missMethyl: an R package for analyzing data from Illumina’s HumanMethylation450 platform (original) (raw)
Abstract
Summary: DNA methylation is one of the most commonly studied epigenetic modifications due to its role in both disease and development. The Illumina HumanMethylation450 BeadChip is a cost-effective way to profile >450 000 CpGs across the human genome, making it a popular platform for profiling DNA methylation. Here we introduce missMethyl, an R package with a suite of tools for performing normalization, removal of unwanted variation in differential methylation analysis, differential variability testing and gene set analysis for the 450K array.
Availability and implementation: missMethyl is an R package available from the Bioconductor project at www.bioconductor.org.
Contact: [email protected]
Supplementary information: Supplementary data are available at Bioinformatics online.
1 Introduction
DNA methylation, one of the most widely studied epigenetic modifications, is crucial for normal embryonic development and is often disrupted in disease. Epigenetic marks can be modified by environmental exposures and are dramatically altered in cancer. Since epigenetic changes are potentially reversible, DNA methylation is an attractive therapeutic target and remains an important area of research.
There are several array and sequencing-based technologies available for profiling DNA methylation. While sequencing costs have declined, it remains expensive for large DNA methylation studies, making arrays a cost-effective alternative. Illumina’s HumanMethylation450 (450K) BeadChip is currently the most popular platform, having been used in many large studies such as The Cancer Genome Atlas, as well as in many epigenome-wide association studies.
Here, we present the missMethyl R package, which contains dedicated tools for performing a range of analyses primarily targeted at 450K arrays. These methods include normalization, removal of unwanted variation (RUV) such as batch effects in a differential methylation analysis, differential variability testing and gene set analysis. Software packages for reading in raw data, pre-processing and differential methylation analysis include minfi (Aryee et al., 2014), methylKit (Akalin et al., 2012) and methylumi. However, missMethyl provides more specialized analysis methods not available elsewhere.
2 Methods
2.1 SWAN: subset-quantile within array normalization
SWAN (Maksimovic et al., 2012) is a within-array normalization method specifically designed for the 450K platform. The 450K array uses a combination of two distinct probe types, Infinium I and II, which are known to display technical differences. The underlying assumption of SWAN is that probes with the same number of CpGs in their probe bodies share similar biology. Thus, their overall intensity distributions should be similar regardless of design type.
The SWAN function takes an object of class MethylSet, RGChannelSet or MethyLumiSet and performs normalization in two steps. First, a subset of Infinium I and II probes that have one, two and three underlying CpGs are randomly selected and quantile normalized, keeping the methylated and unmethylated channels separate. Second, the intensities of the remaining probes are adjusted for each probe type separately using linear interpolation procedures.
The SWAN function outputs a MethylSet object of normalized intensities. Figure 1A, generated using the densityByProbeType function, shows the SWAN-normalized Infinium I and II β value distributions of a single array from a publicly available ageing dataset (Heyn et al., 2012).
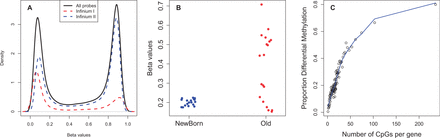
Fig. 1.
Analysis of a publicly available aging methylation dataset. (A) β value distributions for Infinium I and II probes after SWAN, generated using the densityByProbeType function. (B) β values for newborns versus centenarians for a differentially variable CpG (cg00807871) identified using DiffVar. (C) Bias due to differing numbers of CpGs per gene, generated by specifying plot.bias=TRUE in the gometh function
2.2 RUVm: remove unwanted variation
Since 450K arrays are relatively inexpensive, they are widely used in large differential methylation studies. However, large studies tend to be susceptible to batch effects, as well as other unknown sources of technical and biological variation. The missMethyl package offers RUVm as a solution for removing batch effects and unknown unwanted variation from the data (Maksimovic et al., 2015).
To accurately estimate the components of unwanted variation, the two-stage RUVm method relies on negative control probes that are assumed not to be associated with the biological factor of interest. First, a differential methylation analysis using the 613 Illumina negative control probes with RUVinverse is performed. The red and green channel intensities for the 613 negative control probes can be extracted using the getINCs function. From the results of this first analysis, empirical controls are identified, which are then used by the RUVinverse function in the second stage.
The RUVfit function runs this method and takes as input a matrix of M-values, a design matrix, the coefficient to be tested and a vector indicating the negative control probes. The RUVadj function adjusts the variance estimates using empirical Bayes shrinkage and the topRUV function displays the top 10 significantly differentially methylated CpG sites, after adjusting for the unwanted variation.
