used for screening in an unselected group of patients with MR. Conversely, publications that report screening for subtelomeric rearrangements by other techniques than FISH analysis (eg automated fluorescent genotyping16 or comparative genomic hybridisation17) were also excluded. The yield of these different, only recently developed techniques is not yet well comparable and results of different studies cannot be pooled.
Molecular screening was narrowed down to FraX mutation screening. As a result, useful data on the yield of screening for other syndromes, such as methylation analysis of the Prader-Willi/Angelman region in patients with a suggestive phenotype,18 are not reported here. As stated above for specific FISH studies, it was our primary goal to assess the yield of diagnostic investigations in individuals with unexplained MR, and not in those with phenotypic features suggestive of a particular underlying disorder. As the phenotype in fragile X may be unspecific, and results of searches for fragile X syndrome are available from a number of studies of persons with idiopathic MR,19 we have chosen to allow inclusion of this specific molecular investigation technique.
Pilot study
To test the applicability of the standard forms for data extraction and quality assessment (see below), a pilot study using eight articles was performed.20,21,22,23,24,25,26,27The sample was selected so that it encompassed studies in different settings, applied (a combination of) different diagnostic investigations, and comprised articles published in different periods of time between 1966 and 2002. Three reviewers (CDMvK, MCEJ, RCMH) independently evaluated the papers using standard forms. In a subsequent consensus meeting, points of disagreement were discussed and, if needed, the form was adapted to comments.
Data extraction
For each article, a standard form was used to extract details on study design, study population, applied diagnostic investigations, and outcome (yield of techniques; number of established a etiological diagnoses). If important data in a publication were lacking, but in the publication the reader was referred to another paper for these data, then this reference was retrieved and used in evaluating the original paper, even if the reference did not fulfil the selection criteria for this review. In evaluating the methodological qualities however, only the original paper was scored as if this additional information was not available.
Data in articles from earlier investigations, that is, not performed for the purpose of the empirical study, were only included if they had sufficient details and were part of a larger series of results of investigations, performed indeed for the purpose of the study.
Correct categorisation of the grade of MR reported for a study group in an article was sometimes hampered by the use of classification systems that did not coincide with the DSM-IV system used throughout this review. A given study may define ‘mild MR’ as IQ 55–69 and ‘moderate MR’ as IQ 40–54, while the DMS-IV definitions use 50–70 and 35–50, respectively. This made it impossible to determine how many patients in fact did have mild and moderate IQ conforming to the DSM-IV definitions, and prevented determining any meaningful relation between the grade of MR and other variables. As studies may otherwise be useful, and loss of the results only on this ground was not desirable, we reinterpreted the data through the best-possible estimate. Possibly, some classification errors were introduced in this way, but one should realise that IQs derived from formal psychological tests often show intraindividual variations of 5 or more IQ points as well.
Finally, for some anomalies, the causal relation with MR remains uncertain. For chromosome anomalies, this holds for marker chromosomes, inversions, apparently balanced translocations, and some sex chromosome aneuploidies (eg 47,XXX); for dysmorphologic examinations, the likelihood that, for instance, Ehlers–Danlos syndrome or a Poland anomaly represents the cause of MR is unlikely. For the other investigation techniques, similar examples can be given. Some authors clearly acknowledged this.28 Therefore, such diagnoses were scored as having an uncertain causal relation with MR.
Quality assessment
Two reviewers (two of MCEJ, CDMvK, RCMH) independently assessed the quality of each study, conforming to previously published assessment models,4,29,30,31 scoring each of the following items: study design, study group, selection of the study group, clinical relevance of the study group, diagnostic technique applied, description of yield, and diagnoses established.
In consensus meetings, differences in the grading of the methodological quality were assessed for each study, conforming to the steps depicted in the flow chart in Figure 2. The quality was divided in items (Supplementary material Table 1; URL address at the end of the manuscript). Each item could be scored from ‘poor’ to ‘good’. If a study scored ‘poor’ on one of the items Description and Clinical Relevance, it was excluded from the review. If the study scored ‘moderate’ or ‘good’ for each of these two items, then the quality of the technique reported in the study was assessed.
Figure 2
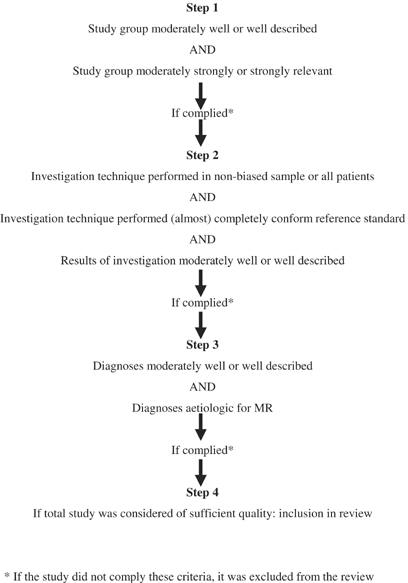
Flow chart of steps in assessing the methodologic quality of a study on an investigation technique.
Only if the diagnostic investigation was applied in all patients, a random sample of the study group, or a nonrandom sample selected based on clearly described criteria, and if it was performed in part or completely, conforming to the reference standard in the year it was performed, and if the results were moderately to well described, the study was included in the next review step. Thus, studies with a good score for Description and Clinical Relevance of the study group might still be excluded due to a poor quality of application of the technique or poor description of the results. For example, for the cytogenetic studies, we used the number of chromosome bands to decide whether a study was sufficiently reliable to report on only numerical chromosome anomalies (<350–400 bands) or also on structural anomalies (>350–400). Around the year 1983, high-resolution banding became widely available.8 We reasoned that a cytogenetic study performed after 1983 would most likely have applied this level of banding, and was therefore sufficiently sensitive to reliably pick-up structural anomalies. Finally, a ‘moderate’ to ‘good’ description of established diagnoses was necessary to allow inclusion in the final review. Although the presence of a list of diagnoses that are indeed (likely to be) aetiologic of MR did improve the quality score of a study, the absence of such a list would not lead to exclusion as long as diagnoses were provided in such a way that reinterpretation was possible.
For inclusion, a study had to score at least ‘moderate’ for six of the seven items. The total quality of the study was appraised as ‘good’, if 5–7 items were scored as ‘good’ and the remaining items as ‘moderate’. In all other cases, the study was classified as ‘moderate’.
Results
Search strategy
The flow diagram in Figure 3 summarises the number of articles accepted and rejected during the selection procedure. The search of the computerised databases identified a total of 4934 citations (Table 1).
Figure 3
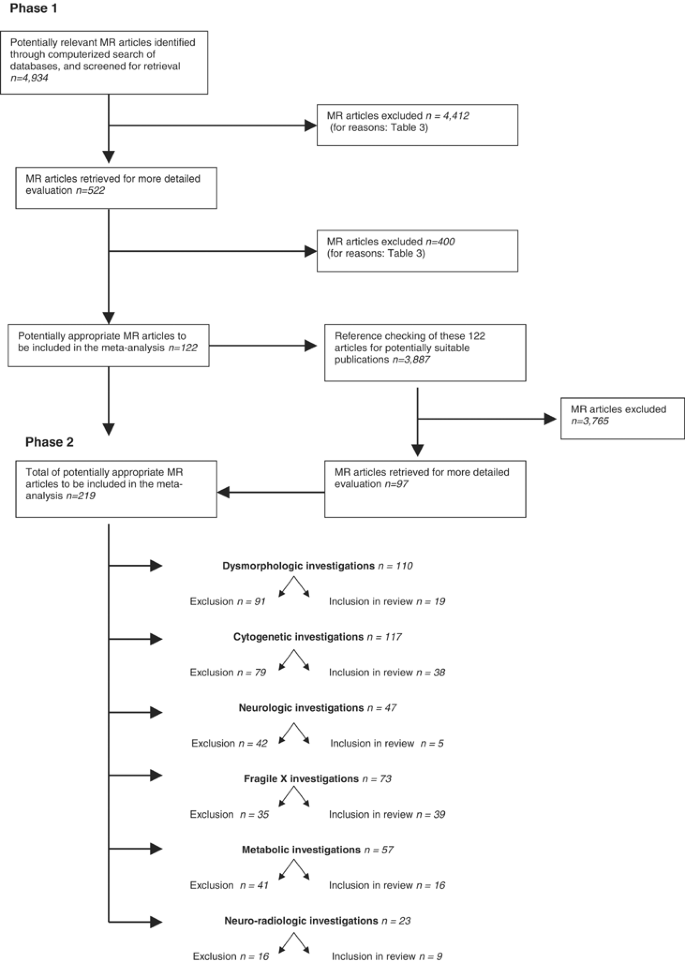
QUORUM flow diagram of publications included and excluded by reviewers during the selection procedure.
