Preterm gut microbiota and metabolome following discharge from intensive care (original) (raw)
Introduction
The preterm gut microbiome has important influences in both the health and disease of neonates1. As a result the focus of previous work has been on the role of the bacterial community on the development of necrotising enterocolitis (NEC) and late onset sepsis (LOS)2,3,4,5,6. While such studies conflict in their findings relating to disease pathogenesis, overall the bacterial diversity in the preterm gut increases over time with a reduction in initial facultative anaerobes including Staphylococcus and Enterococcus and increases in strict anaerobes including Bifidobacterium and Bacteroides7,8. Notably, this increase in strict anaerobes is significantly less than full term, vaginally delivered, breast fed infants over the first 90 days of life9.
The gut microbiome of term infants changes during the first year of life, resulting in an ‘adult-like’ profile between 1–4 years10,11,12. Bifidobacterium is the genus most differentially represented between childhood and adult profiles11. Previous studies suggest that clinical management in early life, such as antibiotic administration, may have profound long terms effects aside from direct gut health, increasing the risk of asthma13,14, allergy15, inflammatory bowel disease16,17 and obesity18,19 in childhood and adulthood. However, there is only limited evidence that links such conditions to modulations in early gut microbiome development20,21,22.
A major knowledge gap remains regarding the functional metabolic consequences in the development of the gut microbiome in preterm infants, especially following discharge from the NICU. This may have important consequences for clinical practise within NICUs, long term health and offer potential interventions that modulate health following discharge. The aim of this study was to explore changes in gut microbiota pre- and post-discharge from the NICU, their relationship to stool metabolomic profiles and any long-term impacts of key neonatal morbidities in preterm infants such as NEC and LOS.
Results
Bacterial profiling of the gut microbiota through the NICU and post discharge
PD stool samples from 29 infants aged 1–3 years were analysed as well as the first and last NICU stool samples from 14 of the 29 patients. The PD gut microbiota of infants previously diagnosed with NEC and/or LOS was not significantly different compared to controls (P = <0.596). Nor was total days of antibiotic exposure, birth mode (caesarean section or vaginal), gestational age (27 weeks), or age of infant at PD sample collection (stratified as either 12–24 or 25–48 month). Thus, we focus our analysis on the development of the preterm gut microbiome through time.
Maximum parsimony based on the Bray-Curtis showed PD samples were significantly different from EN samples (P = 0.001) and LN samples (P = 0.001). Within the NICU samples there was also a significant difference (P = 0.002) between the EN and LN samples. These significant differences are observed in unconstrained PCA, where the NICU samples are relatively conserved compared to the PD samples, which show a much greater variability in the bacterial profiles (Fig. 1; for only NICU samples see Supplementary Figure S1). The most significant shifts in OTUs from EN to LN included a decreased relative abundance of S. aureus (P = <0.001) and increased relative abundance of _Veillonella_ sp. (_P_ = 0.045). Between the LN sample and the PD samples the most significant changes in OTU abundance were decreases in _K. oxytoca_ (_P_ = >0.001), S. aureus (P = >0.001), E. faecalis (P = >0.001) and increases in Akkermansia sp. (P = >0.001), Blautia sp. (P = >0.001), Bacteroides sp. (P = >0.001) and Collinsella sp. (P = 0.002) (Fig. 2A; for individual profiles see Supplementary Figure S2).
Figure 1
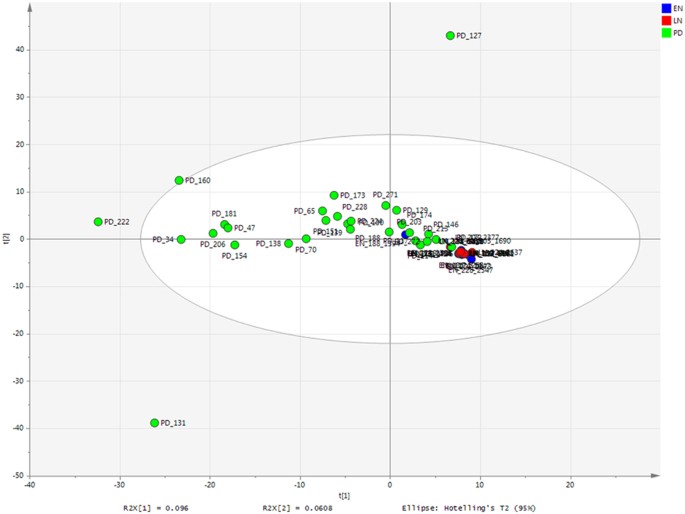
Bacterial profiling PCA of all samples.