2.3 DiffVar: differential variability testing
Thus far, the main focus of many methylation studies has been on detecting CpG sites or regions that are differentially methylated between groups. However, identifying features that differ in terms of variability may also be relevant to understanding disease phenotypes (Hansen et al., 2011). Previously, methods for detecting differential variability in genomics data were not well established or had no dedicated software.
The missMethyl package contains functions to test differential variability in methylation and RNA-Seq data from our previous work (Phipson and Oshlack, 2014). For methylation data, the varFit function takes as input a matrix of M values, β values or a MethylSet object, a design matrix and the coefficients of interest to be tested. If the input is in the form of β values, a logit transformation is applied. For RNA-Seq data, if a DGEList object is supplied, a voom transformation is used (Law et al., 2014), which takes into account the mean-variance relationship observed in count data.
The test is based on Levene’s test for differences in variances and employs the empirical Bayes modelling framework of the limma package (Ritchie et al., 2015). The topVar function outputs the top 10 differentially variable CpGs or genes. Figure 1B shows an example of a significantly differentially variable CpG using DiffVar in the aging dataset.
2.4 gometh: gene set analysis
A differential methylation or differential variability analysis could result in a long list of significant CpGs to interpret. One popular approach to understanding potential gene pathways that are affected is to perform gene set analysis. Although gene set analysis is well established for gene expression experiments, the methodology is ad hoc at best for methylation data. One issue is that the numbers of CpGs associated with each gene on the 450K array ranges from 1 to 1299. Genes with larger numbers of probes are more likely to have significantly differentially methylated CpGs, biasing gene set analysis (Geeleher et al., 2013) (Fig. 1C).
The gometh function modifies the goseq method (Young et al., 2010) specifically for the 450K array by adjusting for the number of CpGs associated with each gene. It takes as input a vector of significant CpGs and calculates the probability of a gene being selected given the number of associated CpGs. A test based on Wallenius’ noncentral hypergeometric distribution is then performed for each gene ontology (GO) category (see Supplementary Material).
The gometh function outputs a dataframe with rows for each GO category tested and various statistics of interest. The topGO function in limma can then be called, which outputs the top 20 GO categories, ranked by p-value.
3 Conclusion
The missMethyl R package contains a suite of functions to perform novel analyses for 450K array data, with new functions likely to be added as they are developed. The functions have been written to complement the limma package and are compatible with data objects from minfi, methylumi and edgeR. In addition, missMethyl is well documented and freely available from Bioconductor.
Funding
This work was supported by Victorian State Government Operational Infrastructure Support and National Health and Medical Research Council grants APP1051481 and APP1051402 to A.O.
Conflict of Interest: none declared.
References
et al. . (
2012
)
methylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles
.
Genome Biol.
,
13
,
R87
.
et al. . (
2014
)
Minfi: a flexible and comprehensive Bioconductor package for the analysis of infinium DNA methylation microarrays
.
Bioinformatics
,
30
,
1363
–
1369
.
et al. . (
2013
)
Gene-set analysis is severely biased when applied to genome-wide methylation data
.
Bioinformatics
,
29
,
1851
–
1857
.
et al. . (
2011
)
Increased methylation variation in epigenetic domains across cancer types
.
Nat. Genet.
,
43
,
768
–
775
.
et al. . (
2012
)
Distinct DNA methylomes of newborns and centenarians
.
Proc. Natl. Acad. Sci. USA
,
109
,
10522
–
10527
.
et al. . (
2014
)
Voom: precision weights unlock linear model analysis tools for RNA-seq read counts
.
Genome Biol.
,
15
,
R29
.
et al. . (
2012
)
SWAN: Subset-quantile within array normalization for illumina infinium HumanMethylation450 BeadChips
.
Genome Biol.
,
13
,
R44
.
et al. . (
2015
)
Removing unwanted variation in a differential methylation analysis of illumina humanmethylation450 array data
.
Nucleic Acids Res.
,
43
,
e106
.
(
2014
)
DiffVar: a new method for detecting differential variability with application to methylation in cancer and aging
.
Genome Biol.
,
15
,
465
.
et al. . (
2015
)
Limma powers differential expression analyses for RNA-sequencing and microarray studies
.
Nucleic Acids Res.
,
43
,
e47
.
et al. . (
2010
)
Gene ontology analysis for RNA-seq: accounting for selection bias
.
Genome Biol.
,
11
,
R14
.
Author notes
†The authors wish it to be known that, in their opinion, the first two authors should be regarded as joint First Authors.
Associate Editor: John Hancock
© The Author 2015. Published by Oxford University Press. All rights reserved. For Permissions, please e-mail: [email protected]