Development of Obesity and Polycystic Ovary Syndrome in Adolescents (original) (raw)
Article navigation

Review Articles| November 28 2012
aThe Children’s Obesity Clinic, Department of Pediatrics, Copenhagen University Hospital, Holbaek, and
Search for other works by this author on:
aThe Children’s Obesity Clinic, Department of Pediatrics, Copenhagen University Hospital, Holbaek, and
Search for other works by this author on:
bInstitute of Preventive Medicine, Copenhagen University Hospital, Copenhagen, Denmark
Search for other works by this author on:
aThe Children’s Obesity Clinic, Department of Pediatrics, Copenhagen University Hospital, Holbaek, and
Search for other works by this author on:
Horm Res Paediatr (2013) 78 (5-6): 269–278.
Accepted:
October 15 2012
Published Online:
November 28 2012
Abstract
Obesity in adolescents is prevalent worldwide. Polycystic ovary syndrome (PCOS) is often associated with obesity in women, and it has serious metabolic and reproductive health implications. Although PCOS does not become clinically visible until early adolescence, its origins are likely much earlier. Therefore, we reviewed the recent literature regarding the mechanisms linking the development of PCOS and obesity in adolescent girls. We found that excess abdominal adipose tissue (AT) initiates metabolic and endocrine aberrations that are central in the progression of PCOS. As an example, abdominal AT impairs insulin action, which interacts with the progression of hyperandrogenism. In addition, excessive androgen levels lead to impaired glucose uptake, which also contributes to insulin resistance, which again increases the deposition of visceral fat. The body composition is influenced by testosterone, which decreases subcutaneous fat lipolysis and influences adipocyte distribution. These mechanisms may explain why PCOS girls have an increased visceral adipose mass independent of body mass index. Therefore, first-line treatment in adolescent PCOS is often lifestyle intervention to prevent the damaging effects of obesity. Pharmacological treatment of adolescent PCOS is not standardized because the long-term effects in adolescents have not yet been evaluated; therefore, drugs should be prescribed cautiously. Although the complex metabolic interrelationships between obesity and PCOS have yet to be fully understood, the co-occurrence of these conditions in adolescent girls tends to increase the severity of the negative health consequences of each condition.
Introduction
Obesity is a worldwide problem that threatens the lives of adults, adolescents and children. Obesity has many associated comorbidities, such as the well-known components of the metabolic syndrome (MetS), which harm health and functioning in men and women. Worryingly, these health risks appear in overweight and obese childhood, which raises the question of how it challenges their future health and especially whether it impairs physiological growth as well as development during the pubertal transition.
Polycystic ovary syndrome (PCOS) is a disorder of ovarian dysfunction often characterized by relatively high androgen concentrations, metabolic abnormalities and many conditions of the MetS. PCOS is one of the most common endocrine disorders in females, and affects ∼15% [1] of women of reproductive age. In addition to its effect on increasing cardiovascular disease (CVD) risk, PCOS impairs women’s fertility and, due to the distress of its many side effects, it also harms their mental health. The disorder typically emerges during adolescence. Despite this, the prevalence and incidence of the disorder are not extensively evaluated in the adolescent population. It has recently been estimated in two populations that 18.5–26% of adolescent girls have PCOS [2,3], thus making it a relatively common syndrome. In women, the risk of PCOS is increased among those who are obese, and similarly it is elevated among obese adolescents. Although a tendency towards polycystic ovarian morphology (PCOM) and enlarged paratubal cysts is found in obese pubertal adolescents, the condition varies with ethnicity and has low predictive value of hyperandrogenism (HA) in adolescents [3]. Furthermore, there is evidence that features of PCOS even emerge among obese preadolescent girls.
Given that nearly one third of the European pediatric population are overweight and obese [4], it is likely that the incidence of PCOS will increase in the coming years. Therefore, we reviewed the most recent literature regarding PCOS and obesity in adolescents to investigate how the codevelopment of these conditions is currently interpreted.
Polycystic Ovary Syndrome
Clinical Manifestation of PCOS
The clinical symptoms of PCOS in women are heterogeneous and include oligoanovulation (OAV), hirsutism, acne, alopecia, acanthosis nigricans and increased body fat mass with increased abdominal fat deposition [5]. Additionally, many women have a history of premature adrenarche/pubarche. Atypical forms of PCOS also occur, such as HA without anovulatory symptoms. The comorbidities of importance include infertility, MetS, and type 2 diabetes mellitus (T2DM) [6,7,8]. Many, but not all, women with PCOS are also classified as overweight with a body mass index (BMI; kg/m2) >25 or obese (BMI >30). Depending on the study, and hence the source of the population, the estimates of obesity among adult PCOS patients range from 42 to 58% [9,10].
Biochemical Characteristics of PCOS
In general, women with PCOS exhibit lowered levels of sex hormone-binding globulin (SHBG) and elevated levels of circulating free testosterone (cFT), luteinizing hormone (LH)/follicle-stimulating hormone (FSH) ratio, postprandial glucose, fasting insulin, triglycerides, total cholesterol and a homeostatic model assessment index above 3.8, which indicates insulin resistance (IR) [11]. Several of these conditions are known to be CVD risk factors [8]. Since anti-Müllerian hormone (AMH) reflects some aspects of ovarian function, and PCOS is often characterized by increased antral follicles in ovaries, it may be useful in the diagnosis of PCOS [12]. In women, increased serum concentrations of AMH seem to correlate well with three parameters of PCOS, namely HA, OAV, and PCOM, but it is not yet generally included in the biochemical panel [12].
