Elevated Concentrations of CRF in the Locus Coeruleus of Depressed Subjects (original) (raw)
- Original Article
- Published: 12 March 2003
Neuropsychopharmacology volume 28, pages 1328–1335 (2003)Cite this article
- 3055 Accesses
- 127 Citations
- 3 Altmetric
- Metrics details
Abstract
Research evidence that corticotropin-releasing factor (CRF) plays a role in the pathophysiology of major depressive disorder (MDD) has accumulated over the past 20 years. The elevation of lumbar cerebrospinal fluid (CSF) concentrations of CRF decreased responsiveness of pituitary CRF receptors to challenge with synthetic CRF, and increased levels of serum cortisol in MDD subjects support the hypothesis that CRF is chronically hypersecreted in at least the endocrine circuits of the hypothalamic–pituitary–adrenal (HPA) axis and may also involve other CRF brain circuits mediating emotional responses and/or arousal. One such circuit includes the excitatory CRF input to the locus coeruleus (LC), the major source of norepinephrine in the brain. Furthermore, there are now reports of decreased levels of CRF in lumbar CSF from MDD patients after symptom relief from chronic treatment with antidepressant drugs or electroconvulsive therapy. Whether this normalization reflects therapeutic effects on both endocrine- and limbic-associated CRF circuits has not yet been effectively addressed. In this brief report, we describe increased concentrations of CRF-like immunoreactivity in micropunches of post-mortem LC from subjects with MDD symptoms as established by retrospective psychiatric diagnosis compared to nondepressed subjects matched for age and sex.
Similar content being viewed by others
INTRODUCTION
Since the discovery of corticotropin-releasing factor (CRF) by Vale and colleagues in 1979, the behavioral role of CRF in mediating fear and arousal responses, in addition to its physiological endocrine role in regulating release of adrenocorticotropin (ACTH) and other pro-opiomelanocortin (POMC) products from the anterior pituitary, has been firmly established in a variety of laboratory animals, including primates. Infusion of synthetic CRF into specific brain regions of several animal species elicits behaviors similar to fear and anxiety (Koob and Britton, 1990; Butler et al, 1990) and alteration of endogenous CRF concentrations has been demonstrated in several brain regions after chronic or acute exposure of laboratory animals to stressful stimuli that result in behavioral responses associated with fear and anxiety (Chappell et al, 1986; Bissette, 2001). Among these brain regions is the locus coeruleus (LC). This mesencephalic nucleus contains noradrenergic neuronal cell bodies that communicate with hypothalamic, limbic, and cortical targets and receive excitatory input from CRF postsynaptic terminals, among others. Thus, increased synaptic availability of CRF in the LC elicits fearful and anxious behaviors concomitant with increased activity of LC noradrenergic neurons, and chronic exposure to stress increases CRF synaptic availability in the LC.
Major depressive disorder (MDD) was first shown to involve the peripheral components of the hypothalamic–pituitary–adrenal (HPA) axis when Sachar et al (1973) demonstrated increased serum cortisol at all hours of the circadian rhythm in endogenously depressed subjects compared to nondepressed subjects. Carroll et al (1981) extended this finding to include early escape from suppression of pituitary secretion of ACTH in two-thirds of melancholic subjects after suppression of the HPA axis by acute administration of the synthetic glucocorticoid dexamethasone. Holsboer and his collaborators demonstrated that CRF receptors at the pituitary were less sensitive to stimulation with synthetic CRF in depressed subjects than those in nondepressed subjects or after relief of depressive symptoms (Heuser et al, 1994). Thus, the pituitary and adrenal components of the HPA axis often act as if they were subject to increased stimulation by CRF during depression and these alterations are largely normalized by treatments that produce remission of depressive symptoms (Figure 1).
Figure 1
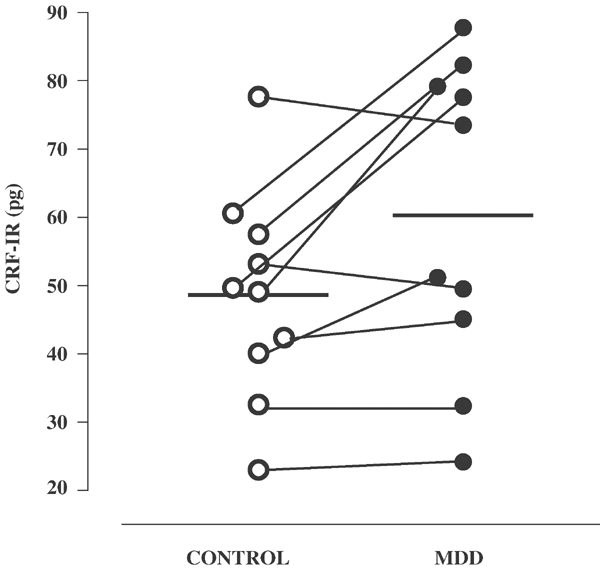
The average concentration of CRF in micropunched LC from depressed subjects compared to nondepressed controls. Difference is significant at the _p_⩽0.019 level (two-tailed, paired _t_-test, df=9).
Direct assessment of CRF in human central nervous system compartments has been necessarily limited to cerebrospinal fluid (CSF) and post-mortem brain examinations. A four-fold increase in the numbers of CRF mRNA expressing neurons in the hypothalamus of post-mortem brain from depressed subjects relative to normal controls has been reported (Raadsheer et al, 1994). Several reports of increased levels of CRF in lumbar CSF of depressed subjects have been published to date and have generally agreed that CRF is elevated during depressive episodes (Owens et al, 2000; Kasckow et al, 2001; Keck and Holsboer, 2001), although CSF obtained from the subarachnoid space over several hours has been reported to show decreased CRF concentrations in eucortisolemic depressed subjects relative to controls (Geracioti et al, 1997). The two published studies (Nemeroff et al, 1991; Kling et al, 1994) where pre- and post-treatment lumbar CSF samples from depressed subjects were obtained before and after electroconvulsive therapy (ECT) and the two published studies using this paired CSF sample design after chronic treatment with fluoxetine (Debellis et al, 1993) and amitriptyline (Heuser et al, 1998) have also demonstrated normalization of CRF levels in CSF after depressive symptom relief. Strong evidence for the involvement of CRF circuits in the production of depressive symptoms is also found in reports of CRF receptor antagonists having antidepressant effects (Zobel et al, 2000). Thus, some CRF circuits in the brain that contribute to lumbar CSF levels of CRF are apparently hyperactive during depressive symptom presence and it seems likely that at least some components of depressive symptoms may be caused by this putative hypersecretion of CRF.
