NF-κB–inducing kinase plays an essential T cell–intrinsic role in graft-versus-host disease and lethal autoimmunity in mice (original) (raw)
NIK-deficient CD4+ T cells do not cause lethal GVHD. To test whether NIK is required in a T cell–intrinsic manner for allogeneic responses, we sorted naive CD4+ T cells from aly/aly mutant mice or littermate controls and transferred them into sublethally irradiated MHC class II mismatched congenic (H-2bm12×CD45.1)F1 recipients. Transfer of T cells with 2 normal copies of NIK caused lethal GVHD in 100% of recipient mice, which was preceded by rapid weight loss starting 2 weeks after transfer (Figure 1). In stark contrast, aly/aly T cells caused no GVHD lethality or weight loss. Interestingly, recipients of heterozygous aly/+ T cells showed an intermediate phenotype, experiencing delayed disease and a 70% mortality rate. The importance of NIK levels seen here mirrors recent reports showing that loss of a single copy of NIK can rescue TNF receptor–associated factor 2 and TNF receptor–associated factor 3 knockout mice from perinatal lethality (20, 21). The failure of NIK-deficient T cells to induce lethal GVHD is likely a result of poor accumulation of allospecific T cells in the recipients, as illustrated in a separate experiment in which we transferred a smaller, nonlethal number (1 × 105) of naive CD4+ T cells into sublethally irradiated (H-2bm12×CD45.1)F1 recipients. In this case, despite lack of overt disease, control CD4+ T cells accumulated in mismatched recipients, and 10-fold more control CD4+ T cells than aly/aly T cells were recovered from recipient spleens 1 month after transfer (Table 1).

NIK-deficient CD4+ T cells do not cause lethal GVHD. 1 × 106 naive +/+, aly/+, or aly/aly CD4+ T cells were injected intravenously into sublethally irradiated (H-2bm12×CD45.1)F1 recipients. (A) Survival is combined data from 4 independent experiments; each genotype was represented in 2–4 experiments. P < 0.05, +/+ versus aly/+ and aly/+ versus aly/aly; log-rank test. (1 mouse in the aly/aly group died only 4 days after transfer, which is much earlier than GVHD onset in this model). (B) Body weight data (± SEM) are representative of 2 independent experiments.

NIK-deficient T cell accumulation after transfer into (H-2bm12×CD45.1)F1 MHC mismatched recipients
NIK-deficient T cells respond normally to TCR stimulation. Some reports have found diminished T cell responsiveness to TCR signals in the absence of NIK, which could explain the failure of aly/aly CD4+ T cells to induce GVHD (15). We tested the ability of naive aly/aly and control CD4+ T cells to respond in vitro to TCR stimulation with or without CD28 costimulation. aly/aly and control T cells upregulated OX40, CD69, and CD25 to the same extent (Figure 2A). Moreover, aly/aly T cells proliferated as well as or better than controls in all conditions (Figure 2B). Similarly, with TCR plus CD28 costimulation, aly/aly T cells produced normal amounts of IL-2, and in the absence of CD28 costimulation, aly/aly T cells produced only slightly less IL-2 (Figure 2C). Upon addition of IL-12 to cultures, aly/aly T cells produced IFN-γ at levels comparable to those of control T cells (Supplemental Figure 1; supplemental material available online with this article; doi:10.1172/JCI44943DS1), which indicates that NIK is not required for Th1 polarization. These results are similar to recent reports that also used sorted naive NIK-deficient CD4+ T cells (16, 19) and suggest that the inability of aly/aly T cells to cause GVHD is not likely to be caused by signaling defects downstream of the TCR.