Clinical Diagnostic Criteria in Women
The diagnostic criteria for PCOS come from The 2003 Rotterdam consensus workshop [13]. It defined PCOS as a syndrome of ovarian dysfunction, diagnosed when fulfilling two out of the three following criteria: (1) menstrual dysfunction/OAV; (2) enlarged polycystic ovaries (on ultrasonography); (3) clinical and/or biochemical HA. Additionally, however, other sets of criteria have been proposed [14]. These criteria, listed in table 1 differ in terms of which and how many conditions are necessary for a diagnosis. Nonetheless, these three sets of criteria are in agreement that to establish a diagnosis of PCOS in adult women, conditions with similar phenotypes should be excluded [13]. Because of the broad spectrum of clinical symptoms, these criteria are recommendations rather than standardized for diagnosis.
Table 1
Different sets of diagnostic criteria for PCOS in adult women [14]
![Different sets of diagnostic criteria for PCOS in adult women [14]](https://karger.silverchair-cdn.com/karger/content_public/journal/hrp/78/5-6/10.1159_000345310/2/m_000345310_t01.gif?Expires=1730209668&Signature=0DyhV946riab1jPOHbO0B2-OZbUBl7DR96mq8f-dtQfcNTIOW8a-GHNXWK-OisdRzBlKnIi88dk9pJNXjlb45lJn50Zyt-LbbQxO5XhO6lPHIz7~LlW0zV4DSKzxwGEgIzs52QflbgOCLlZHx-3lINojs5yju1kVqc3CBQRw0mmEXQ2i9QCFG9LkxwCXrg63h2EvXppyxy6tnRhPXRE38pooB0EYwipC4C~oE-l2fQrN4SvNm74os2gWqJsTANRI2~YReBs5k2SfQSyp-dXCm7g~Yme50jYy~bar4XMFH-W7c073CM9VoX414UGDLk7OugKQam1vhkkEcXf3ZXxwEQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
PCOS in Adolescents
A Diagnostic Challenge
There is debate about the appropriateness of using the Rotterdam criteria for a diagnosis of PCOS in adolescents since they were developed for use in women. Other criteria for diagnosing PCOS come from the NIH and AES. Just like the Rotterdam criteria, they also include HA as a diagnostic parameter, and these criteria have been proposed for use in adolescents. In adolescence, the normal changes during puberty can bias the interpretation of the biochemical measures typically used in the diagnosis of PCOS. For example, during normal pubertal development in girls, hypothalamic pulsatile secretions of gonadotropin-releasing hormone (GNRH) are established, and these become dynamic and cyclic. During puberty, the growth hormone (GH)-mediated release of insulin-like growth factor-1 increases, which in turn decreases insulin sensitivity and increases insulin secretion [15]. The resulting hyperinsulinemia (HI) decreases liver production of SHBG, which increases the bioavailability of steroids, resulting in raised cFT. Thus, in adolescent girls elevated levels of circulating androgens are not necessarily a sign of HA per se. Additionally, the reproductive capacity of adolescents also undergoes non-pathological changes during pubertal development that are influenced by the secretion of gonadotropins. These changes are expressed by irregular menstrual patterns and PCOM on ultrasonography, and may even persist for 2 years after menarche [2,15,16,17]. Because all three sets of criteria include abnormal ovulatory patterns and PCOM, the diagnosis in adolescence remains challenging. Taken together, these biochemical and physiological aspects of pubertal development suggest that the diagnosis of PCOS in adolescents should be assigned using different criteria from those in women.
Diagnostic Criteria in Adolescents
Given these unique aspects of pubertal development, diagnosing PCOS in adolescent girls is a challenge. In a sample of 250 adolescent girls, PCOS was best characterized by clinical HA (hirsutism) and/or biochemical HA and oligomenorrhea [18]. Other groups have suggested that menstrual patterns are classified as pathological in adolescents when menstruation is absent for ≥90 days, or if cycles persist for ≥45 days [19]. Biochemically, HA can be determined by levels of cFT, and dehydroepiandrosterone in adolescent girls [19]. Physiologically, ovarian volume and antral follicle count can be assessed by transabdominal ultrasonography, although these measures may be operator dependent and also of limited use in adolescents [2]. Transvaginal or transrectal ultrasonography in adolescents is questionable or even unethical in young girls prior to sexual debut. Much like in adult women, the diagnostic reliability of AMH in adolescents has yet to be determined [20].
Biological Origin
The etiology of PCOS remains largely unknown, but it is thought to be multifactorial, i.e. influenced by epigenetic, genetic and environmental contributions [17,21].
Prenatal developmental circumstances have been observed to influence the development of PCOS in adult monkeys by in utero exposure to supraphysiological levels of androgens [22]. Interestingly, this discovery in nonhuman primates has recently been explained by epigenetic alterations which predispose to PCOS [23].
The FTO gene on chromosome 16q12.2 is an obesity-related gene that is of particular interest because single nucleotide polymorphisms (SNPs) at this site are found to have a strong association with increased BMI, greater weight and T2DM. In particular, the A allele is associated with greater BMI. A meta-analysis compared FTO SNPs, rs9939609 in particular, between women with PCOS and the general population. This analysis was in accordance with previous studies and showed that each added copy of the allele (A/C) was associated with an increase in BMI. Further, it showed that there was a greater effect of the FTO gene on weight in women with PCOS as compared to the general population [24]. Therefore, every added A allele is associated with higher BMI, and also PCOS women with the FTO gene had a greater BMI compared to women without PCOS. These results suggest that there is a genetic component to PCOS; however, it has yet to be fully elucidated.
The ‘thrifty phenotype hypothesis’ has received much attention because it postulates that T2DM, CVD and MetS are under the influence of fetal programming. Although this ancient metabolic programming has anabolic advantages during starvation, during exposure to an abundance of food supplies it contributes to metabolic disturbances like IR and PCOS [25].