Coloured according to time point. EN-LN P = 0.002; EN-PD P = 0.001; LN-PD P = 0.001.
Figure 2
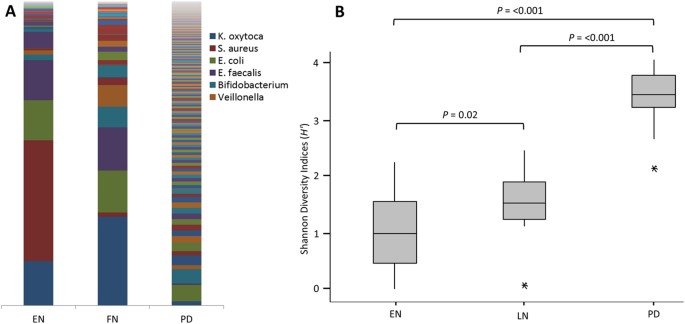
Bacterial community and diversity across the three sample points.
(A) Bar Chart showing the average bacterial profile at each time point (individual profiles in Supplementary Figure S2). Legend restricted to most abundant OTUs. (B) Box plot analysis of the Shannon Diversity index (_H_′) at each time point. EN-Early NICU, LN-Late NICU, PD-Post Discharge.
A significant increase in the Shannon Diversity index (_H_′) of PD samples compared with EN (P = <0.001) and LN (P = <0.001) and between the two NICU time points (P = 0.02) was also observed (Fig. 2B). Phylogenetic investigation of communities by reconstruction of unobserved states (PICRUSt) was implemented to predict functional content of the bacterial OTUs23. This showed that despite greater variation in of the bacterial community, the functional profiles of the PD samples had greater conserved functionality compared to the NICU samples (Fig. 3).
Figure 3
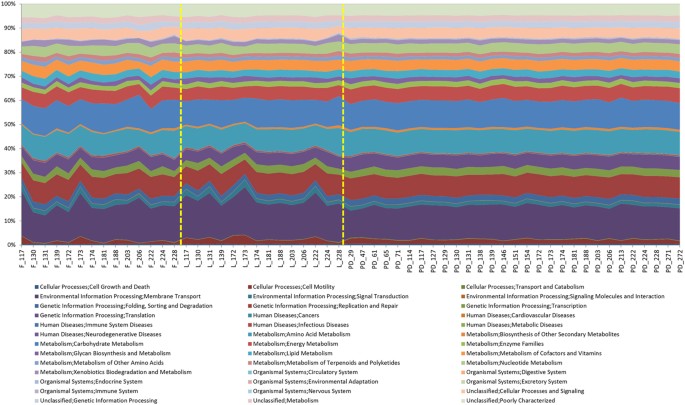
Area chart showing PICRUSt analysis of sequence data.
Inferred metagenomic analysis using the data from the next generation sequencing to determine relative abundances of KEGG pathways encoded in the gut microbiota.
A novel visual inspection analysis of the bacterial profiles supports the above. Visual analysis of EN and LN groups confirmed a decrease of S. aureus in LN samples, as well as an increase of Veillonella sp (Supplementary Figure S3). The plot also indicates that there may be a decrease of K. oxytoca in LN samples although this was not significant in the statistical analysis. Visual analysis of the LN and PD samples showed PD samples are generally more diverse with higher levels in more OTUs (Supplementary Figure S4). However, A decrease in K. oxytoca, S. aureus and E. faecalis occurred from LN to PD, as well as Clostridium sensu stricto, although this was not significant in the main statistical analysis. As confirmed by the statistical analysis Akkermansia sp, Blautia sp, Bacteoides sp and Collinsella sp (are all increased in PD samples. However, these represent overall trends and not all samples followed this pattern.