Screening all titles of all publications allowed exclusion of 4412 studies clearly not related to the objective of the present study (Table 3). Of the remaining 522 papers, abstracts were considered in the selection procedure applying the phase 1 inclusion criteria, which were met by a total of 122 publications.
Screening of all references (_n_=3887) of the 122 relevant papers in a similar manner resulted in an additional 97 studies. Thus, the total number of articles that entered the phase 2 selection procedure and form the basis of this review was 219. Table 3 indicates for each of the different investigation techniques the reasons for exclusion using phase 2 selection criteria.
Pilot study
Based on the phase 2 selection criteria, three of the total of eight publications in the pilot study were excluded from the review due to the following reasons: major goal was not elucidation of aetiology of MR;24 patient selection criteria unclear;25 number of patients with MR <25.26. One publication20 was included only for reporting numerical anomalies in MR patients, as the G-banding at that time (1971) was not of sufficient quality to detect small structural anomalies.
Selected articles
Supplementary material Tables 1 and 2 (available only on the Internet; URL address at the end of the manuscript) list the qualitative and quantitative data of all articles included in this review. Supplementary material Table 1 lists the quality of each individual article regarding the description of the patient group under study, the investigation technique, the description of the results of the investigation, the accuracy of the diagnoses, and the clinical relevance of the study. Supplementary material Table 2 provides detailed information of each individual study, grouped by investigation technique used. The major data provided from each study are year of study, study design, setting, selection of study group, characteristics of study groups including the degree of MR, details about investigation technique, number and percentages of detected anomalies, and number and percentages of detected diagnoses. If needed, specific remarks on each study were added. In the different sections of Supplementary material Table 2, the number of ‘abnormalities detected’ may differ from the numbers ‘diagnoses made’, as not all detected abnormalities are of aetiologic diagnostic significance (see also Data extraction).
Relation between diagnostic yield and setting, MR severity, gender
Table 4 shows the relation between the frequency of anomalies detected by each of the techniques on the one hand and the setting, MR severity, and gender on the other hand. The latter is listed for the totals only, as the number of studies in the separate categories was too small to allow analysis. Determination of the sex ratios of the abnormality frequencies was only possible for part of the studies, because some studies did not specify the gender distribution in relation to cytogenetic investigations results.
Table 4 Crosstables summarising diagnostic yield of the specific investigations per degree of MR and per type of setting
For the investigation technique Dysmorphologic examination, no such crosstables could be gathered, as the different studies either performed the examinations in such a different way or the data were presented in such a different manner that pooling of results was not feasable or useful. Only for two specific items within a general dysmorphological evaluation a limited number of similar results became available. The first is the presence within families of other family members with MR. This was reported in 10 studies.59,63,65,80,92,99,106,113,114 The median number of families in which relatives with MR were present was 15.0% (range 7.5–46%). However, it was generally not stated to which extent the family was surveyed for MR nor to which extent it was reported in the publication. The population based study of Hou et al80 reported that 15% of the families in a large population have a positive history of MR; the degree of familial relationship was not indicated. More or less the same holds for the second item, consanguinity. This was reported in eight studies.23,65,74,96,105,113,114,115 The median percentage of families with consanguinity was 9.1% (range 0.7–85.5%). As, again, it was usually not stated to which extent consanguinity was surveyed or reported, and also because of the different ethnic backgrounds of the study populations, it is not possible to draw meaningful general conclusions from these figures.
There was a limited number of studies that reported on the percentage of patients with abnormal physical features in their population: Ohdo et al56 reported the presence in 55.0%, Majnemer et al58 in 44.5%, Hunter64 in 39.4%, and Van Karnebeek et al65 in 81.9%.
Discussion
Limitations and merits of the review
The inclusion or exclusion of a given publication in this review does not in a simple way relate to the scientific value of an individual study: we had to exclude many valuable publications as they did not meet the specific goals of the present study. One example is the publication by Flint and co-workers in Nature Genetics, which added fundamental new knowledge on the detection of subtelomeric rearrangements in patients with idiopathic MR.116 Although this study is of significant importance, we were unable to include it in this review as the description of selection of patients was insufficient to provide reliable data on frequency of subtelomeric anomalies in unselected MR patients. Nor was it possible to determine the relation between yield of subtelomeric screening and clinical setting or specific characteristics of the studied group, such as MR severity and gender. Another example is the group of comprehensive studies that report a complete diagnostic work-up of an MR study group. Studies from this group of publications often had to be excluded because it was impossible to determine the exact yield of the individual additional investigations, in part because it was not specified in how many patients each of the investigations was performed.117,118
A second limitation of our study is the fact that we are able to report only the positive findings of an investigation; none of the studies described the presence and value of ‘negative findings’ in the diagnostic process. In clinical practice, the exclusion of a specific anomaly (a ‘negative finding’) can be as useful as a positive one in establishing an aetiological diagnosis. It will require a separate study designed specifically for establishing the value of ‘negative findings’ to investigate this further.
A positive quality of the study is the broad character of the search strategy and the minute study selection process, minimising the chance of overlooking important publications. We allowed the inclusion of studies published in languages other than English; articles published during a long time interval, which allowed the inclusion of valuable studies dating back to more than 30 years; publications indexed in different databases, so that articles not listed in one electronic database but only in another were not overlooked; and publications with unusual MeSH headings not retrieved from the databases but found through screening of bibliographies of potentially useful articles. This resulted in the ‘detection’ of several publications of unusual quality,57,80 which seem to have been overseen by many authors of earlier reviews and texts.
The present study also tries to provide an estimate of the quality of studies. Quality is a multidimensional concept, which relates to the design, conduct, and analysis of a study, its clinical relevance, and quality of reporting.30 Assessment of quality is necessary to limit bias in the results of this systematic review, to gain insight into potential comparisons, and guide the interpretations of the findings.4 As there are a only small number of studies that have tried to measure quality,4 knowledge and experience in this area are limited. As quality assessments will always include some degree of clinical and methodological judgement, its results should be interpreted with some caution.
The similarities and differences with the only other comprehensive assessment in this area, the review of the American Academy of Neurology2 merits a more detailed discussion. In this extraordinary paper, an evidence-based evaluation of the literature on diagnostic studies in individuals with MR is described. There are a number of limitations in this article however. One is the restriction to only papers in the English language. In the present studies, 197 of the 219 were English, the other 22 were non-English. A major issue – in our opinion – is the absence of details on the criteria used to select papers for the review. The authors mention all keywords of their search but do not mention how they selected papers out of the retreived citations. The use of one of their keywords (mental retardation) in one literature database (Pubmed) gives 57 641 hits, indicating the importance of these inclusion and exclusion criteria. It was not reported whether the selection of papers was performed by a single investigator or by different investigators, and whether these worked independently or not.
These issues are important to prevent selection bias in the field; such bias can lead to both overestimation and underestimation of the virtues of a given diagnostic test. A last point of concern is that in their presentation of the results of individual studies, the difference in setting of the study or the degree of MR was not consistently reported. We have been unsuccesful in our efforts to gather this information from the authors of this article, and therefore cannot meaningfully compare their results with the present review. Despite these comments, the paper is of considerable value, and the results are in many ways similar to our results.