NIK-deficient CD4+ T cells respond normally to TCR signaling. Sorted naive T cells (CD4+CD25–CD44lo) were stimulated with plate-bound anti-CD3 with (A–C) or without (B and C) anti-CD28. (A) Upregulation of OX40, CD69, and CD25 was assessed on day 2. Gray represents Ox40–/– (for OX40) or +/+ unstimulated (for CD69 and CD25); thin line represents +/+ stimulated; bold line represents aly/aly stimulated. (B) Proliferation was measured by tritiated thymidine incorporation on days 3 and 4. (C) IL-2 was measured by ELISA of culture supernatants on days 3 and 4. Data (± SD) are representative of 1 (A) or 2 (B and C) independent experiments.
NIK is required for OX40-mediated costimulation of CD4+ T cells. In B cells and lymphoid organ stromal cells, NIK acts downstream of TNFRs such as BAFFR and LTβR. Activated T cells also express TNFRs that function as important costimulatory molecules, and several, such as OX40, CD30, HVEM, and 4-1BB, have previously been shown to contribute to GVHD in mouse models (22). In particular, OX40 is essential for lethality when B6 CD4+ T cells are transferred into irradiated H-2bm12 recipients (23), and we confirmed that naive Ox40–/– CD4+ T cells did not cause lethal GVHD in our similar model (Supplemental Figure 2). Therefore, we considered the possibility that the inability of NIK-deficient T cells to mediate lethal GVHD might be owing to a requirement for NIK downstream of OX40 and perhaps other TNFRs.
To test this hypothesis, we used a model of GVHD previously described by our lab, in which costimulation by agonist anti-OX40 antibody produces lethal GVHD in unirradiated (H-2bm12×CD45.1)F1 recipients of B6 CD4+ T cells. Without irradiation of recipients and without anti-OX40, donor CD4+ T cells proliferate and accumulate, but are rendered anergic and do not acquire effector function (24), because in the absence of irradiation damage and inflammation, costimulatory molecules are not upregulated (22). The OX40 stimulus causes alloreactive donor T cells to differentiate to IFN-γ producers. It acts directly on donor cells, as shown by lack of a response by Ox40–/– donor T cells transferred alone into in WT (H-2bm12×CD45.1)F1 recipients (24). We tested naive aly/aly or littermate control donor CD4+ T cells in this model to assess whether NIK is essential for OX40-mediated costimulation. When naive CD4+ T cells were cotransferred with control antibody and assayed 5 days later, aly/aly and control CD4+ T cells behaved identically: both proliferated more than 7 times, as assessed by complete CFSE dilution (data not shown); both expanded to similar numbers (Table 1); and neither upregulated CD25 or produced IFN-γ upon ex vivo stimulation (Figure 3). In contrast, coadministration of agonist anti-OX40 antibody had very disparate effects on control versus aly/aly donor T cells. In recipients of control CD4+ T cells, anti-OX40 increased cell accumulation 3-fold (Table 1), induced massive CD25 upregulation (Figure 3A), and induced effector differentiation such that donor cells produced copious IFN-γ upon ex vivo stimulation with IL-12 and IL-18 or with PMA and ionomycin (Figure 3, B and C). Moreover, approximately 20% of control T cells made IFN-γ even without stimulation (Figure 3, B and C). In contrast, anti-OX40 administration had almost no effect on aly/aly T cells, as assessed by cell numbers, CD25 upregulation, and IFN-γ production (Table 1 and Figure 3). We conclude that NIK is essential for OX40 to costimulate and impart a lethal phenotype to responding alloreactive CD4+ T cells.

NIK-deficient CD4+ T cells do not respond to OX40 signaling. 7 × 106 naive aly/aly or aly/+ CD4+ T cells were injected intravenously into unirradiated (H-2bm12×CD45.1)F1 recipients along with 50 μg agonist anti-OX40 or control antibody. Spleens were harvested 5 days later and stained immediately for CD25 (A) or stimulated for 5 hours with medium alone, IL-12 and IL-18, or PMA and ionomycin and then stained for intracellular IFN-γ (B and C). (A and B) Plots are gated on CD45.1– CD4+ (transferred) cells; numbers reflect percent CD25+ or IFN-γ+ gated cells. Data are representative of 3 mice per group in 1 representative experiment of 3. (C) Percent IFN-γ+ CD45.1– donor CD4+ T cells. Data (± SEM) are representative of 3 independent experiments.
Because NIK is required for normal thymic epithelial cell development, we more stringently tested the T cell–intrinsic role of NIK using aly/aly or control T cells obtained from BM chimeras in which the BM recipients (and thus thymic stroma) were WT. aly/aly T cells that developed in a NIK sufficient thymus also failed to respond to costimulation through OX40 by upregulating CD25 or acquiring the capacity to secrete IFN-γ (Supplemental Figure 3).
In addition to costimulating effector T cells, OX40 may also augment immune responses in some situations by inhibiting Treg differentiation (25–27). We tested whether this function of OX40 also relies on signaling through NIK by inducing Treg differentiation in vitro in the presence or absence of agonist anti-OX40 antibody. OX40 inhibited induced Treg (iTreg) differentiation by approximately 40% in NIK-sufficient CD4+ T cells, whereas it had no effect on iTreg induction in aly/aly CD4+ T cells (Supplemental Figure 4).
OX40 activates noncanonical NF-κB in a NIK-dependent manner. NIK is necessary to activate the noncanonical NF-κB pathway downstream of a limited number of TNFRs, including BAFFR, LTβR, and CD40. To assess whether OX40 also depends on NIK to activate the noncanonical NF-κB pathway, we examined canonical and noncanonical NF-κB signaling downstream of OX40 in aly/aly and littermate control T cells. We stimulated purified naive aly/aly or control CD4+ T cells with anti-CD3 and anti-CD28, and after 48 hours we added control antibody or agonist anti-OX40 for 5 minutes or 20 hours to assess NF-κB activation. Stimulation with anti-OX40 for 5 minutes induced equivalent IκBα phosphorylation in T cells of both genotypes (Figure 4A), which indicates that OX40 initiates robust canonical NF-κB signaling, as previously reported (28), and that this pathway is NIK independent.