Much like the ‘thrifty’ hypothesis, Ibáñez et al. [26] have sought to explain the development of IR in the presence or absence of obesity through the ‘adipose tissue expandability’ (ATE) hypothesis. The ATE hypothesis suggests that fat mass storage capacity is set in early life (age <2 years) and again around puberty (age 10–18 years). Therefore, the amount of subcutaneous adipocytes is different in each individual. These differences might explain why not all obese girls develop androgen excess whereas others do [27]. Intriguingly, low-birth weight girls who experience spontaneous catch-up growth develop higher dehydroepiandrosterone and lower SHBG together with increased visceral adiposity between 6 and 8 years of age, even though they are not overweight. This may represent a metabolic adaptation that persists into adolescence and adulthood, and therefore exposes them to a higher risk of developing PCOS [26].
PCOS and Obesity
Until recently, PCOS has been recognized as a hyperandrogenic disorder originating from hypothalamic-pituitary gonadotropin secretion or ovarian dysfunction [28]. Lately, however, at least some cases of PCOS have been regarded as a disorder of metabolic origin that impairs reproductive function [5]. Since obesity, particularly in the abdominal region, is found in ∼50% [9,29] of women with PCOS, and because obesity appears in mid-childhood and increases during puberty [6,16,27], excess adiposity has generated a great deal of attention. Furthermore, the clinical phenotype and the development of PCOS are thought to be reinforced by obesity [2,7,27,30], especially among those who are genetically prone to the disorder [7]. These observed associations highlight the importance of disentangling the relationship between obesity and PCOS, and in particular their common metabolic disorders.
PCOS and Adolescent Obesity
The interrelationships between obesity, PCOS and metabolic abnormalities are complex. Even in childhood, excess BMI is associated with an increased risk of a wide range of medical complications [31,32,33]. Of particular interest are those related to the degree of IR, as IR seems instrumental in the codevelopment of PCOS and T2DM in obese adolescent girls [11]. Another factor of importance is HA as it exacerbates physiological changes of androgens during puberty, and one effect may be the initiation of precocious pubarche in girls which in turn, is a risk factor for PCOS [14]. Visceral fat also plays an independent role, and it is thought to exacerbate these hormonal alterations in adolescent girls [34]. Further, in adolescent girls, HA and/or ovarian cysts are associated with overweight or obesity [3]. Given this complexity, many investigations have sought to determine how these factors are linked together and what roles they play in the development of obesity and PCOS.
To try and understand which comes first, obesity or PCOS, studies have investigated the temporal relationships in girls and adolescents. One study found that girls with a high BMI in childhood had an increased risk of oligomenorrhea and a diagnosis of PCOS in young adulthood (age 24), yet the possibility that features of PCOS were already present in these girls cannot be excluded [6]. A prospective study on 244 randomly selected postmenarchal girls [2] from a large population-based birth cohort investigated the influence of obesity on the development of abnormal ovarian morphology. It found PCOM in 61.1% of the obese girls but only in 32.1% of the normal-weight girls, suggesting that obesity is a contributing factor. In a study of 493 girls [6], researchers investigated if PCOS (or its features) in adolescents is predictive of later class III obesity (BMI ≥40). Despite not using pelvic ultrasonography, PCOS was diagnosed by the Rotterdam criteria in 12 (40%) of 30 oligomenorrheic girls at age 14 years. Of these girls, 33% displayed class III obesity by 24 years of age versus 8.4% of girls without PCOS. Other predictors of class III obesity included low SHBG, oligomenorrhea, high childhood insulin levels, increased cFT and MetS, all of which are recognized as PCOS phenotypes [6]. These studies illustrate that obesity and PCOS are correlative in their pathogenesis, and also suggest that the detection of either obesity or PCOS features in adolescence should be monitored closely considering their long-term health risks.
IR, Obesity and PCOS
Among obese adolescents, IR is commonly detected and appears similarly in relation to adolescent PCOS [3,17]. Likewise, insulin-lowering treatment in PCOS with metformin has a positive effect on peripheral insulin sensitivity, PCOM, androgen levels, and obesity [35]. This raises the possibility that IR and HI form a critical link between adolescent obesity and the development of PCOS. Therefore, the endocrinological and metabolic aberrations caused by androgen excess in PCOS might be the explanation for IR and increased adipose tissue (AT). Further studies are warranted as it still remains unclear which comes first: IR, obesity or PCOS.
Hyperandrogenemia Enhances Fat Storage
A key characteristic of PCOS is excessive levels of androgens. Aside from their effects on ovarian function and phenotypical features, like alopecia, they also influence where fat is stored in the body. This is partly explained by impaired glucose metabolism and IR [36]. In women with PCOS, there is a different distribution of AT than in women without PCOS. The AT tends to be distributed viscerally, and these women have greater upper body fat accumulation in a pattern that is similar to that of men [37]. The different AT distribution is attributed to the influence of steroids, their metabolism, and the expression of tissue-specific steroid receptors [38]. As this visceral fat is biologically active [39,40], it likely further contributes to the metabolic and endocrine disturbances in PCOS.
Androgens Decrease Lipolysis in Subcutaneous AT
There is great individual variability in the effect of androgens on the accumulation of visceral fat in men and women [41]. Most consistently [37,38], it is observed that androgens influence adipocyte function and distribution by the inhibition of adipocyte differentiation, which modulates lipolysis and lipogenesis [37,42]. Also different compartments of fat tissue exhibit phenotypic heterogeneous dysfunction in PCOS [42,43]. As an example, catecholamine-induced lipolysis is higher in visceral fat than subcutaneous fat [42], which is a possible link to the increased accumulation of fat in this specific depot in women with PCOS. Depressed expression of β2-adrenoceptors and hormone-sensitive lipase in subcutaneous fat might explain why the lipolysis is lower [42].