Number of available studies
The total number of articles reporting the results of one or more diagnostic investigations in MR patients that seem appropriate for inclusion in this review was considerable. Yet, if one considers the number of citations that had to be screened in order to obtain publications truly relevant to our aim, the yield of our searches seems low. The low ‘hit’ rate of the computerised searches may be explained in part by the broad character of our search, which in turn was due to lack of accepted terms and MeSH headings exactly fitting the aims of this review. For example, articles reporting prenatal screening for well-known causes of MR such as Down syndrome or articles on the developmental follow-up of neonates who had suffered hypoxic insults also appeared in our search results. Further, there exist few other observational systematic reviews focused on disease aetiology, thus offering little information for optimising the effectiveness and efficiency of our search strategies. The high number of useful publications found through reference checking (and not computerised searches) suggests that either our search strategies or the quality of indexation of articles on this subject is suboptimal. We conclude that, although many articles did not meet the criteria for our review, the final number of publications of sufficient quality included in this review is considerable, and should allow for reliable conclusions.
Yield of investigation techniques
Chromosome anomalies
A first conclusion that can be drawn from this review is that chromosome anomalies have been detected in all MR study groups. On average, the frequency of detected aberrations was around one in every 10 investigated patients. In general, cytogenetic studies therefore are a valuable diagnostic technique in studying individuals with MR. There is a considerable variation, however, in the frequency of detected aberrations, especially for numerical anomalies.
Numerical anomalies. Analysis of the relationship between yield of chromosome studies and clinical setting was hampered by the uneven distribution of studies among the different settings. Most studies were performed in institutions, which on average will comprise more severely retarded patients than schools and population surveys. The median frequency of anomalies was higher in individuals with moderate to severe MR than those with borderline or mild MR, and differences herein could well explain the variation in the frequency of detected chromosome anomalies in different settings. However, also in the group of borderline to mild MR, the number of cytogenetic anomalies is still considerably high, and allows the recommendation of routine karyotyping.
The relationship between gender and detection frequency of numerical anomalies is reported by a total of 15 studies on 6601 persons, which provides sufficient data to draw conclusions. In total, a slight male predominance is reported (M:F=7.2%:5.6%). The difference is too small, however, to imply an increased likelihood of detecting numerical anomalies in males; we recommend that cytogenetic investigations should be performed regardless of gender.
Obviously, the inclusion or exclusion of individuals with only clinically suspected Down syndrome in a study group influenced the reported yield of numerical anomalies. Whether or not the presence of other dysmorphologic features influenced detection frequency was not systematically assessed in this review, as differences in the applied nomenclature and classification of such features between studies precluded reliable analyses. A recent study in a group of MR children from our group did provide evidence of the hypothesis frequently brought forward by Dr John Opitz, that individuals with aneuploidy show more and more widespread minor anomalies.65 As chromosomal aberrations have been found in persons with a very mild phenotype,14 karyotyping should still be performed in all MR patients without other obvious causes, regardless of the number of abnormal physical findings.
Finally, numerical anomalies affected autosomes considerably more often than sex chromosomes, with a median frequency of 6.5 vs 0.4%. The most obvious explanation lies in the high number of persons with Down syndrome. The nine studies reporting on the relation between the severity of MR in patient and the type of chromosome affected, all show that numerical anomalies of the sex chromosomes occur foremost in borderline to mild MR, while numerical anomalies of the autosomes are detected mostly in patients with more severe MR. This distribution concurs with previous reports.
Structural anomalies. There is less variation in detection frequency of structural anomalies than of numerical anomalies. The pick-up rate of the former anomalies depends largely on the resolution of the cytogenetic investigation: more anomalies will be missed using 350–400 bands compared to higher band numbers. Although studies reporting FISH investigations screening for (submicroscopic) structural rearrangements involving the chromosome ends were included in the present review, those involving interstitial regions were not. Only the advent of molecular karyotyping will provide reliable data on the frequency of structural rearrangements in MR individuals.
As for setting, the numbers per category are small, warranting caution when interpreting the data. The highest average of median frequency was reported by the five studies performed in an outpatient clinic, while the frequency reported by 11 institutional studies was almost three-fold lower, similar to studies performed in schools and population setting. Similar to the detection frequency of numerical anomalies, structural anomalies were reported more often in patients with moderate to profound MR than in those with a more mild MR grade. Again, frequencies in both groups are sufficient to advocate cytogenetic investigations regardless of MR severity. The relation between anomaly frequency and gender in all studies reliably reporting on gender distribution showed a female predominance (P<0.05). Structural anomalies more often affected the autosomes than the sex chromosomes in all studies except one, which reported an equal ratio of affected chromosome type. This female predominance in structural anomalies was also found for the subtelomeric FISH studies (see below). There is no good explanation for this finding. Theoretically, one can speculate that structural anomalies occur more often in females than in males due to (1) genes on the sex chromosomes which regulate the three-dimensional DNA structure, thereby influencing crossover frequencies or other _trans_-actions between chromosomes; (2) the decreased degree of condensation during meiosis in females,119 resulting in more frequent abnormal pairing of chromosome regions, for instance in those harbouring olfactory receptor-gene clusters.120,121 (3) Structural anomalies occur in both genders with equal frequency, but female predominance is explained by an increased prenatal or early postnatal survival of females with such an aberration. Empirical motivation of an increased prenatal survival in females is difficult because the sex ratio of spontaneous abortions remains largely unclear; one study reported a slightly higher frequency in males (XY/XX ratio=1.03).122 As for the postnatal survival of preterm and/or low birth-weight infants, a male disadvantage has been reported in several studies.123,124 The mechanisms underlying the increased mortality (and morbidity) rate in these newborn males remain largely unknown, but are in support of the latter explanation. (4) Finally, the presently found female predominance may be the result of chance alone, as the number of studies on which this conclusion is based is still limited. The sex distribution of structural autosome anomalies deserves more attention in future research.
Numerical and structural anomalies. The variation in the total number of chromosome anomalies is of course the sum of the separate variations in numerical and in structural anomalies. The above conclusions also apply.
FISH analysis of subtelomeric regions. For subtelomeric FISH studies, the results of the review are based on a relatively small number of studies. We found that the frequency of subtelomeric rearrangements in unselected MR patients may be lower than previously reported. As suggested earlier,65 this investigation should be selectively applied until more efficient, less-expensive techniques become available. There was a marked variation between positive test results of studies. Explanations for differences in detection frequency are extensively discussed elsewhere.65 In the present review, the number of studies per category of setting and MR severity is too small to allow firm conclusions about the relation of detection frequency with these two variables. As for gender, all three studies reporting reliable data on the frequency of rearrangements in males vs females again suggest a female predominance (P<0.05).
Fragile X studies
Studies for the fragile X syndrome were performed using cytogenetic techniques in former times, but after detection of the gene defect almost all studies used molecular techniques. The number of studies comparing the yield of both types of investigation techniques is small24 and indicated that the yield is comparable. In the present review, we found that the yield of cytogenetic techniques to detect fragile X syndrome yielded a mean result of 5.4%, while molecular studies yielded only 2.0%. The cytogenetic studies were performed in a different period, during which still many patients with fragile X had to be detected. This may have created a bias during patient selections. Furthermore, patients with other fragile sites at the distal long arm of the X chromosome may be mistaken for patients with fragile X syndrome.
The most reliable study that screened an unselected group of patients is the study by De Vries et al.19,86 They reported on a large group of institution population and found fragile X by molecular techniques in 0.7%. Most other studies did not use unselected study groups, but groups selected on either X-linked MR, the presence (or absence) of macro-orchidism or other phenotypical features of fragile X syndrome, the absence of Down syndrome, or the absence of any known cause for MR. The reader is referred to Supplementary material Table 2 for further detailed information on studies of each selected study.
Several studies used phenotype checklists to increase their yield.19,77,81,82,84,91,125 Despite many differences in the nature of the checklists, this was usually successful, although not always.91 Checklists may ask for several items regarding the face (ear size, mandible size), brain growth (skull circumference), testicle size, skin constitution, degree of MR, behaviour, and family history for MR. Especially, the presence of normocephaly or macrocephaly, and a positive family history seem to be the most valuable criteria (Supplementary material Table 2). If a checkist becomes too long, it becomes cumbersome in general practice and its use decreases. This demands a limited set of criteria for future lists.
The yield of studies in study groups with a more pronounced degree of MR is reported to be, in general, larger (4.1%) compared to those in the borderline–mild MR group (1.0%). This is to be expected as, in general, fragile X gives rise to a more pronounced degree of MR in males.