OX40 activates canonical and noncanonical NF-κB pathways in CD4+ T cells, and the noncanonical pathway depends on NIK. Purified naive CD4+ T cells were stimulated with anti-CD3 plus anti-CD28, and anti-OX40 or control antibody was added 48 hours later. (A and B) Whole-cell extracts were assessed for phosphorylated IκBα (pIκBα) and p100/p52 by Western blot. BD, below level of detection. In B, separated blots are from different gels run at the same time. Note the differing y axis scales in the left and right graphs of B. (C) Nuclear extracts were assessed by EMSA with the indicated supershifting antibodies. Single asterisks denote c-Rel/RelA heterodimers and RelA homodimers; double asterisks denote nonspecific bands. For each set of panels (top, middle, bottom) all 8 lanes were run on the same gel but were noncontiguous. Data are representative of 2 independent experiments.
We examined the effect of OX40 ligation on noncanonical NF-κB activation by assessing p100 processing in whole-cell extracts. Naive CD4+ T cells contained low levels of p100 (Figure 4B). As previously reported (16), naive aly/aly T cells expressed less p100 than control T cells, but by 48 hours after TCR stimulation, p100 levels were the same between genotypes (Figure 4B and Supplemental Figure 5). Although p100 expression increased upon TCR stimulation in both control and aly/aly T cells, processing to p52 was minimal at 24 and 48 hours; 20 hours later (i.e., 68 hours of TCR stimulation), the p52/p100 ratio had increased in both control and aly/aly cultures (Figure 4B), showing that after 2–3 days of in vitro stimulation, CD4+ T cells processed p100 in a partially NIK-independent manner. However, agonist anti-OX40 antibody augmented p100 processing in control T cells, but had no effect on aly/aly T cells (Figure 4B). Thus, although TCR stimulation alone induced both NIK-dependent and NIK-independent p100 processing, OX40 further increased p100 processing, and this effect was entirely NIK-dependent.
To assess downstream events in the NF-κB pathway, we performed EMSA on nuclear extracts from OX40-stimulated aly/aly and control cells. We found maximal DNA binding activity 48 hours after TCR stimulation, which then declined by 20 hours later with control antibody (Figure 4C, top). Anti-OX40 treatment maintained NF-κB binding, and this effect was similar between control and aly/aly T cells. This reflects the dominant contribution of canonical NF-κB activation downstream of the TCR and OX40. Supershift analysis using anti–c-Rel antibody or combined anti–c-Rel and anti-RelA antibodies allowed discrimination of discrete RelA and RelB bands, indicative of canonical and noncanonical NF-κB signaling, respectively. OX40 stimulation upregulated RelA DNA binding activity in both aly/aly and control T cells (Figure 4C, middle). In contrast, OX40 stimulation upregulated RelB DNA-binding activity only in control, but not in aly/aly, CD4+ T cells (Figure 4C, middle and bottom). Together, these data showed that OX40 activated both canonical and noncanonical NF-κB pathways in T cells, but only activation of the noncanonical NF-κB pathway required NIK. By extension, these data indicate that the seemingly minor contribution of RelB to total NF-κB activity may be essential to the function of OX40 in vivo.
NIK overexpression in T cells causes lethal T cell–mediated inflammation and iTreg-mediated suppression. Given the essential role for NIK in our model of OX40-induced GVHD, we asked whether overexpression of NIK in the absence of OX40 or other TNFR costimulation was sufficient to recapitulate this effect. To this end, we obtained conditional NIK transgenic mice in which a single copy NIK-IRES-GFP transgene preceded by a loxP-flanked stop cassette is inserted into the ubiquitously expressed ROSA-26 locus (referred to herein as NIKtg mice; ref. 29). We crossed NIKtg mice with CD4-Cre transgenic mice — generating mice referred to herein as CD4-NIKtg — in order to turn on expression of the NIK transgene specifically in T cells, as confirmed by flow cytometry (Supplemental Figure 6). Expression of the NIK transgene in T cells caused an upregulation of NF-κB2 (p100 plus p52) and an increase in processing of p100 to p52 that was similar to the level of p100 processing seen in WT B cells (Supplemental Figure 7). Unexpectedly, CD4-NIKtg mice died before weaning. They were born at a normal Mendelian frequency of 50%, but began to lose weight rapidly around day 20 (Figure 5A), developed loose stools, and quickly became moribund, requiring euthanasia between days 23 and 30 (Figure 5B). The lungs of all moribund CD4-NIKtg mice examined had moderate to massive lymphocytic infiltration (7 of 7), as did the colons of 6 of 7 CD4-NIKtg mice (Figure 5C). Age-matched littermate control organs were all normal (4 of 4).