Metabolomic profiling of the preterm gut through the NICU and post discharge
PLS-DA of metabolomic profiling data showed the grouping by time point to be highly predictive (EN R2VY = 0.85 and Q2VY = 0.73, LN R2VY = 0.84 and Q2VY = 0.67 and PD R2VY = 0.96 and Q2VY = 0.93, where <0.5 represents well modelled reproducible variables) (Fig. 4). Following removal of noise (metabolites clustered around the origin and not associated with a given group), the metabolite profiles cluster almost exclusively based on time point, with the exception of EN samples from patients 139 and 172 which group with the LN samples (Fig. 5). However, analysis of only PD samples supervised according to NICU demographics including birth mode, gestational age, age at sample collection, days of antibiotics, or diagnosis of NEC and/or LOS did not show robustly modelled variables and were thus deemed not significant (e.g. Supplementary Figure S5), in accordance with the next generation sequencing data.
Figure 4
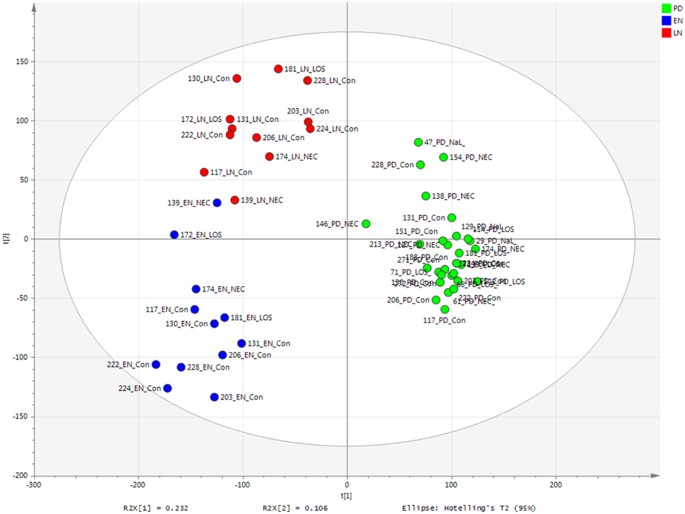
PLS-DA score scatter plot of Metabolomics raw data (R2Y = 89%).
PLS-DA loadings-EN R2 0.85, Q2 0.73; LN R2 0.84, Q2 0.67; PD R2 0.96 and Q2 0.93.
Figure 5
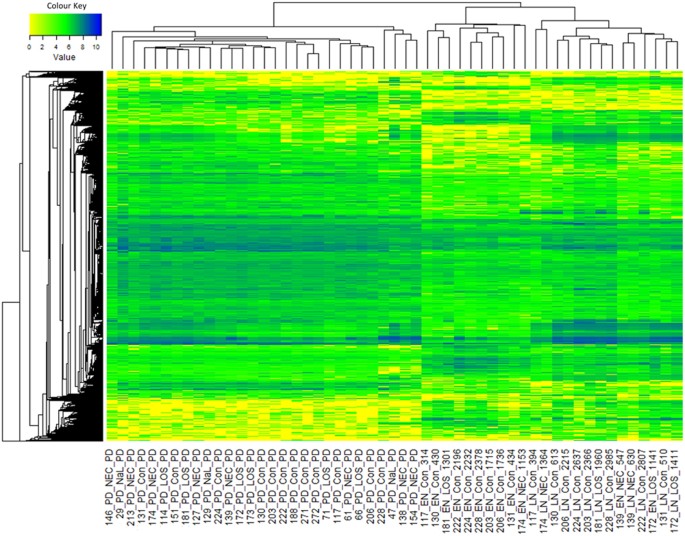
Metabolomics heatmap of all samples.
All values were log10 transformed.
With the threshold used in SIEVE, 51,058 components (metabolites) were detected in total with the majority (47,631) being detected in all samples, regardless of time point (Supplementary Figure S6a). However, a general trend showed the lowest overall intensity in the EN samples and the highest intensity in PD samples. Although relatively low numbers of metabolites were shown to be specific to each group, there were 1,492 components only present in the LN and PD groups and a further 473 components only present in the PD group (Supplementary Figure S6b). No accurate and robust identification of significant metabolites associated with each time point could be determined.
Discussion
The aim of this study was to explore a crucial knowledge gap in the longitudinal development of the preterm gut microbiome following discharge from the NICU. We utilised two complementary technologies in bacterial 16S sequencing and metabolomics to more completely characterise microbiome changes. Because of the long timeframe needed to accrue sufficient numbers of very preterm infants surviving NEC or LOS, the PD samples in this study came from infants with ages ranging from 1–3 years, but all were consuming normal solid diets. We explored potential differences between the PD samples stratified by age and detected no significant difference and thus analysed them collectively.