Recent post-mortem studies demonstrate abnormalities in the amounts of specific proteins in the LC in depression and suicide. For example, elevated amounts of tyrosine hydroxylase (Zhu et al, 1999; Ordway et al, 1994) and reduced amounts of norepinephrine (NE) transporter binding (Klimek et al, 1997) in the LC have been reported for major depressive subjects when comparing to psychiatrically normal control subjects. Amounts of other proteins in the LC, such as neuron-specific enolase (Ordway et al, 1994), monoamine oxidase A (Ordway et al, 1999), and noradrenergic neuron number (Zhu et al, 1999; Ordway et al, 1994) appear to be unchanged. Since treatments that elevate the activity of the LC are capable of producing elevation of tyrosine hydroxylase and reductions of NE transporter, it has been suggested that such findings indicate that alteration in LC biochemistry in MDD result from an increased demand for NE, possibly as a result of increased excitatory input. CRF is a potential candidate transmitter that could contribute to LC dysfunction in MDD.
In order to test the hypothesis that certain specific CRF circuits, which are activated by chronic stress, are also similarly changed in MDD, we measured CRF concentrations in post-mortem LC from 10 drug-free subjects with depressive symptoms at the time of death as established by psychiatric autopsy at the Case Western Reserve Brain Bank in Cleveland, OH. These subjects were matched by age and sex with 10 psychiatrically normal subjects.
METHODS
Tissue Collection and Psychiatric Autopsy
Human brain tissue was obtained at autopsy at the Cuyahoga County Coroner's Office, Ohio, in accordance with an approved Institutional Review Board protocol. Subjects were refrigerated on arrival at the coroner's office and were coded to protect identities. Causes of death were determined by the coroner (see Table 1). Information on the lifetime and current (within last month) psychiatric status, psychotropic medication, and illicit drug use of all subjects was obtained in structured clinical interviews (Andreasen et al, 1977) by a trained interviewer with the next of kin. The interview used was the Schedule for Affective Disorders and Schizophrenia: Lifetime version (SADS-L; Endicott and Spitzer, 1978) supplemented by questions from the Diagnostic Interview Schedule (DIS-III-R; Robbins et al, 1989) to make diagnoses compatible with the Diagnostic and Statistical Manual of Mental Disorders, revised third edition (DSM-III-R; American Psychiatric Association, 1987). Accurate assessment of the number of depressive episodes was difficult in many cases: only one major depressive subject had evidence of only a single episode (active at the time of death) of major depression. Control subjects were individuals who died from natural causes (eg heart disease), accidents (not drivers in a single car fatalities), or homicides and who had no psychiatric diagnosis at the time of death, as indicated by results of interviews of next of kin. Medical histories of control subjects included cardiovascular disease (_n_=6), diabetes (_n_=2), and obesity (_n_=1). Medical histories of MDD subjects included lymphoma (_n_=1), cancer (_n_=2), cataracts (_n_=1), arthritis (_n_=1), arteriosclerosis (_n_=1), diabetes (_n_=1), and obesity (_n_=1). Evaluation of drug and alcohol abuse and dependency was assessed using the DSM-III-R. Information on smoking history was also collected in the interview. Nonsmokers were people with no history of cigarette smoking or tobacco chewing. Axis I diagnoses were made by a psychiatrist (HYM) and a clinical psychologist (JCO), based on data gathered from the structured interview and, when available, hospital and doctor's records. A toxicology screen on blood, bile, and urine from all of the subjects was performed by the Cuyahoga County Coroner's Office as described previously (Klimek et al, 1997; Ordway et al, 1997). Qualitative and quantitative assays were used to detect the following compounds or classes of compounds: ethanol, barbiturates, benzodiazepines, sympathomimetic drugs, and many antidepressant and antipsychotic drugs and their metabolites. Although antidepressant drugs were not detected in any of the subjects selected for the study, it is noteworthy that three of the depressed subjects had an antidepressant drug prescribed within the last month prior to death.
Table 1 Vital Data of Subjects
At the time of autopsy, blocks (approximately 2.5 × 2.5 × 2.5 cm3) of brain tissue were dissected and placed on a cardboard square. The cardboard square has a simple map on it that indicates the brain region and its neuroanatomical coordinates, for example, medial, lateral, rostral, caudal to aid orientation of the block. Immediately after dissection, each tissue block was dipped in 2-methylbutane (−50°C) and frozen on dry ice, marking the end of the post-mortem interval, and stored at −83°C. Post-mortem brain tissues containing the LC were obtained from 10 subjects diagnosed with major depression and 10 age-matched psychiatrically normal controls (Table 1).
Dissection
Tissue blocks of human dorsal pons containing the LC were dissected as described previously (Klimek et al, 1997; Ordway et al, 1997) using a cryostat microtome (−15°C). Blocks were sectioned sequentially in a transverse plane perpendicular to the floor of the fourth ventricle throughout the entire length of the LC beginning near its rostral end. The rostral border of the LC was defined by the frenulum and the caudal border was the caudal extent of the LC (at the level of the motor nucleus of trigeminal nerve), defined as the point at which neuromelanin-containing cells in the LC region were no longer visible. LC nuclei were dissected from tissue sections using 5 mm bore disposable biopsy punch (Sklar Instruments, West Chester, PA), centering the punch over the compact group of neuromelanin-containing cells (visible with the naked eye). Eight punches of LC nuclei from four 40 μm sections were collected at approximately 5–7 mm caudal from the frenulum for each subject. The eight punches of each subject were pooled and stored in an Eppendorf microfuge tube at −83°C until assayed.