There is a number of studies for fragile X syndrome in females.19,44,47,49,50,53,70,76,77,79,81,83,84,88,89,91,93,94,126 Many of the studies have selected their study group (see individual studies in Supplementary material Table 2), and even after such selection the yield in females was often low. The most reliable study is again the study by De Vries et al,19,86 who reported a yield of 0.3% in girls. This yield increased with use of clinical preselection criteria, a positive family history being the most obvious one.
Metabolic investigation
The results of this systematic review on metabolic investigations in persons with MR are limited. One reason is the relatively small number of relevant publications that were available for this study. A second reason is that the nature of the various studies differed considerably: the metabolic pathways that were investigated in one study could completely differ from those in other studies. This lack of a standardised screening protocol precludes any comparison. Only for screening studies for phenylketonuria (PKU) sufficient studies were available to allow a crosstabulation (Table 4).
In general, the yield of metabolic studies is low. If one considers the results of the different studies for metabolic disorders in general, results varied from 0.2 to 8.4% (median 1.0%). The higher figures are from countries where specific entities are more common, such as aspartylglycosaminuria in Finland,59 or the figures were reached by applying checklist criteria,64,65,105 sometimes in a highly inbred population.105 In such specific study populations, the yield may be high. There is one population study97 studying all school children with MR in special schools and institutes for the mentally retarded that showed a metabolic abnormality in 1.0% of the study population; this figure must be an under-estimation of the true frequency as children below 4 years were underrepresented in this study. In other populations, neonatal screening programmes for metabolic disorders identify children with often treatable causes of MR.
A preselection of cases using a stepwise or checklist (like in fragile X syndrome) approach has been reported infrequently. One study126 using a stepwise approach showed an increase of yield to 13.6%. Items in such an approach may be dysmorphologic symptoms, hepatosplenomegaly, and ophthalmologic and neurologic findings.
Neurologic investigation
In the paper from the American Academy of Neurology,2 the results of diagnostic yield of clinical neurological studies were not mentioned as such, but only the results of EEG studies and neuroimaging were reported. The diagnostic yield of EEG studies fell outside the scope of our review as EEG studies in itself are rarely if ever diagnostic. For diagnostic purposes, there are no clues that EEG studies are indicated.2 This does not mean that EEG studies are not useful in some children with MR: Shevell et al2 demonstrated that available literature data indicate a yield of 4.4% for epilepsy-related diagnoses, and suggested that an EEG should be obtained in children with a history or examination features suggesting the presence of epilepsy.
Most clinicians will agree that a careful neurological investigation of every child with MR is obligatory. In our literature search, it appeared difficult to obtain evidence for the utility of this, as many papers did not differentiate their results according to the results of clinical neurological examination. There is however a limited number of papers available (Table 4 and Supplementary material Table 2). As expected, all show a remarkably high yield in the settings for which data are available, that is, in outpatient clinic studies and population-based surveys, irrespective of the degree of MR. The total yield of aetiological diagnoses in all studies is 42.9%. This figure does not yet include the value of neurological exams in providing indications for other diagnostic studies such as neuroimaging or molecular analyses. Therefore, the true diagnostic yield of a neurological examination is probably higher. We conclude that every child with MR, irrespective of setting and degree of retardation, should undergo a basic, clinical neurological exam.
Neuroradiological studies
The different neuroimaging modalities that entered the present study included cranial CT scanning and brain MRI studies. The results in Table 4 refer to the number of abnormalities found in such studies. In 30.0% of all studies, brain abnormalities were reported. None of the studies reported on the value of the absence of any neuroradiological abnormality for a diagnostic work-up. Therefore, the true value for finding abnormalities or the absence of abnormalities must be higher. MRI studies were reported to be more sensitive compared to CT scanning, as expected.65,111,127
If the neuroimaging was performed ‘on indication’, that is, in cases with abnormal brain size or a focal neurological finding, the results increased. Shevell et al2 reported that in an earlier study by their group128 the percentage of abnormalities was 13.9% if performed on a ‘screening basis’, but increased to 41.2% if performed on an ‘indicated basis’; the authors discussed other studies on smaller number of patients that showed similar results.
If one allows only completely diagnostic results to be tabulated, the results are much less impressive: Kjos et al110 reported 3.9% diagnoses in patients who had no known cause for their MR and followed no progressive or degenerative course, Bouhadiba et al112 found 0.9% diagnoses in patients with neurological symptoms, and four other studies64,108,109,111 found no diagnosis on the basis of the neuroimaging alone. Three studies reported the results of unselected patients: Majnemer and Shevell58 found a diagnosis by this type of investigation in 0.2% of patients, Stromme63 in 1.4%, and Van Karnebeek et al65 in 2.2% of patients.
For all of the above studies, the yields are somewhat lower in the patients with a borderline-mild MR compared to a moderate–profound retardation, although the differences are often small.
Neuroimaging is at present possible only in patients who remain immobile for a longer period of time. Many patients with MR, especially the younger ones, will not be able to accomplish this, and will need some type of sedation for such studies. This makes the burden to the patient for performing neuroimaging studies much higher as compared to the physical exam, urine sampling, and blood sampling that is needed for the other diagnostic tests. This influences the desired diagnostic yield needed before one will perform a neuroradiological study. The increase in diagnostic yield in neuroimaging on an indicated basis (ie brain size abnormality, focal neurological finding, symptoms at history taking or physical exam indicating a higher chance for a brain anomaly such as a phacomatose) is important in this respect. With the advent of new and faster imaging modalities, the burden to patients may decrease and the indication for neuroimaging may change. At present, we recommend neuroimaging on an indicated basis only.
Dysmorphological investigations
The utility of a dysmorphological examination was difficult to evaluate. The main reason was that a very limited number of studies reported on the specific results of such examinations, while probably most if not all studies did make use of the presence or absence of unusual features at physical surface investigation, in part to steer further diagnostic studies. Numerical results on the resulting diagnostic yield cannot be derived from these studies. In larger groups of patients, the number of two or more dysmorphic features found was from high (39.4%,64 44.5%58) to very high (55.0%,56 81.9%65). Obviously, these figures depended heavily on the definition of dysmorphic features, and the way the dysmorphological exam was performed.65 As in neurological exams, the figures on dysmorphic features do not represent the number of aetiological diagnoses made using this investigation technique, and also do not indicate how often the finding of dysmorphic features formed the reason to initiate further metabolic, cytogenetic, molecular, neuroradiological, or other investigations. One study reported about this in detail: Van Karnebeek et al65 reported on the importance of physical examination, including paediatric neurological, and dysmorphological examinations, and found this to have been essential for the diagnosis in 62% and contributory in 79%, respectively. No distinction between the three parts of physical exam was made. Shevell et al128 reported similar findings. In conclusion, taking a good clinical history and performing a detailed physical examination by a trained specialist remain the basis of every aetiological study in children with MR.
In performing literature reviews, one often encounters differences in the way the results of investigations are reported. In the present study, the largest differences were found in the way dysmorphological exams were reported: the definitions used, the extensiveness of the physical exam itself, and the terms used in descriptions varied widely. We conclude that there is definitely a need for more standardisation of reporting dysmorphological examinations.
Recommendations from this study
In both current clinical practice and in future empirical studies aimed at detecting the aetiological diagnosis in children with MR, the following guidelines can be formulated (each time it is indicated if the guideline is based on the present systematic literature search):
- In each child, a detailed clinical history should be taken and physical examination should be performed, irrespective of setting or degree of MR (basis: this study). This physical exam should also include both a detailed paediatric neurological exam and a dysmorphological exam (basis: this study). Therefore, a good clinical history and physical exam by a trained specialist remain the basis of aetiological studies in persons with MR.
- In each child, standard cytogenetic studies should be performed, irrespective of setting, degree of MR, and the presence (or not) of dysmorphic features (basis: this study). Only if noncytogenetic causes of MR are evident through history taking and physical exam, this can be omitted.
- FISH analysis for subtelomeric rearrangements should at present be used on stringent selection criteria, as can be found in the available checklists (basis: this study). If more efficient and less-expensive techniques become widely available, studies for subtelomeric (and possibly also interstitial) rearrangements may become indicated in each child.