The mechanisms causing IR in PCOS have many similarities with those seen in relation to visceral adiposity [44]. Accumulation of fat in the visceral compartments is contradictory with the fact that lipolysis in this depot is higher than in subcutaneous fat depots, unless these high rates of fat mobilization are matched by high rates of fat deposition. Dicker et al. [42] proposed that increased lipolysis is important in the development of IR in PCOS. Excess free fatty acids derived from lipolysis/hydrolysis of acylglycerol in adipocytes accumulate in the hepatic portal veins, and this induces hepatic dysfunction [45]. This contributes to elevated glucose secretion, provokes pancreatic insulin secretion and glucose uptake in AT [38,45]. Androgens are generally thought to have a direct influence on the metabolic aberrations in PCOS, though some studies indicate differently [46].
Hyperandrogenemia and IR
As women with PCOS have both HA and IR, it is important to understand how these conditions are associated (fig. 1). To understand how HA influences IR, researchers examined the effects of supraphysiological concentrations of testosterone on subcutaneous adipocytes from women [36]. It was found that testosterone induced IR in subcutaneous adipocytes through the activation of androgen receptors. The effect of insulin on glucose metabolism was impaired due to defects in phosphorylation of a downstream protein, protein kinase-Cζ, which normally mediates the effect of insulin on glucose transport.
Fig. 1
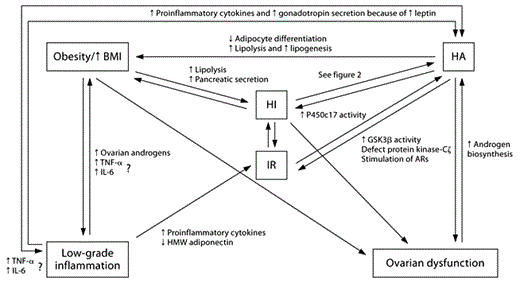
Interacting mechanisms in obesity and HA. HA and obesity have many similar molecular mechanisms that influence and exacerbate the path of each condition. The origin is individual and therefore varies. Obesity: In presence of excess AT, lipolysis increases, which causes pancreatic secretion of insulin and might eventually cause IR. AT also causes low-grade inflammation through the release of a wide range of mediators that influence body distribution (through activation of androgen synthesis) and increase release of proinflammatory mediators (TNF-α and IL-6). Obesity is often related to ovarian dysfunction due these aforementioned mechanisms especially through its effects on insulin secretion. Low-grade inflammation is associated with decreased release of HMW adiponectin and causes increased release of proinflammatory mediators; both conditions are associated with IR. Increased secretion of leptin causes gonadotropin secretion, and together with proinflammatory cytokines this causes increased androgen synthesis. HI: Continuous HI causes development of IR, increases fat deposition and influences ovarian function through its direct stimulation of hormonal biosynthesis. HI also influences HA, as described in figure 2. HA is associated with HI, but this relation is not clearly elaborated, but might be related to increased deposition of AT and increased lipolysis during HA. HA decreases adipocyte differentiation and increases lipolysis and lipogenesis in subcutaneous fat, and causes deposition of visceral fat. HA is associated with increased activity in GSK3β, defects in protein kinase-Cζ and stimulation of androgen receptors (ARs). Increased production of androgens affects the hypothalamic-pituitary-ovarian axis and causes ovarian dysfunction, and might induce secretion of adipocytokines. Ovarian dysfunction: Dysfunctional ovaries might directly cause increased androgen biosynthesis, and therefore contribute to the pathogenesis and severity of HA and deposition of AT.
Fig. 1
Interacting mechanisms in obesity and HA. HA and obesity have many similar molecular mechanisms that influence and exacerbate the path of each condition. The origin is individual and therefore varies. Obesity: In presence of excess AT, lipolysis increases, which causes pancreatic secretion of insulin and might eventually cause IR. AT also causes low-grade inflammation through the release of a wide range of mediators that influence body distribution (through activation of androgen synthesis) and increase release of proinflammatory mediators (TNF-α and IL-6). Obesity is often related to ovarian dysfunction due these aforementioned mechanisms especially through its effects on insulin secretion. Low-grade inflammation is associated with decreased release of HMW adiponectin and causes increased release of proinflammatory mediators; both conditions are associated with IR. Increased secretion of leptin causes gonadotropin secretion, and together with proinflammatory cytokines this causes increased androgen synthesis. HI: Continuous HI causes development of IR, increases fat deposition and influences ovarian function through its direct stimulation of hormonal biosynthesis. HI also influences HA, as described in figure 2. HA is associated with HI, but this relation is not clearly elaborated, but might be related to increased deposition of AT and increased lipolysis during HA. HA decreases adipocyte differentiation and increases lipolysis and lipogenesis in subcutaneous fat, and causes deposition of visceral fat. HA is associated with increased activity in GSK3β, defects in protein kinase-Cζ and stimulation of androgen receptors (ARs). Increased production of androgens affects the hypothalamic-pituitary-ovarian axis and causes ovarian dysfunction, and might induce secretion of adipocytokines. Ovarian dysfunction: Dysfunctional ovaries might directly cause increased androgen biosynthesis, and therefore contribute to the pathogenesis and severity of HA and deposition of AT.
Similarly, subcutaneous adipocytes from women with PCOS were studied to see what might cause IR in relation to PCOS [47]. Interestingly, the adipocytes from women with PCOS displayed enhanced glycogen synthase kinase-3β (GSK3β) action. As a consequence, insulin-stimulated glucose transport was impaired and IR developed. Furthermore, overexpression of GSK3β promoted androgen biosynthesis through direct stimulation of P450c17 enzyme activity [47]. Results from these two studies illustrate that continuous exposure to androgens impairs insulin action and contributes to the development of IR [36].