CRF RADIOIMMUNOASSAY
Micropunches from each of the four regions of the LC were extracted in 1 N ice-cold HCl during homogenization with an Ultraturex microhomogenizer. After centrifugation at 4°C, supernatant aliquots were placed in 10 × 75 mm borosilicate glass tubes and frozen while the pellets were dissolved in 1 N NaOH for subsequent protein assay. Frozen supernatant aliquots were lyophilized with a Savant system and reconstituted in RIA assay buffer. The CRF radioimmunoassay is described in Bissette (2001) and a rabbit antiserum raised against rat/human CRF at the National Institutes of Health, Bethesda, MD, is used. Displacement assay conditions were used and under these conditions the sensitivity was 1.25 pg/tube with an IC50 of 10 pg. Radioactive tracer was prepared with the Bolton–Hunter chloramine T reaction using Tyr0-CRF.
Using an antisera concentration that bound 30% of this tracer, the assay was run across 4 days. On day 1, CRF antisera was added to the lyophilized duplicate micropunch extracts reconstituted in 200 μl of RIA buffer. Standard curves were prepared in RIA buffer with synthetic rat/human CRF in serial dilutions from 5120 to 0.625 pg per tube. After incubation at 4°C for 36 h, 20 000 cpm of the tracer in 50 μl RIA buffer was added per tube and incubated at 4°C for 12 h. On the third day, 20 μl of goat anti-rabbit antisera diluted 3 : 1 in an RIA buffer was added to precipitate the primary antiserum/tracer complex and tubes were incubated at 4°C for 24 h. On the fourth day, tubes were centrifuged at 4°C for 20 min in a Sorvall 3B preparative centrifuge, supernatants were vacuum aspirated and pellets were counted for 2 min each on a Wallac Rackgamma gamma counter with 70% efficiency. Proteins were assayed in the tissue pellets by a Technicon automated system using the Folin-phenol reagent and total sample protein was divided into total sample CRF content to yield units of pg CRF per mg protein.
STATISTICAL ANALYSIS
Average CRF concentrations were compared between the matched pairs of subjects with a paired sample _t_-test after ANOVA. Level of significance was set at _p_=0.05 for this analysis.
RESULTS
The age of control subjects ranged from 26 to 78 years (57±5 years) and the age of depressed subjects ranged from 23 to 87 years (61±7 years), yielding a mean difference of 4±4 years that was not statistically significant. Post-mortem intervals of control and depressed subjects ranged from 11 to 27 h (18±2 h) and from 5 to 30 h (19±2 h), respectively. Both ages and post-mortem intervals were not significantly different between major depressive and control subjects. There were no significant correlations in either study group between age or post-mortem delay and levels of CRF immunoreactivity (data not shown). CRF immunoreactivity was evaluated in 10 males (four major depressive and six control subjects) and 10 females (six major depressive and four control subjects). There was no significant difference in the CRF immunoreactivity between males and females across both the study groups (data not shown).
Of the four anterior–posterior regions of the LC that were micropunched, only the second region had equal numbers of samples from all 20 subjects, therefore only this region of the LC was compared between the matched pairs of subjects for CRF concentrations. The average concentration of CRF in this region of the LC was 48.6 pg/mg protein in the controls (range 23.0–77.7 pg) and the average concentration of CRF in the MDD subjects was 60.3 pg/mg protein (range 24.2–87.8 pg), which was significant at the _p_⩽0.019 level (paired _t_-test after ANOVA, _t_=2.852, df=9).
DISCUSSION
These data indicate that the LC concentrations of CRF are increased in human post-mortem brain from subjects with MDD at the time of death. The subjects chosen for the tissue used in this study were among the best that the current technology can provide. The absence of psychiatric drugs in the majority of subjects, matching the few subjects with drugs present between control and depressed groups and the care taken to establish retrospectively depressive symptoms at the time of death in the depressed subjects make these tissues among the better characterized of those available for post-mortem human psychiatric research. These efforts help insure that the differences in LC concentrations of CRF between the two groups of subjects are most likely because of the presence of depressive symptoms and do not accrue from agonal stress at the time of death. The area from which these micropunches were derived correspond to the region of the LC where immunohistochemical studies have demonstrated the highest concentrations of CRF staining fibers and terminals (Austin et al, 1995). Even so, it was necessary to pool from 8 to 10 micropunches to have enough CRF to measure accurately with freshly prepared radioactive tracer in a displacement radioimmunoassay using a sensitive and specific antisera to CRF. A preliminary immunohistochemical evaluation of CRF in the pontine region of suicides with depressive symptoms has documented an average 27% increase in CRF immunoreactivity in the depressed group of 12 matched pairs of control and depressed subjects (Austin and Murphy, 2000), and this agrees well with our finding of an average 20% increase in CRF protein in our depressed subjects.