- Molecular studies for fragile X syndrome have a lower yield than earlier expected, but may still be performed in all boys with MR (basis: this study). The chances for finding fragile X syndrome are higher in children with a more expressed degree of MR, and in an outward clinic setting (basis: this study). The use of checklists or simple criteria may increase the yield considerably; the two most powerful criteria seem to be the presence of a positive family history for MR and the absence of microcephaly (basis: this study). Studies in girls should not be performed routinely, but only if positive clues are present, of which a positive family history for MR is the most important.
- Metabolic studies should not be performed as the first diagnostic study in each child, but in the absence of clues for other causes, the yield is still of a sufficiently high level to allow testing (basis: this study). The use of checklists improves the yield considerably. Further development of such checklists is desirable. The nature of metabolic pathways studied in children with MR should become more standardised internationally.
- Neuroradiological studies have a high yield for brain abnormalities, but a low yield for establishing aetiological diagnoses (basis: this study). Neuroradiology should not be performed in each child, also because of the burden of neuroimaging for the retarded child. If specific symptoms are present (abnormal brain size, focal neurological finding, symptoms at clinical history taking or physical exam that indicate a higher chance for a brain anomaly, such as a phacomatosis), neuroimaging is indicated. The yield is higher in patients with a more expressed degree of MR, and MRI scanning has a higher yield compared to CT scanning (basis: this study). The development of new and faster modalities may change this recommendation.
- The yield of specific neurological and dysmorphological studies should be studied in more detail (basis: this study). Investigation techniques and reporting of results of dysmorphological studies should become more standardised internationally.
- The cause for the higher yield in both structural and numerical chromosome anomalies in females needs further evaluation.
- The value for the diagnostic process of the absence of abnormalities, for instance, in neuroimaging should be further studied.
- If algorithms are developed using the above recommendations, they should be evaluated in clinical practice using various settings and study populations with various degrees of MR.
Final remark
If more and more reliable empirical data become available on diagnostic studies in children with MR, this should allow clinicians to weigh the benefits of performing specific investigations like the resolution of diagnostic uncertainty, prevention of further investigations, improved possibilities for genetic counselling, clinical management, and ultimately prevention and treatment, against the disadvantages, such as discomfort for the individual subjected to the testing procedures, anxiety for parents awaiting test results, and increase in costs. Studies dedicated to these subjects should be encouraged.
References
- Curry CJ, Stevenson RE, Aughton D et al: Evaluation of mental retardation: recommendations of a Consensus Conference: American College of Medical Genetics. Am J Med Genet 1997; 72: 468–477.
Article CAS PubMed Google Scholar - Shevell MI, Ashwal S, Donley D et al: Practice parameter: evaluation of the child with global developmental delay. Neurology 2003; 60: 367–380.
CAS PubMed Google Scholar - Clarke M : The QUORUM statement. Lancet 2000; 355: 756–757.
CAS PubMed Google Scholar - Clark M, Oxman AD : Cochrane Reviewers Handbook 4.1.4, Cochrane Library,: 2001, Vol 4. Update Sofware. Updated Quarterly.
- Luckasson R, Coulter DL, Polloway EA : Mental Retardation: Definition, Classification and Systems of Supports. Washington, DC: American Association on Mental Retardation, 1992.
Google Scholar - World Health Organization: Fifteenth report of the WHO Expert Committee on Mental Health. Geneva: World Health Organization, 1968.
- American Psychiatric Association: Diagnostic and Statistical Manual of Mental Disorders. Washington, DC: American Psychiatric Association, 1994, 4th edn.
- Dutrillaux B : High-resolution chromosome staining. Pathologica 1983; 75: 207–209.
PubMed Google Scholar - Knight SJ, Horsley SW, Regan R et al: Development and clinical application of an innovative fluorescence in situ hybridization technique which detects submicroscopic rearrangements involving telomeres. Eur J Hum Genet 1997; 5: 1–8.
CAS PubMed Google Scholar - Sutherland GR : Fragile sites on human chromosomes: demonstration of their dependence on the type of tissue culture medium. Science 1977; 197: 265–266.
CAS PubMed Google Scholar - Fu YH, Kuhl DP, Pizzuti A et al: Variation of the CGG repeat at the fragile X site results in genetic instability: resolution of the Sherman paradox. Cell 1991; 67: 1047–1058.
Article CAS PubMed Google Scholar - Schaefer GB, Bodensteiner JB : Evaluation of the child with idiopathic mental retardation. Pediatr Clin N Am 1992; 39: 929–943.
CAS Google Scholar - Hennekam RCM : Abnormal mental development; in Hennekam RCM (ed): Baillière's Clinical Paediatrics. London: Baillière Tindal, 1998, Vol 6: pp 317–322.
Google Scholar - Gorlin RJ, Cohen MM, Hennekam RCM : Syndromes of the Head and Neck. Oxford: Oxford University Press, 2001.
Google Scholar - Ligon AH, Beaudet AL, Shaffer LG : Simultaneous, multilocus FISH analysis for detection of microdeletions in the diagnostic evaluation of developmental delay and mental retardation. Am J Hum Genet 1997; 61: 51–59.
CAS PubMed PubMed Central Google Scholar - Rio M, Molinari F, Heuertz S et al: Automated fluorescent genotyping detects 10% of cryptic subtelomeric rearrangements in idiopathic syndromic mental retardation. J Med Genet 2002; 39: 266–270.
CAS PubMed PubMed Central Google Scholar - Kirchhoff M, Rose H, Duno M, Gerdes T, Lundsteen C : Screening of dysmorphic and mentally retarded subjects with high resolution comparative genomic hybridization. Eur J Hum Genet 2002; 10: 65–65.
- Buchholz T, Jackson J, Robson L, Smith A : Evaluation of methylation analysis for diagnostic testing in 258 referrals suspected of Prader–Willi or Angelman syndromes. Hum Genet 1998; 103: 535–539.
CAS PubMed Google Scholar - De Vries BB, van den Ouweland AM, Mohkamsing S et al: Screening and diagnosis for the fragile X syndrome among the mentally retarded: an epidemiological and psychological survey. Collaborative Fragile X Study Group. Am J Hum Genet 1997; 61: 660–667.
CAS PubMed PubMed Central Google Scholar - Aitken J, Brunton M, Jacobs PA, Price WH, MacColl K : Chromosome studies on male patients at a mental subnormality hospital. Clin Genet 1971; 2: 338–346.
CAS PubMed Google Scholar - Battaglia A, Bianchini E, Carey JC : Diagnostic yield of the comprehensive assessment of developmental delay/mental retardation in an institute of child neuropsychiatry. Am J Med Genet 1999; 82: 60–66.
CAS PubMed Google Scholar - Brett EM, Hoare RD : An assessment of the value and limitations of air encephalography in children with mental retardation and with epilepsy. Brain 1969; 92: 731–742.
CAS PubMed Google Scholar - Durkin MS, Hasan ZM, Hasan KZ : Prevalence and correlates of mental retardation among children in Karachi, Pakistan. Am J Epidemiol 1998; 147: 281–288.
CAS PubMed Google Scholar - Hofstee Y, Arinami T, Hamaguchi H : Comparison between the cytogenetic test for fragile X and the molecular analysis of the FMR-1 gene in Japanese mentally retarded individuals. Am J Med Genet 1994; 51: 466–470.
CAS PubMed Google Scholar - Serur D, Jeret JS, Wisniewski K : Agenesis of the corpus callosum: clinical, neuroradiological and cytogenetic studies. Neuropediatrics 1988; 19: 87–91.
CAS PubMed Google Scholar - Skjeldal OH, Sponheim E, Ganes T, Jellum E, Bakke S : Childhood autism: the need for physical investigations. Brain Dev 1998; 20: 227–233.
CAS PubMed Google Scholar - Wellesley D, Hockey A, Stanley F : The aetiology of intellectual disability in Western Australia: a community-based study. Dev Med Child Neurol 1991; 33: 963–973.
CAS PubMed Google Scholar - Soudek D, Sroka H : Chromosomal variants in mentally retarded and normal men. Clin Genet 1979; 16: 109–116.
CAS PubMed Google Scholar - Begg C, Cho M, Eastwood S et al: Improving the quality of reporting of randomized controlled trials. The CONSORT statement. JAMA 1996; 276: 637–639.