A significant reduction in the relative abundance of S. aureus (P = <0.001) and an increased relative abundance of Veillonella sp. (P = 0.045) was found between the EN and LN periods. In accordance with previous studies, the highest levels of S. aureus in EN was found in patients delivered by caesarean section, with bacterial signatures of delivery mode lost in the LN samples prior to discharge24. While the bacterial diversity remained relatively low during NICU stay, there was a significant increase in diversity from EN to LN (P = 0.02), indicating the establishment of more complex gut communities in the NICU, despite restricted exposure to microorganisms8. It has previously been suggested that preterm infants undergo delayed establishment of the gut microbiome with lower overall diversity over the first 3 months of life25. However, the bacterial diversity was found to significantly increase following discharge (P = <0.001), with diversity scores comparable to those of full term infants20.
PCA analysis of the bacterial profiles showed that the NICU samples cluster together independent of time point, however the PD samples show a high degree of variation. It is noteworthy that significant changes between the relative abundance of species occurring during the NICU period are comparably minor compared to the much great complexity and individual nature of an infant gut microbiota following discharge. The most significant decreases in relative abundance PD corresponded to K. oxytoca (P = >0.001), S. aureus (P = >0.001) and E. faecalis (P = >0.001), which are organisms typically associated with the NICU environment6,26. The significant reduction in these organisms probably represents increased exposure to non-hospital environmental microbes and a more complex diet10. The most significant increases in relative abundance in PD samples corresponded to Akkermansia sp. (P = >0.001), Blautia sp. (P = >0.001), Bacteroides sp. (P = >0.001) and Collinsella sp. (P = 0.002). Importantly, these genus are all common constituents of the healthy adult gut microbiota and their presence further suggests that the potentially vulnerable preterm gut is able to achieve a flora comparable to that of a healthy full term infant11. The reduction of previously abundant NICU organisms and increases in organism more common to the healthy adults gut microbiota is interesting and might represent the influence of more complex diet. For example, members of the genus Bacteroides are capable of utilising plant derived polysaccharides and by the PD stage of life in this study infants will be consuming complex solid diets27. Furthermore, Blautia was recently only detected following weaning in a piglet model28.
In contrast to the bacterial data, the metabolomic profiles showed a relatively high degree of comparability between the PD samples. The majority of metabolites were detected at each time point, suggesting functional aspects of preterm gut microbiome development is uniform, as oppose to undergoing significant stepwise changes after discharge. An increase in the number and intensity of identified metabolites correlated with increased life stages25. This is similar to data using gas chromatography MS (GCMS), where the number of detected volatile organic compounds (VOCs) were shown to increase with increasing age in a comparable cohort of infants29.
The gradual progression of the gut metabolome corresponds with the microbiota data presented here and in previous studies, where the bacterial richness increased overtime, rather than new species replacing existing species4. The striking observation of PD samples to have conserved metabolic function profiles, both for specific inferred bacterial function (PICRUSt analysis) and overall gut function (LCMS analysis), despite distinct and variable gut microbiota profiles, is important. This is in accordance with previous studies using metagenomic analysis which show un-weaned infants (i.e. not consuming complex semi-solid foods) had a relatively simple gut microbiota, but high variation in functional composition in comparison to weaned infants and adults who had more a complex gut microbiota but greater functional uniformity, regardless of age or sex30,31. Such data suggests that the human gut requires a relatively conserved suite of functions and the actual species colonizing the host and their relative abundance can vary, providing the individual has the overall functional capacity demonstrated here.
No significant long-term modifications to the gut microbiota were found in response to all patient variables tested. The age of the infant at PD sampling did not have a significant effect on the gut microbiome, further suggesting the potential importance of sentinel events much earlier in infancy, such as weaning. We further show that diagnosis with serious disease (NEC and/or LOS) and the associated increased in antibiotic exposure did not have significant long-term effects on the gut microbiome. Further studies with larger numbers of infants are needed to describe how early alteration of the gut microbiome effects long-term inferences on the host, such as obesity19.