The physiological and psychiatric results of such increased concentrations of CRF in the LC cannot be accurately deduced without more knowledge of the likely mechanism behind these concentration changes. Increased regional concentrations of a neuropeptide can be because of increased synthesis that exceeds physiological release and degradation or, alternatively, can be because of decreased release and/or degradation relative to synthesis and storage. In the former case, CRF mRNA levels should also increase and, if release is sustained, should be associated with locally reduced numbers of postsynaptic CRF receptors. This receptor ‘downregulation’ was originally reported for CRF receptors in the frontal cortex of suicides (Nemeroff et al, 1988). However, decreased numbers of CRF receptors were not confirmed in the two later studies (Leake et al, 1990; Hucks et al, 1997), although in none of these studies was depressive symptoms documented at the time of death as in the present study. In the hypothetical case of decreased release of CRF resulting in increased storage of presynaptic CRF, a regional increase in CRF-binding protein and, eventually, increased numbers of CRF receptors might be expected. Because multiple examples of both of these putative mechanisms have been seen in other situations of regionally altered neuropeptide concentrations, without knowing what the CRF receptor responses were and what the status of CRF synthesis was at the time of death in these subjects, it is not possible to specify positively the mechanism behind the increased LC concentrations of CRF in these depressed subjects with absolute certainty.
However, knowing the physiological effects of CRF in the LC and knowing what the neuroanatomic projections of the LC are, as well as the behavioral effects of CRF on LC neurons in laboratory animals, one can make some reasonable deductions about the current finding.The LC is the major source of NE to the cortex, hippocampus, and cerebellum and the hypothalamic projection of the LC provides excitatory innervation to CRF neurons of the paraventricular nucleus based upon immunohistochemical evidence (Alonso et al, 1986; Petrov et al, 1993). Intracerebroventricular administration of CRF increases release of NE as measured by microdialysis probes sited in the medial hypothalamus (Lavicky and Dunn, 1993). This stimulatory effect of NE on CRF release has been observed in rat hypothalamic explants and was blocked by beta-adrenergic receptor antagonists, but not alpha adrenergic receptor blocking drugs (Tsagarakis et al, 1988). However, others (Suda et al, 1987) have reported decreased release of hypothalamic CRF after incubation of isolated hypothalamus with nanomolar concentrations of NE that were blocked by both alpha- and beta-adrenergic antagonists. Destruction of noradrenergic efferents to the hypothalamus with a neurotoxin such as 6-hydroxy dopamine (Feldman and Weidenfeld, 1993) or brain-stem hemisection (Pacak et al, 1993) attenuates activation of the HPA axis by stressors such as bright light, loud noise, and immobilization. While the LC contributes to hypothalamic NE, microdialysis studies have indicated that medullary sources of NE are more important activators of hypothalamic NE release by stressors (Pacak et al, 1995).
The LC receives CRF input from the central nucleus of the amygdala, in addition to other limbic brain regions, including bed nucleus of the stria terminalis and hypothalamic subregions (Valentino et al, 1992). The LC also receives excitatory CRF input from other sources (Valentino et al, 1996), such as substance P and glutamate. CRF afferents to the LC are topographically organized with respect to the type of information that is conveyed (van Bockstaele et al, 1998) and CRF terminals form direct contacts with noradrenergic dendrites (Van Bockstaele et al, 1996). In general, pontine-medullary CRF projections to the LC coordinate cognitive and autonomic responses to internal physiologic challenges, while limbic CRF projections mediate LC activation by external stressors having emotional content (Koob, 1999). Limbic CRF neurons project to the peri-LC area, particularly to the rostrolateral peri-LC. CRF, injected i.c.v. or directly into the LC, activates LC neurons and enhances NE release in projection areas (Smagin et al, 1995). Internal and external stressors activate the LC via CRF, including colonic distension, hypotensive challenge, and footshock. LC activation by these stressors is blocked by CRF antagonists (Curtis et al, 1994; Lechner et al, 1997; Melia et al, 1992; Valentino et al, 1991). Interestingly, CRF immunostaining is robustly increased in the LC by acute and chronic stress in rats and by nonshock, psychological stress (Makino et al, 1999). Based on these animal studies, it may be argued that the increased concentrations of CRF in the LC of depressed subjects may be because of pre-mortem stress-induced elevations of LC CRF and a depression-associated elevation in pre-mortem LC activity. By extension, some of the symptoms of depression may be due a CRF-induced overdrive of LC neurons.
Other evidence of elevated activity of the LC in major depression has been reported. For example, levels of tyrosine hydroxylase immunoreactivity are elevated in the LC of major depressive subjects and suicide victims compared to normal control subjects (Zhu et al, 1999). In animals, tyrosine hydroxylase mRNA is increased in the LC even after a single stressor, although increases in tyrosine hydroxylase protein require repeated stressors (Melia et al, 1992; Nestler et al, 1999; Sabban and Kvetnansky, 2001). Interestingly, repeated administration of CRF results in the upregulation of tyrosine hydroxylase in the LC (Melia and Duman, 1991). Furthermore, stress-induced upregulation of TH in the LC is antagonized by coadministration of a CRF antagonist (Melia and Duman, 1991). Hence, elevations of CRF in the LC of depressed subjects demonstrated here may underlie the neurobiological cause for elevated concentrations of tyrosine hydroxylase in depression. These conclusions presume that the increased concentrations of CRF in the LC of these depressed subjects are because of increased release of CRF onto LC neurons, which as mentioned above is not without alternative explanation. However, the behavioral and physiological literature findings make a strong case for such an interpretation.
Firing rates of LC neurons are increased by tasks requiring vigilance (Robbins and Everitt, 1995) and by stressful stimuli (Abercrombie and Zigmond, 1995). The anxiogenic properties of stressful stimuli are thought to be due, in large part, to such activation of LC neurons (Charney et al, 1995). Electrophysiological evidence has established that CRF-containing nerve terminals synapse with LC neurons and microiontophoretic application of CRF increases the firing rate of the LC neurons (Foote and Aston-Jones, 1995; Valentino and Aston-Jones, 1995; Page and Abercrombie, 1999). This effect of CRF on LC neuronal firing rates is potentiated by two weeks of daily cold exposure (Jedema et al, 2001) or in animals subjected to a forced swim test (Curtis et al, 1999), indicating that chronic physiological stressors can modulate the effect of CRF on the LC. However, the effects of CRF on LC firing rates can be desensitized by 1 or 5 days of prior (30 min) footshock stress (Curtis et al, 1995) or intracerebral application of CRF (Conti and Foote, 1996), indicating different responses of LC firing rates to CRF depending upon the quantity and quality of stressor exposure. The ability of CRF to induce behavioral correlates of fear and anxiety (defensive withdrawal) after intraventricular delivery is potentiated by infusion into the LC region (Butler et al, 1990) and two weeks of chronic, unpredictable stress produces increased concentrations of CRF in the LC of laboratory rats (Chappell et al, 1986). A CRF receptor antagonist applied to the LC attenuates the defensive withdrawal induced by prior immobilization (Smagin et al, 1996). Thus, it may be argued that the increased concentrations of CRF in the LC of these depressed patients may be because of the stressful aspects of depressive symptoms and, by extension, that some of the symptoms of depression may be because of CRF overdrive of these LC neurons.