Excess Insulin Has Multiple Effects
In PCOS, HI may have unique effects (see fig. 2). Studies in mice have revealed that liver and muscle cells exhibit IR during continuous HI, whereas insulin receptors in pituitary and ovarian cells stay sensitive, an adaptation that increases pituitary hormone secretion and ovarian androgen production [48]. This observation has been used as a model to explain the insulinemic contribution to advanced androgen production and progress to PCOS in women. Accordingly, HI contributes to HA by the direct stimulation of steroidogenesis in ovarian theca and granulosa cells [49]. Up to a certain level, excess insulin stimulates hypothalamic GNRH secretion, and therefore induces gonadotropin secretion (particularly LH) from pituitary cells, which in turn stimulates androgen production in the ovaries [50]. When HI is established, the pituitary is prompted to secrete large amounts of LH, which tends to increase the ratio of LH/FSH [14,28]. LH stimulates the synthesis of androgens in the ovaries, and a lack of FSH impairs the aromatization of androgens to estrogens in granulosa cells. These changes cause growth of the small ovarian follicles, and they hinder the maturation necessary for development of the dominant follicle which then manifests as polycystic ovaries [51].
Fig. 2

Influence of excess insulin on HA. Insulin stimulates pancreatic secretion of SHBG which increases the concentration of free circulating testosterone. During HI, the hypothalamus and pituitary remain sensitive to insulin and increase pulse frequency of GNRH secretion, which causes elevation of the LH/FSH ratio, and this induces ovarian androgen production. Also, insulin directly stimulates ovarian hormone production. These mechanisms all contribute to the development of HA.
Fig. 2
Influence of excess insulin on HA. Insulin stimulates pancreatic secretion of SHBG which increases the concentration of free circulating testosterone. During HI, the hypothalamus and pituitary remain sensitive to insulin and increase pulse frequency of GNRH secretion, which causes elevation of the LH/FSH ratio, and this induces ovarian androgen production. Also, insulin directly stimulates ovarian hormone production. These mechanisms all contribute to the development of HA.
HA is further exacerbated because of decreased SHGB secretion caused by insulin. Therefore obesity-related HI is an important contributor to ovarian production of androgens, ovulatory dysfunction and PCOM in PCOS.
Obesity-Related Low-Grade Chronic Inflammation May Affect the Pathophysiology of PCOS
Leptin/Adiponectin
In adolescents and women with PCOS, the adipocyte-produced and -released adipocytokines including leptin, adiponectin, visfatin, monocyte chemoattractant protein-1, and retinol-binding protein 4 are altered [29,52,53]. As an example, a case-control study [29] (31 women with PCOS and 57 controls) showed that levels of leptin in PCOS patients were elevated, whereas adiponectin levels were unchanged or even lowered. This observation is interesting since leptin is able to stimulate gonadotropin secretion, a mechanism that relates to early puberty and is often seen in PCOS and obese girls [15]. Adiponectin is closely related to insulin sensitivity, and IR is connected with lowered adiponectin concentrations [29]. Total adiponectin is not predictive of PCOS, but high-molecular-weight (HMW) adiponectin, which is part of plasma adiponectin that forms multimers, has an inverse association with IR and the inflammatory state from abdominal obesity. Interestingly, HMW adiponectin is lowered in PCOS women independently of BMI and IR, an indirect effect that is possibly mediated by androgens through changes in body composition [54]. Therefore, the combination of abdominal adiposity and androgens (in PCOS) might be additive because both conditions lower HMW adiponectin.
Proinflammatory Cytokines
In both adolescent and adult women, several proinflammatory mediators, including tumor necrosis factor-α (TNF-α) and interleukin-6 (IL-6), are elevated in obesity and PCOS [55,56,57]. Whether these cytokines are elevated because of excess AT or because of mechanisms induced by HA in PCOS is of interest.
HA is thought to induce low-grade inflammation by intensifying the sensitivity to glucose-induced oxidative stress, which is caused by the activation of reactive oxygen species that promote the initiation of TNF-α and IL-6 gene transcription. This response is seen in PCOS patients but not in lean reproductive-age women [55,58]. TNF-α induces IR through decreased expression of glucose transporter type 4 [59] which reduces the transport of glucose and contributes to the development of HI. Liver production of the acute phase reactant C-reactive protein is induced by IL-6, and it has been associated with cardiovascular events including IR, T2DM and MetS [60]. Besides their potential influence on CVD, proinflammatory mediators stimulate steroidogenic enzymes in the ovaries which accelerate androgen production and induce central obesity through the activation of the hypothalamic-pituitary-adrenal axis which again leads to IR that again might, in turn, initiate HA [56,57]. Therefore, the degree of both obesity and PCOS influences the inflammatory state which exacerbates the severity of PCOS. Despite this, adipocytokines are not believed to be the primary instrumental factor in the development of androgen excess but rather reflect the development of abdominal adiposity observed in PCOS [26].
Treatment
Lifestyle interventions are the first-line treatment for PCOS, especially when it is accompanied by obesity. This suggested priority is based on the fact that reduction of central fat ameliorates the PCOS phenotypes, inter alia improved cyclicity and resumption of ovulation [40]. Also weight loss improves the status of cardiovascular risk factors accompanied by a decreased intima media thickness [61]. The effects of lifestyle intervention on normal-weight PCOS patients, however, could not be identified in the literature. Nonetheless, it is plausible that these women could also benefit from these types of interventions. In addition, it is of great importance to address and improve the psychological aspects of the condition (i.e. self-image, depression). Further, given the negative effects of PCOS on reproductive function [13], these issues need to be discussed with the patient and/or their parents. This consequence is not obvious, and it is typically not the reason why adolescents or their parents seek treatment for obesity, especially because the phenotypic outcome (anovulation) is not clinically visible before entry into puberty.
In a sample of 59 obese girls with PCOS aged 12–19 years, 26 girls reduced their body weight after 1 year of lifestyle intervention consisting of physical exercise, nutritional education, and behavioral guidance. These patients decreased their cFT, insulin concentrations and homeostatic model assessment and increased SHBG concentrations during the intervention [61]. Similar results have been reported in other studies [30,40,62]. The fundamental challenge in relation to lifestyle treatment is how to sustain the effect, i.e. maintain children/adolescents at a lower or normal weight, and stay physically active and healthy over longer periods of time. A recent study in our clinic [63] showed that it is possible to reduce the degree of obesity in 62.5% of children and youths in up to 2 years of obesity treatment with a relatively high retention irrespective of baseline adiposity, age, puberty, and social class and with no initial eligibility criteria.