CAS PubMed Google Scholar - Juni P, Altman DG, Egger M (eds): Assessing the quality of controlled clinical trials; in Systematic Reviews in Health Care: Meta-analysis in Context. London: BMJ Books, 2001, 2nd edn.
Google Scholar - Verhagen AP, de Vet HC, de Bie RA, Boers M, van den Brandt PA : The art of quality assessment of RCTs included in systematic reviews. J Clin Epidemiol 2001; 54: 651–654.
CAS PubMed Google Scholar - Midenet M, Noel B, Quack B : Etude cytogenetique de la population d'un etanlissement recevant des enfants arrieres mentaux profonds. Revue Neuropsychiatr Infantile 1970; 18: 249–258.
CAS Google Scholar - Moser HW, Wolf PA : The nosology of mental retardation: including the report of a survey of 1378 mentally retarded individuals at the Walter E. Fernald State School. Birth Defects (Orig Artic Ser) 1971; 7: 117–134.
CAS Google Scholar - Newton MS, Jacobs PA, Price WH, Woodcock G, Fraser IA : A chromosome survey of a hospital for the mentally subnormal. 1. Sex chromosome abnormalities. Clin Genet 1972; 3: 215–225.
CAS PubMed Google Scholar - Newton MS, Cunningham C, Jacobs PA, Price WH, Fraser IA : Chromosome survey of a hospital for the mentally subnormal. 2. Autosome abnormalities. Clin Genet 1972; 3: 226–248.
CAS PubMed Google Scholar - Warter S, Bon C, Ebtinger R, Clavert J : Cytogenetic study of 350 mental deficient patients at medico-pedagogic institutions. Ann Med Psychol 1977; 1: 51–61.
CAS Google Scholar - Jacobs PA, Matsuura JS, Mayer M, Newlands IM : A cytogenetic survey of an institution for the mentally retarded: I. Chromosome abnormalities. Clin Genet 1978; 13: 37–60.
CAS PubMed Google Scholar - Ally FE, Grace HJ : Chromosome abnormalities in South African mental retardates. S Afr Med J 1979; 55: 710–712.
CAS PubMed Google Scholar - Grace HJ, Ally FE, Nelemans AP, Kint B : A cytogenetic study of a mentally retarded population in South Africa. S Afr Med J 1979; 55: 707–709.
CAS PubMed Google Scholar - Narahara K : A cytogenetic study of children with clinically unclassifiable multiple congenital malformations and mental retardation. Acta Med Okayama 1981; 35: 343–355.
CAS PubMed Google Scholar - Moghe M, Patel ZM, Peter JJ, Ambani LM : Cytogenetic studies in a selected group of mentally retarded children. Hum Genet 1981; 58: 184–187.
CAS PubMed Google Scholar - Kodama Y : Cytogenetic and dermatoglyphic studies on severely handicapped patients in an institution. Acta Med Okayama 1982; 36: 383–397.
CAS PubMed Google Scholar - Proops R, Mayer M, Jacobs PA : A study of mental retardation in children in the Island of Hawaii. Clin Genet 1983; 23: 81–96.
CAS PubMed Google Scholar - Op't Hof J, Venter PA, Du Toit JL et al: A multidisciplinary diagnostic/genetic study on the 105 patients with mental retardation of the E.S. Le Grange School. J Ment Defic Res 1985; 29: 37–47.
PubMed Google Scholar - Nielsen KB, Dyggve HV, Knudsen H, Olsen J : A chromosomal survey of an institution for the mentally retarded. Study of 476 karyotypes with banding techniques and clinical assessment of patients with chromosome anomalies. Dan Med Bull 1983; 30: 5–13.
CAS PubMed Google Scholar - Fryns JP, Kleczkowska A, Kubien E, Van den Berghe H : Cytogenetic findings in moderate and severe mental retardation. A study of an institutionalized population of 1991 patients. Acta Paediatr Scand Suppl 1984; 313: 1–23.
CAS PubMed Google Scholar - Lamont MA, Dennis NR, Seabright M : Chromosome abnormalities in pupils attending ESN/M schools. Arch Dis Child 1986; 61: 223–226.
CAS PubMed PubMed Central Google Scholar - Dereymaeker AM, Fryns JP, Haegeman J, Deroover J, van den Berghe H : A genetic–diagnostic survey in an institutionalized population of 158 mentally retarded patients. The Viaene experience. Clin Genet 1988; 34: 126–134.
CAS PubMed Google Scholar - Hagerman R, Berry R, Jackson III AW et al: Institutional screening for the fragile X syndrome. Am J Dis Child 1988; 142: 1216–1221.
CAS PubMed Google Scholar - Asthana JC, Sinha S, Haslam JS, Kingston HM : Survey of adolescents with severe intellectual handicap. Arch Dis Child 1990; 65: 1133–1136.
CAS PubMed PubMed Central Google Scholar - Chudley AE, Ray M, Evans JA, Cheang M : Possible association of rare autosomal folate sensitive fragile sites and idiopathic mental retardation: a blind controlled population study. Clin Genet 1990; 38: 241–256.
CAS PubMed Google Scholar - Fryns JP, Volcke PH, Haspeslagh M, Beusen L, Van den Berghe H : A genetic diagnostic survey in an institutionalized population of 262 moderately mentally retarded patients: the Borgerstein experience. J Ment Defic Res 1990; 34: 29–40.
PubMed Google Scholar - Volcke Ph, Fryns JP, Pyck K, Van den Berghe H : Ervaringen met systematisch etiologisch onderzoek in een schoolpopulatie van 57 kinderen met lichte mentale handicap en/of zwakzinnigheid. Tijdschr Geneesk 1991; 47: 1359–1364.
Google Scholar - Wuu KD, Chiu PC, Li SY et al: Chromosomal and biochemical screening on mentally retarded school children in Taiwan. Jpn J Hum Genet 1991; 36: 267–274.
CAS Google Scholar - Haspeslagh M, Fryns JP, Holvoet M et al: A clinical, cytogenetic and familial study of 307 mentally retarded, institutionalized, adult male patients with special interest for fra(X) negative X-linked mental retardation. Clin Genet 1991; 39: 434–441.
CAS PubMed Google Scholar - Ohdo S, Sonoda T, Ohba K, Hayakawa K : Etiologic and pathogenetic study of mental retardation with multiple congenital anomalies. Acta Paediatr Jpn 1992; 34: 144–150.
CAS PubMed Google Scholar - Graham SM, Selikowitz M : Chromosome testing in children with developmental delay in whom the aetiology is not evident clinically. J Paediatr Child Health 1993; 29: 360–362.
CAS PubMed Google Scholar - Majnemer A, Shevell MI : Diagnostic yield of the neurologic assessment of the developmentally delayed child. J Pediatr 1995; 127: 193–199.
CAS PubMed Google Scholar - Matilainen R, Airaksinen E, Mononen T, Launiala K, Kaariainen R : A population-based study on the causes of mild and severe mental retardation. Acta Paediatr 1995; 84: 261–266.
CAS PubMed Google Scholar - Schaap C, Schrander-Stumpel CT, Colla-Pijkels ET et al: A genetic diagnostic survey in an institutionalized population of 116 moderately to severely retarded male patients: the Rekem experience. Genet Couns 1995; 6: 251–258.
CAS PubMed Google Scholar - Devriendt K, Holvoet M, Fryns JP : Etiologisch–diagnostisch onderzoek bij 66 volwassen personen met mentale handicap verblijvend in een bezigheidstehuis. Tijdschr Geneesk 1998; 54: 921–925.
Google Scholar - Lantigua-Cruz A, Mora F, Arechaederra M et al: Etiological characterization of 512 severely mentally retarded institutionalized patients in Havana. Community Genet 1999; 2: 184–189.
CAS PubMed Google Scholar - Stromme P : Aetiology in severe and mild mental retardation: a population-based study of Norwegian children. Dev Med Child Neurol 2000; 42: 76–86.
CAS PubMed Google Scholar - Hunter AG : Outcome of the routine assessment of patients with mental retardation in a genetics clinic. Am J Med Genet 2000; 90: 60–68.