Because several lines of evidence indicate that CRF projections to the median eminence are hypersecreting CRF into the pituitary portal system with subsequent dysregulation of the HPA axis in MDD (Roy, 1992; Keck and Holsboer, 2001), it is tempting to speculate that the LC projection to the hypothalamus may be one of the excitatory inputs into this endocrine CRF circuit. Furthermore, the close apposition of the LC to the ventricular system may allow increased CRF release in the area of the LC to contribute to the elevated CRF concentrations found in the lumbar CSF of depressed subjects, as is strongly suspected for contributions from hypothalamic CRF also. While CRF levels in lumbar CSF from depressed patients (Nemeroff et al, 1984; Banki et al, 1987) or completed suicides (Arato et al, 1989) has been repeatedly demonstrated to be elevated, CRF concentrations in lumbar CSF from subjects with a prior suicide attempt have been reported either to be decreased (Traskman-Bendz et al, 1992; Brunner et al, 2001) or unchanged (Westrin et al, 2001), possibly because of the absence of depressive symptoms at the time of CSF withdrawal. Thus, a case can now be made for hypersecretion of CRF in both the LC and hypothalamus of depressed patients potentially contributing to elevated lumbar CSF levels of CRF.
Perhaps the most important contribution that this finding of increased CRF in the LC provides for depression research is the validation of laboratory animal models of chronic stress as surrogates for at least some of the neurochemical changes now confirmed in humans. The advent of imaging techniques and the revolution in molecular biology has combined to make animal models suspect in investigations of neurochemical substrates of depressive symptoms in recent years. Yet the inability of current imaging techniques to provide such data for most neurotransmitters in humans and the vast uncertainties associated with extrapolating mRNA concentrations to synaptic availability of the protein product leaves few alternatives for conducting such animal model research. Clear evidence of congruence between human and animal responses in CRF circuits innervating the LC has now provided a way to assess the relevance of animal models to human MDD, at least for this neuropeptide neurotransmitter.
References
- Abercrombie ED, Zigmond MJ (1995). Modification of central catecholaminergic systems by stress and injury: Functional significance and clinical implications. In: Bloom FE, Kupfer DJ (eds) Psychopharmacology: The Fourth Generation of Progress. Raven Press: New York. pp 355–362.
Google Scholar - Alonso G, Szafarczyk A, Balmerfrezol M, Assenmacher I (1986). Immunocytochemical evidence for stimulatory control by the ventral noradrenergic bundle of parvocellular neurons of the paraventricular nucleus secreting corticotropin-releasing hormone and vasopressin in rats. Brain Res 397: 297–307.
Article CAS PubMed Google Scholar - American Psychiatric Association (1987). Diagnostic and Statistical Manual of Mental Disorders, 3rd edn (revised). APA Press, Washington, DC.
- Andreasen NC, Endicott J, Spitzer RL, Winokur G (1977). The family history method using diagnostic criteria. Reliability and validity. Arch Gen Psychiatry 34: 1229–1235.
Article CAS PubMed Google Scholar - Arato M, Banki CM, Bissette G, Nemeroff CB (1989). Elevated CSF CRF in suicide victims. Biol Psychiatry 25: 355–359.
Article CAS PubMed Google Scholar - Austin MC, Murphy HA (2000). Corticotropin-releasing hormone immunoreactivity is increased in pontine nuclei of depressed suicide victims. Soc Neurosci Abstr 26: 2312 867.3.
Google Scholar - Austin MC, Rice PM, Mann JJ, Arango V (1995). Localization of corticotropin-releasing hormone in the human locus coeruleus and pedunculopontine tegmental nucleus: an immunocytochemical and in situ hybridization study. Neuroscience 64: 713–727.
Article CAS PubMed Google Scholar - Banki CM, Bissette G, Arato M, O'Connor L, Nemeroff CB (1987). Cerebrospinal fluid corticotropin-releasing factor-like immunoreactivity in depression and schizophrenia. Am J Psychiatry 144: 873–877.
Article CAS PubMed Google Scholar - Bissette G (2001). Effects of sertraline on regional neuropeptide concentrations in olfactory bulbectomized rats. Pharmacol Biochem Behav 69: 1–13.
Article Google Scholar - Brunner J, Stalla GK, Stalla J, Uhr M, Grabner A, Wetter TC, Bronisch T (2001). Decreased corticotropin-releasing hormone (CRH) concentrations in the cerebrospinal fluid of eucortisolemic suicide attempters. J Psychiatr Res 35: 1–9.
Article CAS PubMed Google Scholar - Butler PD, Weiss JM, Stout JC, Nemeroff CB (1990). Corticotropin-releasing factor produces fear-enhancing and behavioral activating effects following infusion into the locus coeruleus. J Neurosci 10: 176–183.
Article CAS PubMed PubMed Central Google Scholar - Carroll BJ, Feinberg M, Greden JF, Tarika J, Albala AA, Haskett RF, James NM, Kronfol Z, Lohr N, Steiner M, deVigne JP, Young E (1981). A specific laboratory test for the diagnosis of melancholia: standardization, validation, and clinical utility. Arch Gen Psychiatry 38: 15–22.