Pharmacological Treatment
Pharmacological treatment may be added to further ameliorate PCOS symptoms, regarding cosmetic issues, metabolic disturbances (glucose and lipid status) and menstrual irregularities (to restore fertility). Oral contraceptives (estrogen-progestin combination pill) and insulin sensitizers (metformin) are currently accepted as effective in adolescents [14,28] either in combination with lifestyle intervention or when intervention fails. Recently, a study revealed a positive regulatory effect on endocrine biochemical markers and visceral adiposity in non-obese PCOS women after polytherapy consisting of pioglitazone (a PPARγ agonist), flutamide (androgen receptor blocker), and metformin in combination with estrogen-progestagen [64]. However, a further examination of pharmacological treatment modalities is beyond the scope of this review. Although promising, polytherapies such as these have not been evaluated in adolescents. Also, as the long-term metabolic effects of estrogen-progestin pills are not well studied, this should be kept in mind when treating adolescents. Therefore, pharmacological treatment for young girls should be undertaken with great consideration, and should be decided on a case to case basis balancing the indication and potential complications from the disease against drug-induced side effects.
Conclusion
As greater numbers of adolescent girls are becoming overweight and obese, they are potentially facing a greater risk of developing PCOS. The origins of PCOS are multifactorial, and likely there is a genetic component. Obesity and PCOS share many common features in their pathogenesis, with IR and HI playing key roles. Nonetheless, it remains unknown which condition comes first, and research is needed to disentangle the causal pathways among these factors. Although the complex metabolic interrelationships between obesity and PCOS have yet to be fully understood, the co-occurrence of these conditions in adolescent girls tends to increase the severity of the negative health consequences of each condition. Given the serious consequences of PCOS on women’s cardiovascular, reproductive and mental health, it is of great importance to detect girls who are obese in adolescence (and even younger), because counseling and treatment in an early stage of the metabolic disturbances may prevent the future deleterious effects on their health.
References
Consensus on women’s health aspects of polycystic ovary syndrome (PCOS). Hum Reprod 2012;27:14–24.
Hickey M, Doherty DA, Atkinson H, Sloboda DM, Franks S, Norman RJ, et al: Clinical, ultrasound and biochemical features of polycystic ovary syndrome in adolescents: implications for diagnosis. Hum Reprod 2011;26:1469–1477.
Muolokwu E, Sanchez J, Bercaw JL, Sangi-Haghpeykar H, Banszek T, Brandt ML, et al: Paratubal cysts, obesity, and hyperandrogenism. J Pediatr Surg 2011;46:2164–2167.
Lobstein T, Frelut ML: Prevalence of overweight among children in Europe. Obes Rev 2003;4:195–200.
Yildiz BO, Azziz R: Ovarian and adipose tissue dysfunction in polycystic ovary syndrome: report of the 4th special scientific meeting of the Androgen Excess and PCOS Society. Fertil Steril 2010;94:690–693.
Glueck CJ, Morrison JA, Daniels S, Wang P, Stroop D: Sex hormone-binding globulin, oligomenorrhea, polycystic ovary syndrome, and childhood insulin at age 14 years predict metabolic syndrome and class III obesity at age 24 years. J Pediatr 2011;159:308–313.e2.
Baker J, Holm J-C: Projected cardiovascular impact of obesity in children and adolescents: will obesity increase the cardiovascular risk of women to that of men? Curr Cardiovasc Risk Rep DOI: 10.1007/s12170-012-0230-8.
Amato MC, Verghi M, Galluzzo A, Giordano C: The oligomenorrhoic phenotypes of polycystic ovary syndrome are characterized by a high visceral adiposity index: a likely condition of cardiometabolic risk. Hum Reprod 2011;26:1486–1494.
Azziz R, Woods KS, Reyna R, Key TJ, Knochenhauer ES, Yildiz BO: The prevalence and features of the polycystic ovary syndrome in an unselected population. J Clin Endocrinol Metab 2004;89:2745–2749.
Azziz R, Sanchez LA, Knochenhauer ES, Moran C, Lazenby J, Stephens KC, et al: Androgen excess in women: experience with over 1000 consecutive patients. J Clin Endocrinol Metab 2004;89:453–462.
Toscani MK, Mario FM, Radavelli-Bagatini S, Spritzer PM: Insulin resistance is not strictly associated with energy intake or dietary macronutrient composition in women with polycystic ovary syndrome. Nutr Res 2011;31:97–103.
Lin Y-H, Chiu W-C, Wu C-H, Tzeng C-R, Hsu C-S, Hsu M-I: Antimüllerian hormone and polycystic ovary syndrome. Fertil Steril 2011;96:230–235.
Revised 2003 consensus on diagnostic criteria and long-term health risks related to polycystic ovary syndrome. Fertil Steril 2004;81:19–25.
Bremer AA: Polycystic ovary syndrome in the pediatric population. Metab Syndr Relat Disord 2010;8:375–394.
Burt Solorzano CM, McCartney CR: Obesity and the pubertal transition in girls and boys. Reproduction 2010;140:399–410.
Nair MKC, Pappachan P, Balakrishnan S, Leena ML, George B, Russell PS: Menstrual irregularity and poly cystic ovarian syndrome among adolescent girls – a 2 year follow-up study. Indian J Pediatr DOI: 10.1007/s12098-011-0432-y.
Sir-Petermann T, Codner E, Pérez V, Echiburú B, Maliqueo M, Ladrón de Guevara A, et al: Metabolic and reproductive features before and during puberty in daughters of women with polycystic ovary syndrome. J Clin Endocrinol Metab 2009;94:1923–1930.