Although this study shows the preterm gut microbiome develops in a similar fashion to healthy full term infants, it is still unclear at which age an infant gut microbiome stabilises and remains comparable throughout adulthood. One emerging hypothesis is that the introduction of semi-solid foods is a key event where the gut microbiome develops into its adult form. Thus further work should focus sampling around and subsequently to these events as this crucial period may represent a ‘window of opportunity’ at which modification of the gut microbiota (eg with probiotics/prebiotics etc.) can influence long-term health32,33. Future follow-up work using carefully collected cohorts of preterm and term infants with detailed pheno- and entero-typing and demographic data throughout life presents the exciting possibility to explore how the early gut microbiome development influences growth, development and health and disease throughout maturity.
In summary, we show that the preterm gut microbiome undergoes significant changes following discharge from the NICU, with an increase in diversity and accumulation of organisms comparable to that of term infants, regardless of earlier disease and limited environmental exposure. Furthermore, while increased complexity and individualisation of the gut microbiota correlated with increased age, the overall functional properties of the gut metabolome become more stable and conserved between infants.
Methods
Ethics statement
Ethics approval was obtained from the County Durham and Tees Valley Research Ethics Committee and written informed parental consent was obtained. The methods were carried out in accordance with the approved guidelines.
Infants and samples
Infants who had enrolled to our SERVIS study were approached in a selective manner and asked to provide a stool sample in later infancy. Infants were selected on the basis of previous disease state (NEC and/or LOS) and gestationally matched controls. Fifty-five infants were identified based on all previous NEC (n = 20) and LOS (n = 15) cases and sufficient controls (n = 20). Of the 56, 29 contributed a post discharge stool (8 NEC, 6 LOS, 3, NEC and LOS and 12 controls) between 1 and 3 years of age (Table 1). Post discharge (PD) stool samples were sent using standard postal systems and stored at −20 °C immediately upon receipt and then at −80 °C.
Table 1 Patient demographics.
Additional analysis was carried out on an early NICU (EN) and late NICU (LN) stool pre discharge from 14 infants (n = 28), where detailed sequencing data was available from patients involved in another study (MG-RAST accession numbers 4603816.3–4603844.3). 16S rRNA bacterial profiling was performed on all samples in the study. Metabolomics was carried out on all samples, with the exception of the NICU samples from patients 173 and 188, where insufficient informative sample remained. The unit has standardised feeding practices, antibiotic and antifungal use and no infant was supplemented with probiotics during their NICU stay.
16S rRNA Bacterial profiling
Nucleic acid extraction of stool was carried out on 100 mg of sample using the PowerLyzer™ PowerSoil® DNA Isolation Kit (MoBio, CA, USA) in accordance with the manufacturer’s instructions. Bacterial profiling utilised the 16S rRNA gene targeting variable region 4 (V4) and was carried out by NU-OMICS (Northumbria University) based on the Schloss wet-lab MiSeq SOP and raw fastq data were processed using Mothur (version 1.31.2) as previously described34. Chimeric sequences were detected by Chimera.uchime and removed from downstream analysis. Alignment was generated via the Silva database35. A cutoff of 70 was applied to assign sequences to the trainset9_032012 resulting in 5,496,973 reads. All sequences from PD samples were deposited in MG-RAST under the accession numbers 4516545.3–4516585.3.
LCMS/MS metabolomic profiling
Water, methanol and acetonitrile (ACN) were liquid chromatography mass spectrometry (LCMS) grade (VWR). Metabolites were extracted from 100 mg stool, homogenised in 1 mL cold 80% methanol by vortexing for 15 min at 4 °C. The suspension was then centrifuged at 10,000 × g for 10 min at 4 °C and lyophilised in a freeze dryer before storage at −80 °C. Samples were re-suspended in 1 mL of initial start phase buffer (5% ACN). Stool metabolite profiling was performed using reverse-phase ultra-performance LCMS tandem mass-spectrometry (UPLC-MS/MS). An Accucore C18 column (2.6 μm, 150 × 2.1 mm) was used at 40 °C with a 3.0 μl injection and 300 μl/min flow rate throughout. A multi-step LC gradient was used with 5% ACN increasing to 95% ACN over 22 minutes, with a further 95% ACN for 3 minutes followed by a final 5 minutes re-equilibration at 5% ACN. Samples were run in triplicate and the order of samples in each triplicate sequence was randomised. A blank consisting of LCMS grade water underwent the same procedure and an aliquot of every sample was used as a pool. Prior to each run a blank and 5 pools were processed to equilibrate the system and then blanks and pools were processed periodically every 10 samples. A Q-Exactive (Thermo) was used for the MS and subsequent data dependant MS/MS. Metabolomic profiling was performed using HESI with high resolution (70,000) positive and negative switching. The mass range was set from 100–1000 m/z. MS/MS was subsequently employed with data dependency based on the metabolites of interest.