Article CAS PubMed Google Scholar - Chappell PB, Smith MA, Kilts CD, Bissette G, Ritchie J, Anderson C, Nemeroff CB (1986). Alterations in corticotropin-releasing factor-like immunoreactivity in discrete rat brain regions after acute and chronic stress. J Neurosci 6: 2908–2914.
Article CAS PubMed PubMed Central Google Scholar - Charney DS, Bremner JD, Redmond DE (1995). Noradrenergic substrates for anxiety and fear: clinical associations based on preclinical research. In: Bloom FE, Kupfer DJ (eds) Psychopharmacology: The Fourth Generation of Progress. Raven Press: New York. pp 387–396.
Google Scholar - Conti LH, Foote SL (1996). Reciprocal cross-desensitization of locus coeruleus electrophysiological responsivity to corticotropin-releasing factor and stress. Brain Res 722: 19–29.
Article CAS PubMed Google Scholar - Curtis AL, Grigoriadis DE, Page ME, Rivier J, Valentino RJ (1994). Pharmacological comparison of two corticotropin-releasing factor antagonists: in vivo and in vitro studies. J Pharmacol Exp Ther 268: 359–365.
CAS PubMed Google Scholar - Curtis AL, Pavcovich LA, Grigoriadis DE, Valentino RJ (1995). Previous stress alters corticotropin-releasing factor neurotransmission in the locus coeruleus. Neuroscience 65: 541–550.
Article CAS PubMed Google Scholar - Curtis AL, Pavcovich LA, Valentino RJ (1999). Long-term regulation of locus coeruleus sensitivity to corticotropin-releasing factor by swim stress. J Pharmacol Exp Ther 289: 1211–1219.
CAS PubMed Google Scholar - DeBellis MD, Gold PW, Geracioti TD, Listwak SJ, Kling MA (1993). Association of fluoxetine treatment with reductions in CSF concentrations of corticotropin-releasing hormone and arginine vasopressin in patients with major depression. Am J Psychiatry 150: 656–657.
Article CAS Google Scholar - Endicott J, Spitzer RL (1978). A diagnostic interview: the schedule for affective disorders and schizophrenia. Arch Gen Psychiatry 35: 837–844.
Article CAS PubMed Google Scholar - Feldman S, Weidenfeld J (1993). Hypothalamic norepinephrine depletion inhibits CRF-41 release following neural stimuli. Neuroreport 5: 258–260.
Article CAS PubMed Google Scholar - Foote SL, Aston-Jones GS (1995). Pharmacology and physiology of central noradrenergic systems. In: Bloom FE, Kupfer DJ (eds) Psychopharmacology: The Fourth Generation of Progress. Raven Press: New York. pp 335–346.
Google Scholar - Geracioti TD, Loosen PT, Orth DN (1997). Low cerebrospinal fluid corticotropin-releasing hormone concentrations in eucortisolemic depression. Biol Psychiatry 42: 165–174.
Article CAS PubMed Google Scholar - Heuser I, Bissette G, Dettling M, Schweiger U, Gotthardt U, Schmider J, Lammers C-H, Nemeroff CB, Holsboer F (1998). Cerebrospinal fluid concentrations of corticotropin-releasing hormone, vasopressin and somatostatin in depressed patients and healthy controls: response to amitriptyline treatment. Depression Anxiety 8: 71–79.
Article CAS PubMed Google Scholar - Heuser I, Yassouridis A, Holsboer F (1994). The combined dexamethasone/CRH test: a refined laboratory test for psychiatric disorders. J Psychiatr Res 28: 341–356.
Article CAS PubMed Google Scholar - Hucks D, Lowther S, Crompton MR, Katona CL, Horton RW (1997). Corticotropin-releasing factor binding sites in cortex of depressed suicides. Psychopharmacology 134: 174–178.
Article CAS PubMed Google Scholar - Jedema HP, Finlay JM, Sved AF, Grace AA (2001). Chronic cold exposure potentiates CRF-evoked increases in electrophysiologic activity of locus coeruleus neurons. Biol Psychiatry 49: 351–359.
Article CAS PubMed Google Scholar - Kasckow JW, Baker D, Geracioti TD (2001). Corticotropin-releasing hormone in depression and post-traumatic stress disorder. Peptides 22: 845–851.
Article CAS PubMed Google Scholar - Keck ME, Holsboer F (2001). Hyperactivity of CRH neuronal circuits as a target for therapeutic interventions in affective disorders. Peptides 22: 835–844.
Article CAS PubMed Google Scholar - Klimek V, Stockmeier CA, Overholser J, Meltzer HY, Kalka S, Dilley G, Ordway GA (1997). Reduced levels of norepinephrine transporters in the locus coeruleus in major depression. J Neurosci 17: 8451–8458.
Article CAS PubMed PubMed Central Google Scholar - Kling MA, Geracioti TD, Licinio J, Michelson D, Oldfield EH, Gold PW (1994). Effects of electroconvulsive therapy on the CRH-ACTH-cortisol system in melancholic depression: preliminary findings. Psychopharmacol Bull 30: 489–494.
CAS PubMed Google Scholar - Koob GF (1999). Corticotropin-releasing factor, norepinephrine, and stress. Biol Psychiatry 46: 1167–1180.
Article CAS PubMed Google Scholar - Koob G, Britton K (1990). Behavioral effects of corticotropin-releasing factor. In: DeSouza EB, Nemeroff CB (eds) Corticotropin-Releasing Factor: Basic and Clinical Studies of a Neuropeptide. CRC Press: Boca Raton, FL, 253–265.
Google Scholar - Lavicky J, Dunn AJ (1993). Corticotropin-releasing factor stimulates catecholamine release in hypothalamus and prefrontal cortex in freely moving rats as assessed by microdialysis. J Neurochem 60: 602–612.