CAS PubMed Google Scholar - Van Karnebeek CD, Koevoets C, Sluijter S et al: Prospective screening for subtelomeric rearrangements in children with mental retardation of unknown aetiology: the Amsterdam experience. J Med Genet 2002; 39: 546–553.
CAS PubMed PubMed Central Google Scholar - Rossi E, Piccini F, Zollino M et al: Cryptic telomeric rearrangements in subjects with mental retardation associated with dysmorphism and congenital malformations. J Med Genet 2001; 38: 417–420.
CAS PubMed PubMed Central Google Scholar - Sismani C, Armour JA, Flint J, Girgalli C, Regan R, Patsalis PC : Screening for subtelomeric chromosome abnormalities in children with idiopathic mental retardation using multiprobe telomeric FISH and the new MAPH telomeric assay. Eur J Hum Genet 2001; 9: 527–532.
CAS PubMed Google Scholar - Anderlid B, Schoumans J, Anneren G et al: Subtelomeric rearrangements detected in patients with idiopathic mental retardation. Am J Med Genet 2002; 107: 275–284.
PubMed Google Scholar - Clarkson B, Pavenski K, Dupuis L et al: Detecting rearrangements in children using subtelomeric FISH and SKY. Am J Med Genet 2002; 107: 267–274.
PubMed Google Scholar - Blomquist HK, Gustavson KH, Holmgren G, Nordenson I, Palsson-Strae U : Fragile X syndrome in mildly mentally retarded children in a northern Swedish county. A prevalence study. Clin Genet 1983; 24: 393–398.
CAS PubMed Google Scholar - Fishburn J, Turner G, Daniel A, Brookwell R : The diagnosis and frequency of X-linked conditions in a cohort of moderately retarded males with affected brothers. Am J Med Genet 1983; 14: 713–724.
CAS PubMed Google Scholar - Froster-Iskenius U, Felsch G, Schirren C, Schwinger E : Screening for fra(X)(q) in a population of mentally retarded males. Hum Genet 1983; 63: 153–157.
CAS PubMed Google Scholar - Kahkonen M, Leisti J, Wilska M, Varonen S : Marker X-associated mental retardation. A study of 150 retarded males. Clin Genet 1983; 23: 397–404.
CAS PubMed Google Scholar - Bundey S, Webb TP, Thake A, Todd J : A community study of severe mental retardation in the West Midlands and the importance of the fragile X chromosome in its aetiology. J Med Genet 1985; 22: 258–266.
CAS PubMed PubMed Central Google Scholar - Arinami T, Kondo I, Nakajima S : Frequency of the fragile X syndrome in Japanese mentally retarded males. Hum Genet 1986; 73: 309–312.
CAS PubMed Google Scholar - Turner G, Robinson H, Laing S, Purvis Smith H : Preventive screening for the fragile X syndrome. N Engl J Med 1986; 315: 607–609.
CAS PubMed Google Scholar - Turner G, Robinson H, Laing S et al: Population screening for Fragile X. N Engl J Med 1986; 339: 1210–1213.
Google Scholar - Butler MG, Singh DN : Clinical and cytogenetic survey of institutionalized mentally retarded patients with emphasis on the fragile-X syndrome. J Intellect Disabil Res 1993; 37: 131–142.
PubMed PubMed Central Google Scholar - Elango R, Verma IC : Fragile X syndrome among children with mental retardation. Indian J Pediatr 1996; 63: 533–538.
CAS PubMed Google Scholar - Hou JW, Wang TR, Chuang SM : An epidemiological and aetiological study of children with intellectual disability in Taiwan. J Intellect Disabil Res 1998; 42: 137–143.
PubMed Google Scholar - Giangreco CA, Steele MW, Aston CE, Cummins JH, Wenger SL : A simplified six-item checklist for screening for fragile X syndrome in the pediatric population. J Pediatr 1996; 129: 611–614.
CAS PubMed Google Scholar - Arvio M, Peippo M, Simola KO : Applicability of a checklist for clinical screening of the fragile X syndrome. Clin Genet 1997; 52: 211–215.
CAS PubMed Google Scholar - Gerard B, Le Heuzey MF, Brunie G et al: Systematic screening for fragile X syndrome in a cohort of 574 mentally retarded children. Ann Genet 1997; 40: 139–144.
CAS PubMed Google Scholar - Mila M, Sanchez A, Badenas C et al: Screening for FMR1 and FMR2 mutations in 222 individuals from Spanish special schools: identification of a case of FRAXE-associated mental retardation. Hum Genet 1997; 100: 503–507.
CAS PubMed Google Scholar - O'Dwyer J, Holmes J, Mueller R, Taylor G : The prevalence of fragile-X syndrome in an institution for people with learning disability. Psychiatr Genet 1997; 7: 115–119.
CAS PubMed Google Scholar - De Vries BB, Mohkamsing S, van den Ouweland AM et al: Screening for the fragile X syndrome among the mentally retarded: a clinical study. The Collaborative Fragile X Study Group. J Med Genet 1999; 36: 467–470.
CAS PubMed PubMed Central Google Scholar - Faradz SM, Buckley M, Lam PT, Leigh D, Holden JJ : Molecular screening for fragile X syndrome among Indonesian children with developmental disability. Am J Med Genet 1999; 83: 350–351.
CAS PubMed Google Scholar - Patsalis PC, Sismani C, Hettinger JA et al: Molecular screening of fragile X (FRAXA) and FRAXE mental retardation syndromes in the Hellenic population of Greece and Cyprus: incidence, genetic variation, and stability. Am J Med Genet 1999; 28: 184–190.
Google Scholar - Pang CP, Poon PM, Chen QL et al: Trinucleotide CGG repeat in the FMR1 gene in Chinese mentally retarded patients. Am J Med Genet 1999; 84: 179–183.
CAS PubMed Google Scholar - Haddad LA, Aguiar MJ, Costa SS, Mingroni-Netto RC, Vianna-Morgante AM, Pena SD : Fully mutated and gray-zone FRAXA alleles in Brazilian mentally retarded boys. Am J Med Genet 1999; 84: 198–201.
CAS PubMed Google Scholar - Gonzalez-del Angel A, Vidal S, Saldana Y et al: Molecular diagnosis of the fragile X and FRAXE syndromes in patients with mental retardation of unknown cause in Mexico. Ann Genet 2000; 43: 29–34.
CAS PubMed Google Scholar - Tan BS, Law HY, Zhao Y, Yoon CS, Ng ISL : DNA testing for Fragile X syndrome in 255 makes from special schools in Singapore. Ann Acad Med Singapore 2000; 29: 207–212.
CAS PubMed Google Scholar - Ruangdaraganon N, Limprasert P, Sura T, Sombuntham T, Sriwongpanich N, Kotchabhakdi N : Prevalence and clinical characteristics of fragile X syndrome at child development clinic, Ramathibodi Hospital. J Med Assoc Thailand 2000; 83: 69–76.
CAS Google Scholar - Tzeng CC, Tzeng PY, Sun HS, Chen RM, Lin SJ : Implication of screening for FMR1 and FMR2 gene mutation in individuals with nonspecific mental retardation in Taiwan. Diagn Mol Pathol 2000; 9: 75–80.
CAS PubMed Google Scholar - Centerwall WR, Ittyerah TR : Phenylketonuria among the mentally retarded in India. Lancet 1966; 2: 193–194.
CAS PubMed Google Scholar - Roberts GE : A survey of mentally retarded children. I. General findings. Dev Med Child Neurol 1966; 8: 26–33.
CAS PubMed Google Scholar - Kutter D, Metz H : The frequency of some oligophrenias due to metabolic diseases in the grand-duchy of Luxembourg. Schweiz Arch Neurol Neurochir Psychiatr 1968; 101: 369–382.
CAS PubMed Google Scholar - Punekar BD : Phenylketonuria: A survey of insitutions for the mentally retardates. Indian J Med Res 1968; 56: 1813–1817.
CAS PubMed Google Scholar - Sinclair S : Etiological factors in mental retardation: a study of 470 cases. Indian Pediatrics 1973; 9: 391–396.
Google Scholar - Gurry DL, Benson J, Simpson M, Helgeland L : Phenylketonuria in children in special and remedial classes. A survey in Perth. Med J Aust 1973; 1: 645–647.