SIEVE (Version 2.2 beta) was used to process the Thermo RAW files by component extraction. All blanks were used for background subtraction. Positive and negative data were processed individually and combined prior to downstream analysis. An intensity threshold of 500,000 was applied detecting a total of 51,058 components (36,405 positive and 14,653 negative). Data dependant MS/MS (ddMS2) analysis of significant components was performed for putative identification and monoisotopic masses were matched against inhouse standards, however accurate metabolite identification could not be obtained.
Statistical analysis
For normalisation of the NGS bacterial profiling data, all samples were subsampled to 35,448 reads and singleton reads were removed. Significance of variables in the NGS data was analysed within Mothur using maximum parsimony based on the Bray-Curtis, where _P_-values of <0.05 were deemed significant. Bacterial profiling data was analysed by principal component analysis (PCA) using SIMCA 13.0 (Umetrics, Stockholm, Sweden)36. Metabolomic profiling data was analysed using partial least squared-discriminatory analysis (PLS-DA). Operational taxonomic units (OTUs) and components were automatically log transformed within SIMCA. Only variables with a VIP of >1 were included and the most significant variables associated with each group were determined based on the variables with error bars above 0 in the coefficient plot37. Multiple iterations of the VIP analysis were conducted for optimal noise reduction (Supplementary Figure S7). To check that data was adhering to multivariate normalities, Hotelling’s T 2 tolerance limits were calculated and set at 0.95. Heatmaps were generated in R using the vegan and gplots packages.
The bacterial profiles of the samples were further visually investigated using a Parallel Coordinates plot38, where each axis represents an OTUs and the bacterial profile of a sample is represented by a polyline intersecting the axes at corresponding bacterial level. The sample profiles are coloured according to sample classification to support comparison between sample groups. The profiles are log transformed and display the 40 most abundant OTUs.
Additional Information
How to cite this article: Stewart, C. J. et al. Preterm gut microbiota and metabolome following discharge from intensive care. Sci. Rep. 5, 17141; doi: 10.1038/srep17141 (2015).
References
- Berrington, J. E., Stewart, C. J., Cummings, S. P. & Embleton, N. D. The neonatal bowel microbiome in health and infection. Curr. Opin. Infect. Dis. 27, 236–43 (2014).
Article Google Scholar - Sim, K. et al. Dysbiosis anticipating necrotizing enterocolitis in very premature infants. Clin. Infect. Dis. ciu822– (2014). 10.1093/cid/ciu822
- Morrow, A. L. et al. Early microbial and metabolomic signatures predict later onset of necrotizing enterocolitis in preterm infants. Microbiome 1, 13 (2013).
Article Google Scholar - Stewart, C. J. et al. Bacterial and fungal viability in the preterm gut: NEC and sepsis. Arch Dis Child Fetal Neonatal Ed 98, F298–303 (2013).
Article Google Scholar - Normann, E., Fahlén, A., Engstrand, L. & Lilja, H. E. Intestinal microbial profiles in extremely preterm infants with and without necrotizing enterocolitis. Acta Paediatr. (2012). 10.1111/apa.12059
- Stewart, C. J. et al. The preterm gut microbiota: changes associated with necrotizing enterocolitis and infection. Acta Paediatr 101, 1121–7 (2012).
Article CAS Google Scholar - La Rosa, P. S. et al. Patterned progression of bacterial populations in the premature infant gut. Proc. Natl. Acad. Sci. 111, 12522–7 (2014).
Article CAS ADS Google Scholar - Stewart, C. J. et al. Development of the preterm gut microbiome in twins at risk of necrotising enterocolitis and sepsis. PLoS One 8, e73465 (2013).
Article CAS ADS Google Scholar - Arboleya, S. & Gueimonde, M. Facultative to strict anaerobes ratio in the preterm infant microbiota. Gut Microbes 3, 583–588 (2012).
Article Google Scholar - Wopereis, H., Oozeer, R., Knipping, K., Belzer, C. & Knol, J. The first thousand days-intestinal microbiology of early life: establishing a symbiosis. Pediatr. Allergy Immunol. 25, 428–38 (2014).