Article CAS PubMed Google Scholar - Leake A, Perry EK, Perry RH, Fairbairn AF, Ferrier IN (1990). Cortical concentrations of corticotropin-releasing hormone and its receptor in Alzheimer type dementia and major depression. Biol Psychiatry 28: 603–608.
Article CAS PubMed Google Scholar - Lechner SM, Curtis AL, Brons R, Valentino RJ (1997). Locus coeruleus activation by colon distention: role of corticotropin-releasing factor and excitatory amino acids. Brain Res 756: 114–124.
Article CAS PubMed Google Scholar - Makino S, Shibasaki T, Yamauchi N, Nishioka T, Mimoto T, Wakabayashi I, Gold PW, Hashimoto K (1999). Psychological stress increased corticotropin-releasing hormone mRNA and content in the central nucleus of the amygdala but not in the hypothalamic paraventricular nucleus in the rat. Brain Res 850: 136–143.
Article CAS PubMed Google Scholar - Melia KR, Duman RS (1991). Involvement of corticotropin-releasing factor in chronic stress regulation of the brain noradrenergic system. Proc Natl Acad Sci 88: 8382–8386.
Article CAS PubMed PubMed Central Google Scholar - Melia KR, Rasmussen K, Terwilliger RZ, Haycock JW, Nestler EJ, Duman RS (1992). Coordinate regulation of the cyclic AMP system with firing rate and expression of tyrosine hydroxylase in the rat locus coeruleus: effects of chronic stress and drug treatments. J Neurochem 58: 494–502.
Article CAS PubMed Google Scholar - Nemeroff CB, Bissette G, Akil H, Fink M (1991). Neuropeptide concentrations in the cerebrospinal fluid of depressed patients treated with electroconvulsive therapy: corticotropin-releasing factor, ß-endorphin and somatostatin. Br J Psychiatry 158: 59–63.
Article CAS PubMed Google Scholar - Nemeroff CB, Owens MJ, Bissette G, Andorn AC, Stanley M (1988). Reduced corticotropin-releasing factor binding sites in the frontal cortex of suicide victims. Arch Gen Psychiatry 45: 577–579.
Article CAS PubMed Google Scholar - Nemeroff CB, Widerlov E, Bissette G, Walleus H, Karlsson I, Eklund K, Kilts CD, Loosen PT, Vale W (1984). Elevated concentrations of CSF corticotropin-releasing factor-like immunoreactivity in depressed patients. Science 226: 1342–1344.
Article CAS PubMed Google Scholar - Nestler EJ, Alreja M, Aghajanian GK (1999). Molecular control of locus coeruleus neurotransmission. Biol Psychiatry 46: 1131–1139.
Article CAS PubMed Google Scholar - Ordway GA, Farley JT, Dilley GE, Overholser JC, Meltzer HY, Balraj EK, Stockmeier CA, Klimek V (1999). Quantitative distribution of monoamine oxidase A in brainstem monoamine nuclei is normal in major depression. Brain Res 847: 71–79.
Article CAS PubMed Google Scholar - Ordway GA, Stockmeier CA, Cason GW, Klimek V (1997). Pharmacology and distribution of norepinephrine transporters in the human locus coeruleus and raphe nuclei. J Neurosci 17: 1710–1719.
Article CAS PubMed PubMed Central Google Scholar - Ordway GA, Streator-Smith K, Haycock J (1994). Elevated tyrosine hydroxylase in the locus coeruleus of suicide victims. J Neurochem 62: 680–685.
Article CAS PubMed Google Scholar - Owens MJ, Nemeroff CB, Bissette G (2000). Neuropeptides: biology and regulation. In: Sadock BJ, Sadock VA (eds) Comprehensive Textbook of Psychiatry. Lippincott, Williams & Wilkins, Philadelphia, PA, Baltimore, MD. pp 60–70.
Google Scholar - Pacak K, Palkovits M, Kopin IJ, Goldstein DS (1995). Stress-induced norepinephrine release in the hypothalamic paraventricular nucleus and pituitary-adrenocortical and sympathoadrenal activity: in vivo microdialysis studies. Front Neuroendocrinol 16: 89–150.
Article CAS PubMed Google Scholar - Pacak K, Palkovits M, Kvetnansky R, Kopin IJ, Goldstein DS (1993). Stress-induced norepinephrine release in the paraventricular nucleus of rats with brainstem hemisections: a microdialysis study. Neuroendocrinology 58: 196–201.
Article CAS PubMed Google Scholar - Page ME, Abercrombie ED (1999). Discrete local application of corticotropin-releasing factor increases locus coeruleus discharge and extracellular norepinephrine in rat hippocampus. Synapse 33: 304–313.
Article CAS PubMed Google Scholar - Petrov T, Krukoff TL, Jhamandas JH (1993). Branching projections of catecholaminergic brainstem neurons to the paraventricular hypothalamic nucleus and the central nucleus of the amygdala. Brain Res 609: 81–92.
Article CAS PubMed Google Scholar - Raadsheer FC, Hoogendijk WJ, Stam FC, Tilders FJ, Swaab DF (1994). Increased numbers of corticotropin-releasing hormone expressing neurons in the hypothalamic paraventricular nucleus of depressed patients. Neuroendocrinology 60: 436–444.
Article CAS PubMed Google Scholar - Robbins L, Cottler L, Keating S (1989). NIMH Diagnostic Interview Schedule, Version III, Revised (DIS-III-R) Department of Psychiatry, Washington University, St Louis, MO.
Google Scholar - Robbins TW, Everitt BJ (1995). Central norepinephrine neurons and behavior. In: Bloom FE, Kupfer DJ (eds) Psychopharmacology: The Fourth Generation of Progress. Raven Press: New York. pp 363–372.
Google Scholar - Roy A (1992). Hypothalamic–pituitary–adrenal axis function and suicidal behavior in depression. Biol Psychiatry 32: 812–816.