CAS PubMed Google Scholar - Holmgren G : Urinary metabolic screening of mentally retarded patients from institutions in the northern part of Sweden. Hum Hered 1973; 23: 548–560.
CAS PubMed Google Scholar - Schmid A, Moser H, Walti U et al: Die Oligophrenie im Kindesalter. Klinische, biochemische und epidemiologische Untersuchungen an 286 Heimkindern. Schweiz Med Wochenschr 1974; 104: 915–923.
CAS PubMed Google Scholar - Turner G : An aetiological study of 1,000 patients with an I.Q. assessment below 51. Med J Aust 1975; 2: 927–931.
CAS PubMed Google Scholar - ICMR: Multicentric study on genetic causes of mental retardation in India. Indian J Med Res 1997; 85: 161–168.
- Farag TI, al Awadi SA, el Badramary MH et al: Disease profile of 400 institutionalized mentally retarded patients in Kuwait. Clin Genet 1993; 44: 329–334.
CAS PubMed Google Scholar - McDonald A : Severely retarded children in Quebec: prevalence, causes and care. Am J Ment Defic 1973; 78: 205–215.
CAS PubMed Google Scholar - Hagberg B, Hagberg G, Lewerth A, Lindberg U : Mild mental retardation in Swedish school children. II. Etiologic and pathogenetic aspects. Acta Paediatr Scand 1981; 70: 445–452.
CAS PubMed Google Scholar - Lingam S, Read S, Holland IM, Wilson J, Brett EM, Hoare RD : Value of computerised tomography in children with non-specific mental subnormally. Arch Dis Child 1982; 57: 381–383.
CAS PubMed PubMed Central Google Scholar - Harbord MG, Finn JP, Hall-Craggs MA, Robb SA, Kendall BE, Boyd SG : Myelination patterns on magnetic resonance of children with developmental delay. Dev Med Child Neurol 1990; 32: 295–303.
CAS PubMed Google Scholar - Kjos BO, Umansky R, Barkovich AJ : Brain MR imaging in children with developmental retardation of unknown cause: results in 76 cases. Am J Neuroradiol 1990; 11: 1040.
Google Scholar - Demaerel P, Kingsley DP, Kendall BE : Isolated neurodevelopmental delay in childhood: clinicoradiological correlation in 170 patients. Pediatr Radiol 1993; 23: 29–33.
CAS PubMed Google Scholar - Bouhadiba Z, Dacher J, Monroc M, Vanhulle C, Menard JF, Kalifa G : Apport de l'IRM cerebrale a l'exploration des retards psychomoteurs de l'enfant. J Radiol 2000; 81: 870–873.
CAS PubMed Google Scholar - Costeff H, Cohen BE, Weller L : Parental consanguinity among Israeli mental retardates. Acta Paediatr Scand 1972; 61: 452–458.
CAS PubMed Google Scholar - Laxova R, Ridler MAC, Bowen-Bravery M : An etiological survey of the severely retarded Hertfordshire children who were born between January 1, 1965 and December 31, 1967. Am J Med Genet 1977; 1: 75–86.
CAS PubMed Google Scholar - Fernell E : Aetiological factors and prevalence of severe mental retardation in children in a Swedish municipality: the possible role of consanguinity. Dev Med Child Neurol 1998; 40: 608–611.
CAS PubMed Google Scholar - Flint J, Wilkie AO, Buckle VJ, Winter RM, Holland AJ, McDermid HE : The detection of subtelomeric chromosomal rearrangements in idiopathic mental retardation. Nat Genet 1995; 9: 132–140.
CAS PubMed Google Scholar - Kaveggia EG, Opitz JM, Pallister PD : Diagnostic/genetic studies in severe mental retardation; in Proceedings of the Second Congress International Association for the Scientific Study on Mental Deficiency. Warsaw: Polish Medical Publishers, 1971, pp 305–312.
Google Scholar - Stevenson RE, Massey PS, Schroer RJ, McDermott S, Richter B : Preventable fraction of mental retardation: analysis based on individuals with severe mental retardation. Ment Retard 1996; 34: 182–188.
CAS PubMed Google Scholar - Bojko M : Human meiosis VIII. Chromosome pairing and formation of the synoptenemal complex in oocytes. Carlsberg Res Commun 1983; 48: 457–483.
Google Scholar - Giglio S, Broman KW, Matsumoto N et al: Olfactory receptor-gene clusters, genomic-inversion polymorphisms, and common chromosome rearrangements. Am J Hum Genet 2001; 68: 874–883.
CAS PubMed PubMed Central Google Scholar - Giglio S, Calvari V, Gregato G et al: Heterozygous submicroscopic inversions involving olfactory receptor-gene clusters mediate the recurrent t(4;8)(p16;p23) translocation. Am J Hum Genet 2002; 71: 276–285.
CAS PubMed PubMed Central Google Scholar - Be C, Velasquez P, Youlton R : Spontaneous abortion: cytogenetic study of 609 cases. Rev Med Chil 1997; 125: 317–322.
CAS PubMed Google Scholar - Shankaran S, Fanaroff AA, Wright LL et al: Risk factors for early death among extremely low-birth-weight infants. Am J Obstet Gynecol 2002; 186: 796–802.
PubMed Google Scholar - Stevenson DK, Verter J, Fanaroff AA et al: Sex differences in outcomes of very low birthweight infants: the newborn male disadvantage. Arch Dis Child (Fetal Neonatal Ed) 2000; 83: F182–F185.
CAS Google Scholar - Hagerman RJ, Amiri K, Cronister A : Fragile X checklist. Am J Med Genet 1991; 38: 283–287.
CAS PubMed Google Scholar - Van Karnebeek CD, Scheper FY, Abeling NG et al.: Etiology of cognitive delay in children referred to a tertiary care centre: A perspective study. Am J Mental Retardat (in the press).
- Papavasiliou AS, Bazigou H, Paraskevoulakos E, Kotsalis C : Neurometabolic testing in developmental delay. J Child Neurol 2000; 15: 620–622.
CAS PubMed Google Scholar - Van Bogaert P, Baleriaux D, Christophe C, Szliwowski HB : MRI of patients with cerebral palsy and normal CT scan. Neuroradiol 1992; 34: 52–56.
CAS Google Scholar - Shevell MI, Majnemer A, Rosenbaum P, Abrahamowicz M : Etiologic yield of suvspecialists' evaluation of young children with global developmental delay. J Pediatr 2000; 136: 593–598.
CAS PubMed Google Scholar
Acknowledgements
We thank Nicole Boluyt and Leontien C Kremer, paediatricians, for the comments on study strategies, and the Nederlandse Vereniging voor Kindergeneeskunde for providing a grant for part of the study.
Author information
Authors and Affiliations
- Department of Paediatrics/Emma Children's Hospital, Academic Medical Center, Amsterdam, The Netherlands
Clara D M van Karnebeek, Arnold G E Leenders, Martin Offringa & Raoul C M Hennekam - Department of Clinical Genetics, Academic Medical Center, Amsterdam, The Netherlands
Clara D M van Karnebeek, Maaike C E Jansweijer & Raoul C M Hennekam
Authors
- Clara D M van Karnebeek
You can also search for this author inPubMed Google Scholar - Maaike C E Jansweijer
You can also search for this author inPubMed Google Scholar - Arnold G E Leenders
You can also search for this author inPubMed Google Scholar - Martin Offringa
You can also search for this author inPubMed Google Scholar - Raoul C M Hennekam
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toRaoul C M Hennekam.
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website (http://www.nature.com.ejhg).
Supplementary information
Rights and permissions
About this article
Cite this article
van Karnebeek, C., Jansweijer, M., Leenders, A. et al. Diagnostic investigations in individuals with mental retardation: a systematic literature review of their usefulness.Eur J Hum Genet 13, 6–25 (2005). https://doi.org/10.1038/sj.ejhg.5201279
- Received: 19 January 2004
- Revised: 02 July 2004
- Accepted: 14 July 2004
- Published: 03 November 2004
- Issue Date: 01 January 2005
- DOI: https://doi.org/10.1038/sj.ejhg.5201279