Article Google Scholar - Ringel-Kulka, T. et al. Intestinal microbiota in healthy U.S. young children and adults–a high throughput microarray analysis. PLoS One 8, e64315 (2013).
Article CAS ADS Google Scholar - Palmer, C., Bik, E. M., DiGiulio, D. B., Relman, D. A. & Brown, P. O. Development of the human infant intestinal microbiota. PLoS Biol 5, 1556–73 (2007).
Article CAS Google Scholar - Risnes, K. R., Belanger, K., Murk, W. & Bracken, M. B. Antibiotic exposure by 6 months and asthma and allergy at 6 years: findings in a cohort of 1,401 US children. Am. J. Epidemiol. 173, 310–8 (2011).
Article Google Scholar - Kozyrskyj, A. L., Ernst, P. & Becker, A. B. Increased risk of childhood asthma from antibiotic use in early life. Chest 131, 1753–9 (2007).
Article Google Scholar - Liu, A. H. & Murphy, J. R. Hygiene hypothesis: Fact or fiction? J. Allergy Clin. Immunol. 111, 471–478 (2003).
Article Google Scholar - Kronman, M. P., Zaoutis, T. E., Haynes, K., Feng, R. & Coffin, S. E. Antibiotic exposure and IBD development among children: a population-based cohort study. Pediatrics 130, e794–e803 (2012).
Article Google Scholar - Virta, L., Auvinen, A., Helenius, H., Huovinen, P. & Kolho, K.-L. Association of repeated exposure to antibiotics with the development of pediatric Crohn’s disease–a nationwide, register-based finnish case-control study. Am. J. Epidemiol. 175, 775–84 (2012).
Article Google Scholar - Trasande, L. et al. Infant antibiotic exposures and early-life body mass. Int. J. Obes. (Lond). 37, 16–23 (2013).
Article CAS Google Scholar - Ajslev, T. A., Andersen, C. S., Gamborg, M., Sørensen, T. I. A. & Jess, T. Childhood overweight after establishment of the gut microbiota: the role of delivery mode, pre-pregnancy weight and early administration of antibiotics. Int J Obes 35, 522–9 (2011).
Article CAS Google Scholar - Abrahamsson, T. R. et al. Low gut microbiota diversity in early infancy precedes asthma at school age. Clin. Exp. Allergy 44, 842–50 (2014).
Article CAS Google Scholar - Bisgaard, H. et al. Reduced diversity of the intestinal microbiota during infancy is associated with increased risk of allergic disease at school age. J. Allergy Clin. Immunol. 128, 646–52.e1–5 (2011).
Article Google Scholar - Kalliomäki, M. & Collado, M. Early differences in fecal microbiota composition in children may predict overweight. Am. J. Clin Nutr. 87, 534–538 (2008).
Article Google Scholar - Langille, M. G. I. et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 31, 814–21 (2013).
Article CAS Google Scholar - Dominguez-Bello, M. G. et al. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc Natl Acad Sci USA 107, 11971–5 (2010).
Article ADS Google Scholar - Arboleya, S. et al. Establishment and development of intestinal microbiota in preterm neonates. FEMS Microbiol. 79, 763–72 (2012).
Article CAS Google Scholar - Smith, B. et al. Investigation of the early intestinal microflora in premature infants with/without necrotizing enterocolitis using two different methods. Pediatr Res 71, 115–120 (2012).
Article CAS ADS Google Scholar - Koropatkin, N., Cameron, E. & Martens, E. How glycan metabolism shapes the human gut microbiota. Nat. Rev. 10, 323–335 (2012).
CAS Google Scholar - Alain, B. Pajarillo, E., Chae, J.-P., P., Balolong, M., Bum Kim, H. & Kang, D.-K. Assessment of fecal bacterial diversity among healthy piglets during the weaning transition. J. Gen. Appl. Microbiol. 60, 140–146 (2014).
Article Google Scholar - Garner, C. E. et al. Analysis of faecal volatile organic compounds in preterm infants who develop necrotising enterocolitis: a pilot study. J Pediatr Gastroenterol Nutr 49, 559–65 (2009).
Article CAS Google Scholar - Turnbaugh, P. J. et al. A core gut microbiome in obese and lean twins. Nature 457, 480–484 (2009).