Article CAS PubMed Google Scholar - Sabban EL, Kvetnansky R (2001). Stress-triggered activation of gene expression in catecholaminergic systems: dynamics of transcriptional events. Trends Neurosci 24: 91–98.
Article CAS PubMed Google Scholar - Sachar EJ, Hellman L, Roffwarg HP, Halpern FS, Fukushima DK, Gallagher TF (1973). Disrupted 24-hour patterns of cortisol secretion in psychotic depression. Arch Gen Psychiatry 28: 19–24.
Article CAS PubMed Google Scholar - Smagin GN, Harris RB, Ryan DH (1996). Corticotropin-releasing factor receptor antagonist infused into the locus coeruleus attenuates immobilization stress-induced defensive withdrawal in rats. Neurosci Lett 220: 167–170.
Article CAS PubMed Google Scholar - Smagin GN, Swiergiel AH, Dunn AJ (1995). Corticotropin-releasing factor administered into the locus coeruleus, but not the parabrachial nucleus, stimulates norepinephrine release in the prefrontal cortex. Brain Res Bull 36: 71–76.
Article CAS PubMed Google Scholar - Suda T, Yajima F, Tomori N, Sumitomo T, Nakagami Y, Ushiyama T, Demura H, Shizume K (1987). Inhibitory effect of norepinephrine on immunoreactive corticotropin-releasing factor release from the rat hypothalamus in vitro. Life Sci 40: 1645–1649.
Article CAS PubMed Google Scholar - Traskman-Bendz L, Ekman R, Regnell G, Ohman R (1992). HPA-related CSF neuropeptides in suicide attempters. Eur J Neuropsychopharmacol 2: 99–106.
Article CAS Google Scholar - Tsagarakis S, Holly JM, Rees LH, Besser GM, Grossman A (1988). Acetylcholine and norepinephrine stimulate the release of corticotropin-releasing factor-41 from the rat hypothalamus in vitro. Endocrinology 123: 1962–1969.
Article CAS PubMed Google Scholar - Valentino RJ, Page M, van Bockstaele E, Aston-Jones G (1992). Corticotropin-releasing factor innervation of the locus coeruleus region: distribution of fibers and sources of input. Neuroscience 48: 689–705.
Article CAS PubMed Google Scholar - Valentino RJ, Page ME, Curtis AL (1991). Activation of noradrenergic locus coeruleus neurons by hemodynamic stress is due to local release of corticotropin-releasing factor. Brain Res 555: 25–34.
Article CAS PubMed Google Scholar - Valentino RJ, Aston-Jones GS (1995). Physiological and anatomic determinants of locus coeruleus discharge: behavioral and clinical implications. In: Bloom FE, Kupfer DJ (eds) Psychopharmacology: The Fourth Generation of Progress. Raven Press: New York. pp 373–386.
Google Scholar - Valentino RJ, Chen S, Zhu Y, Aston-Jones G (1996). Evidence for divergent projections to the brain noradrenergic system and the spinal parasympathetic system from Barrington's nucleus. Brain Res 732: 1–15.
Article CAS PubMed Google Scholar - Van Bockstaele EJ, Colago EE, Valentino RJ (1996). Corticotropin-releasing factor-containing axon terminals synapse onto catecholamine dendrites and may presynaptically modulate other afferents in the rostral pole of the nucleus locus coeruleus in the rat brain. J Comp Neurol 364: 523–534.
Article CAS PubMed Google Scholar - Van Bockstaele EJ, Colago EE, Valentino RJ (1998). Amygdaloid corticotropin-releasing factor targets locus coeruleus dendrites: substrate for the co-ordination of emotional and cognitive limbs of the stress response. J Neuroendocrinol 10: 743–757.
Article CAS PubMed Google Scholar - Westrin A, Ekman R, Regnell, Taskman-Bendz L (2001). A follow-up study of suicide attempters: increase of CSF-somatostatin but no change in CSF-CRH. Eur J Neuropsychopharmacol 11: 135–143.
Article CAS Google Scholar - Zobel AW, Nickel T, Kunzel HE, Ackl N, Sonntag A, Ising M, Holsboer F (2000). Effects of the high-affinity corticotropin-releasing hormone receptor 1 antagonist R121919 in major depression: the first 20 patients treated. J Psychiatr Res 34: 171–181.
Article CAS PubMed Google Scholar - Zhu M-Y, Klimek V, Dilley GE, Haycock JW, Stockmeier CA, Overholser JC, Meltzer HY, Ordway GA (1999). Elevated levels of tyrosine hydroxylase in the locus coeruleus in major depression. Biol Psychiatry 46: 1275–1286.
Article CAS PubMed Google Scholar
Acknowledgements
This work was supported by NIH Grants MH-46692 and MH-45488 and the National Alliance for Research in Schizophrenia and Affective Disorders (NARSAD).
Author information
Authors and Affiliations
- Division of Neurobiology and Behavioral Research, Department of Psychiatry and Human Behavior, University of Mississippi Medical Center, Jackson, MS, USA
Garth Bissette, Violetta Klimek, Jun Pan, Craig Stockmeier & Gregory Ordway
Authors
- Garth Bissette
- Violetta Klimek
- Jun Pan
- Craig Stockmeier
- Gregory Ordway
Corresponding author
Correspondence toGarth Bissette.
Rights and permissions
About this article
Cite this article
Bissette, G., Klimek, V., Pan, J. et al. Elevated Concentrations of CRF in the Locus Coeruleus of Depressed Subjects.Neuropsychopharmacol 28, 1328–1335 (2003). https://doi.org/10.1038/sj.npp.1300191
- Received: 27 September 2002
- Revised: 19 February 2003
- Accepted: 25 February 2003
- Published: 12 March 2003
- Issue Date: 01 July 2003
- DOI: https://doi.org/10.1038/sj.npp.1300191