Bruni V, Dei M, Nannini S, Balzi D, Nuvolone D: Polycystic ovary syndrome in adolescence. Ann N Y Acad Sci 2010;1205:175–184.
Merino PM, Codner E, Cassorla F: A rational approach to the diagnosis of polycystic ovarian syndrome during adolescence. Arq Bras Endocrinol Metabol 2011;55:590–598.
Hart R, Doherty DA, Norman RJ, Franks S, Dickinson JE, Hickey M, et al: Serum antimullerian hormone (AMH) levels are elevated in adolescent girls with polycystic ovaries and the polycystic ovarian syndrome (PCOS). Fertil Steril 2010;94:1118–1121.
Prapas N, Karkanaki A, Prapas I, Kalogiannidis I, Katsikis I, Panidis D: Genetics of polycystic ovary syndrome. Hippokratia 2009;13:216–223.
Eisner JR, Dumesic DA, Kemnitz JW, Colman RJ, Abbott DH: Increased adiposity in female rhesus monkeys exposed to androgen excess during early gestation. Obes Res 2003;11:279–286.
Xu N, Kwon S, Abbott DH, Geller DH, Dumesic DA, Azziz R, et al: Epigenetic mechanism underlying the development of polycystic ovary syndrome (PCOS)-like phenotypes in prenatally androgenized rhesus monkeys. PLoS ONE 2011;6:e27286.
Wojciechowski P, Lipowska A, Rys P, Ewens KG, Franks S, Tan S, et al: Impact of FTO genotypes on BMI and weight in polycystic ovary syndrome: a systematic review and meta-analysis. Diabetologia 2012;55:2636–2645.
Vaag AA, Grunnet LG, Arora GP, Brøns C: The thrifty phenotype hypothesis revisited. Diabetologia 2012;55:2085–2088.
Ibáñez L, Lopez-Bermejo A, Díaz M, Suárez L, de Zegher F: Low-birth weight children develop lower sex hormone binding globulin and higher dehydroepiandrosterone sulfate levels and aggravate their visceral adiposity and hypoadiponectinemia between six and eight years of age. J Clin Endocrinol Metab 2009;94:3696–3699.
de Zegher F, Lopez-Bermejo A, Ibáñez L: Adipose tissue expandability and the early origins of PCOS. Trends Endocrinol Metab 2009;20:418–423.
Buggs C, Rosenfield R: Polycystic ovary syndrome in adolescence. Endocrinol Metab Clin North Am 2005;34:677–705.
Lecke SB, Mattei F, Morsch DM, Spritzer PM: Abdominal subcutaneous fat gene expression and circulating levels of leptin and adiponectin in polycystic ovary syndrome. Fertil Steril 2011;95:2044–2049.
Reinehr T, de Sousa G, Roth CL, Andler W: Androgens before and after weight loss in obese children. J Clin Endocrinol Metab 2005;90:5588–5595.
Bille DS, Chabanova E, Gamborg M, et al: Liver fat content investigated by magnetic resonance spectroscopy in obese children and youths included in multidisciplinary treatment. Clin Obes DOI: 10.1111/j.1758- 8111.2012.00038.x.
Juonala M, Magnussen CG, Berenson GS, Venn A, Burns TL, Sabin MA, et al: Childhood adiposity, adult adiposity, and cardiovascular risk factors. N Engl J Med 2011;365:1876–1885.
Holm J-C, Gamborg M, Neland M, Ward L, Gammeltoft S, Heitmann BL, et al: Longitudinal changes in blood pressure during weight loss and regain of weight in obese boys and girls. J Hypertens 201;30:368–374.
Rossi B, Sukalich S, Droz J, Griffin A, Cook S, Blumkin A, et al: Prevalence of metabolic syndrome and related characteristics in obese adolescents with and without polycystic ovary syndrome. J Clin Endocrinol Metab 2008;93:4780–4786.
Allen HF, Mazzoni C, Heptulla RA, Murray MA, Miller N, Koenigs L, et al: Randomized controlled trial evaluating response to metformin versus standard therapy in the treatment of adolescents with polycystic ovary syndrome. J Pediatr Endocrinol Metab 2005;18:761–768.
Corbould A: Chronic testosterone treatment induces selective insulin resistance in subcutaneous adipocytes of women. J Endocrinol 2007;192:585–594.
Kuk JL, Lee S, Heymsfield SB, Ross R: Waist circumference and abdominal adipose tissue distribution: influence of age and sex. Am J Clin Nutr 2005;81:1330–1334.
Blouin K, Veilleux A, Luu-The V, Tchernof A: Androgen metabolism in adipose tissue: recent advances. Mol Cell Endocrinol 2009;301:97–103.
Abrams P, Levitt Katz LE: Metabolic effects of obesity causing disease in childhood. Curr Opin Endocrinol Diabetes Obes 2011;18:23–27.
Kuchenbecker WKH, Groen H, van Asselt SJ, Bolster JHT, Zwerver J, Slart RHJ, et al: In women with polycystic ovary syndrome and obesity, loss of intra-abdominal fat is associated with resumption of ovulation. Hum Reprod 2011;26:2505–2512.
Blouin K, Boivin A, Tchernof A: Androgens and body fat distribution. J Steroid Biochem Mol Biol 2008;108:272–280.
Dicker A, Rydén M, Näslund E, Muehlen IE, Wirén M, Lafontan M, et al: Effect of testosterone on lipolysis in human pre-adipocytes from different fat depots. Diabetologia 2004;47:420–428.
Arner P: Effects of testosterone on fat cell lipolysis. Species differences and possible role in polycystic ovarian syndrome. Biochimie 2005;87:39–43.
Kabir M, Catalano KJ, Ananthnarayan S, Kim SP, Van Citters GW, Dea MK, et al: Molecular evidence supporting the portal theory: a causative link between visceral adiposity and hepatic insulin resistance. Am J Physiol Endocrinol Metab 2005;288:E454–E461.