Article CAS ADS Google Scholar - Urokawa, K. K. et al. Comparative Metagenomics Revealed Commonly Enriched Gene Sets in Human Gut Microbiomes. DNA Res 14, 169–181 (2007).
Article Google Scholar - Schloss, P. D., Iverson, K. D., Petrosino, J. F. & Schloss, S. J. The dynamics of a family’s gut microbiota reveal variations on a theme. Microbiome 2, 25 (2014).
Article Google Scholar - Fanaro, S. & Vigi, V. Which role for prebiotics at weaning? J Clin Gastroenterol 42, S209–13 (2008).
Article Google Scholar - Kozich, J. J., Westcott, S. L., Baxter, N. T., Highlander, S. K. & Schloss, P. D. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl. Environ. Microbiol. 79, 5112–20 (2013).
Article CAS Google Scholar - Schloss, P. D., Gevers, D. & Westcott, S. L. Reducing the effects of PCR Amplification and Sequencing Artifacts on 16S rRNA-based studies. PLoS One 6, e27310 (2011).
Article CAS ADS Google Scholar - Eriksson, L., Johansson, E. & Kettaneh-Wold, N. Multi-and Megavariate Data Analysis, Part 2, Advanced Applications and Method Extensions. (MKS Umetrics AB, 2006).
- Trivedi, K., D. The application of SIMCA P + in Shotgun Metabolomics analysis of ZIC“ HILIC-MS Spectra of human Urine-experience with the Shimadzu IT-T of and profiling solutions data extraction software. J. Chromatogr. Sep. Tech. 03, 145 (2012).
Google Scholar - Inselberg, A. The plane with parallel coordinates. Vis. Comput. 1, 69–91 (1985).
Article MathSciNet Google Scholar
Acknowledgements
This work was supported by funding from Tiny Lives charity (Newcastle upon Tyne, UK), Newcastle upon Tyne Hospitals NHS Charity and in part by an unrestricted educational grant from Nestle UK. The content is solely the responsibility of the authors.
Author information
Authors and Affiliations
- Faculty of Health and Life Sciences, Northumbria University, Newcastle upon Tyne, United Kingdom
Christopher J. Stewart, Andrew Nelson & Stephen P. Cummings - Newcastle Neonatal Service, Royal Victoria Infirmary, Newcastle upon Tyne, United Kingdom
Tom Skeath, Janet E. Berrington & Nicholas D. Embleton - Department of Computer Science and Digital Technologies, Northumbria University, Newcastle upon Tyne, United Kingdom
Sara J. Fernstad - Department of Microbiology, Freeman Hospital, Newcastle upon Tyne, United Kingdom
Emma C. L. Marrs & John D. Perry
Authors
- Christopher J. Stewart
You can also search for this author inPubMed Google Scholar - Tom Skeath
You can also search for this author inPubMed Google Scholar - Andrew Nelson
You can also search for this author inPubMed Google Scholar - Sara J. Fernstad
You can also search for this author inPubMed Google Scholar - Emma C. L. Marrs
You can also search for this author inPubMed Google Scholar - John D. Perry
You can also search for this author inPubMed Google Scholar - Stephen P. Cummings
You can also search for this author inPubMed Google Scholar - Janet E. Berrington
You can also search for this author inPubMed Google Scholar - Nicholas D. Embleton
You can also search for this author inPubMed Google Scholar
Contributions
C.S. designed the research, contributed funding for the study, conducted the wet lab work, analysed data and wrote the manuscript; T.S. contributed to designing the research, patient consent and collection of stool samples; A.N. conducted data analysis; S.J.F. conducted the data visualization; E.C.L.M. and J.P. contributed to sample collection and database curating; S.P.C. designed the research, supervised the lab work and wrote the manuscript; J.E.B. contributed to designing the research, patient consent and collection of stool and wrote the manuscript. N.D.E. contributed to designing the research, contributed funding for the study, patient consent and collection of stool and wrote the manuscript. All authors contributed to discussions, writing and reviewing of the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article
Cite this article
Stewart, C., Skeath, T., Nelson, A. et al. Preterm gut microbiota and metabolome following discharge from intensive care.Sci Rep 5, 17141 (2015). https://doi.org/10.1038/srep17141
- Received: 08 September 2015
- Accepted: 26 October 2015
- Published: 24 November 2015
- DOI: https://doi.org/10.1038/srep17141