Bergman RN, Ader M: Free fatty acids and pathogenesis of type 2 diabetes mellitus. Trends Endocrinol Metab 2000;11:351–356.
Forrester-Dumont K, Galescu O, Kolesnikov A, Raissouni N, Bhangoo A, Ten S, et al: Hyperandrogenism does not influence metabolic parameters in adolescent girls with PCOS. Int J Endocrinol 2012;2012:1–5.
Chang W, Goodarzi MO, Williams H, Magoffin DA, Pall M, Azziz R: Adipocytes from women with polycystic ovary syndrome demonstrate altered phosphorylation and activity of glycogen synthase kinase 3. Fertil Steril 2008;90:2291–2297.
Wu S, Divall S, Wondisford F, Wolfe A: Reproductive tissues maintain insulin sensitivity in diet-induced obesity. Diabetes 2012;61:114–123.
Ojaniemi M, Tapanainen P, Morin-Papunen L: Management of polycystic ovary syndrome in childhood and adolescence. Horm Res Paediatr 2010;74:372–375.
Moret M, Stettler R, Rodieux F, Gaillard RC, Waeber G, Wirthner D, et al: Insulin modulation of luteinizing hormone secretion in normal female volunteers and lean polycystic ovary syndrome patients. Neuroendocrinology 2009;89:131–139.
Maciel GAR, Baracat EC, Benda JA, Markham SM, Hensinger K, Chang RJ, et al: Stockpiling of transitional and classic primary follicles in ovaries of women with polycystic ovary syndrome. J Clin Endocrinol Metab 2004;89:5321–5327.
Kershaw EE, Flier JS: Adipose tissue as an endocrine organ. J Clin Endocrinol Metab 2004;89:2548–2556.
Cekmez F, Cekmez Y, Pirgon O, Canpolat FE, Aydinöz S, Metin Ipcioglu O, et al: Evaluation of new adipocytokines and insulin resistance in adolescents with polycystic ovary syndrome. Eur Cytokine Netw 2011;22:32–37.
O’Connor A, Phelan N, Tun TK, Boran G, Gibney J, Roche HM: High-molecular-weight adiponectin is selectively reduced in women with polycystic ovary syndrome independent of body mass index and severity of insulin resistance. J Clin Endocrinol Metab 2010;95:1378–1385.
Xiong Y, Liang X, Yang X, Li Y, Wei L: Low-grade chronic inflammation in the peripheral blood and ovaries of women with polycystic ovarian syndrome. Eur J Obstet Gynecol Reprod Biol 2011;159:148–150.
Samy N, Hashim M, Sayed M, Said M: Clinical significance of inflammatory markers in polycystic ovary syndrome: their relationship to insulin resistance and body mass index. Dis Markers 2009;26:163–170.
Alemzadeh R, Kichler J, Calhoun M: Spectrum of metabolic dysfunction in relationship with hyperandrogenemia in obese adolescent girls with polycystic ovary syndrome. Eur J Endocrinol 2010;162:1093–1099.
González F, Nair KS, Daniels JK, Basal E, Schimke JM: Hyperandrogenism sensitizes mononuclear cells to promote glucose-induced inflammation in lean reproductive-age women. Am J Physiol Endocrinol Metab 2012;302:E297–E306.
González F: Inflammation in polycystic ovary syndrome: underpinning of insulin resistance and ovarian dysfunction. Steroids 2012;77:300–305.
Ridker PM, Buring JE, Cook NR, Rifai N: C-reactive protein, the metabolic syndrome, and risk of incident cardiovascular events: an 8-year follow-up of 14,719 initially healthy American women. Circulation 2003;107:391–397.
Lass N, Kleber M, Winkel K, Wunsch R, Reinehr T: Effect of lifestyle intervention on features of polycystic ovarian syndrome, metabolic syndrome, and intima-media thickness in obese adolescent girls. J Clin Endocrinol Metab 2011;96:3533–3540.
Giallauria F, Palomba S, Vigorito C, Tafuri MG, Colao A, Lombardi G, et al: Androgens in polycystic ovary syndrome: the role of exercise and diet. Semin Reprod Med 2009;27:306–315.
Holm J-C, Gamborg M, Bille DS, Gr Nb K HN, Ward LC, Faerk J: Chronic care treatment of obese children and adolescents. Int J Pediatr Obes 2011;6:188–196.
Vinaixa M, Rodriguez MA, Samino S, Díaz M, Beltran A, Mallol R, et al: Metabolomics reveals reduction of metabolic oxidation in women with polycystic ovary syndrome after pioglitazone-flutamide-metformin polytherapy. PLoS ONE 2011;6:e29052.
© 2012 S. Karger AG, Basel
2012
Copyright / Drug Dosage / Disclaimer
Copyright: All rights reserved. No part of this publication may be translated into other languages, reproduced or utilized in any form or by any means, electronic or mechanical, including photocopying, recording, microcopying, or by any information storage and retrieval system, without permission in writing from the publisher.
Drug Dosage: The authors and the publisher have exerted every effort to ensure that drug selection and dosage set forth in this text are in accord with current recommendations and practice at the time of publication. However, in view of ongoing research, changes in government regulations, and the constant flow of information relating to drug therapy and drug reactions, the reader is urged to check the package insert for each drug for any changes in indications and dosage and for added warnings and precautions. This is particularly important when the recommended agent is a new and/or infrequently employed drug.
Disclaimer: The statements, opinions and data contained in this publication are solely those of the individual authors and contributors and not of the publishers and the editor(s). The appearance of advertisements or/and product references in the publication is not a warranty, endorsement, or approval of the products or services advertised or of their effectiveness, quality or safety. The publisher and the editor(s) disclaim responsibility for any injury to persons or property resulting from any ideas, methods, instructions or products referred to in the content or advertisements.