Overexpression of NIK in T cells causes lethal inflammation. (A and B) CD4-NIKtg and age-matched littermate WT control mice were weighed and euthanized when moribund. Data in A show average weights (± SD) from 2 combined litters. (C) Sections of colon and lungs from moribund CD4-NIKtg and age-matched littermate WT control mice were assessed for leukocyte infiltrates by hematoxylin-eosin stain. Scale bars: 100 μm. Data are from 2 combined litters.
The rapid-onset mortality accompanied by lymphocytic infiltration of multiple organs that we observed in CD4-NIKtg mice was similar to the phenotype of Foxp3–/– and FoxP3scurfy mice, which harbor an inactivating mutation in Foxp3 (30). These mice die of T cell–mediated autoimmunity resulting from lack of regulation by Foxp3+ Tregs. To determine whether NIK overexpression interferes with Treg and/or effector T cell development, we assessed lymphocyte populations from CD4-NIKtg mice at day 11, before onset of outward disease symptoms. Spleen and thymus cellularity were similar between CD4-NIKtg and control littermates (Supplemental Figure 8), but peripheral and mesenteric LNs were noticeably larger (data not shown). The proportion of CD8+ T cells was slightly lower in LN and spleen of CD4-NIKtg mice, and the proportion of total CD4+ T cells was the same in spleen and LN and slightly lower in the thymus of CD4-NIKtg mice (Supplemental Figure 8). To our surprise, when we examined CD4+ T cells more closely, we found that a significantly greater percentage of CD4+ T cells was Foxp3+ in CD4-NIKtg lymphoid organs compared with lymphoid organs of control littermates (Figure 6, A and C). Despite this large increase of Foxp3+ T cells, the conventional T cell subsets (CD8+ and Foxp3–CD4+) showed an activated phenotype, as assessed by a 2- to 3-fold increase in the percentage of CD44hi cells (Figure 6, B and D, and data not shown), which suggests they are poorly regulated by Tregs. In CD4-NIKtg mice, both Foxp3+ and Foxp3– T cell populations overexpress NIK, so it was unclear whether CD4-NIKtg Tregs are poor regulators or whether CD4-NIKtg conventional T cells (Tconvs) are resistant to regulation. CD4-NIKtg Foxp3+CD4+ T cells had significantly lower CD44 and Foxp3 expression compared with control littermates (Figure 6, E and F), consistent with poorer Treg function (31). To directly address this possibility, we performed in vitro Treg suppression assays using combinations of WT and CD4-NIKtg Tregs and Tconvs. In this assay, WT Tregs inhibited the ability of WT Tconvs to proliferate in response to polyclonal stimulation with anti-CD3 and APCs. While WT Tregs efficiently suppressed WT Tconvs, showing suppression at a 1:18 Treg/Tconv ratio, CD4-NIKtg Tregs were incapable of suppressing WT Tconvs, even at a 1:1 Treg/Tconv ratio (Figure 6, G and H). In addition, CD4-NIKtg Tconvs were difficult to suppress, showing only modest regulation by WT Tregs, even at the highest 1:2 Treg/Tconv ratio (Figure 6, G and I). This suggested that overexpression of NIK both increases Tconv resistance to Treg-mediated suppression and impairs Treg function.

CD4-NIKtg mice have increased populations of activated T cells and nonfunctional Tregs. (A–F) Organs were harvested from 11-day-old CD4-NIKtg (bold lines and white bars) and age-matched littermate WT control mice (narrow lines and black bars) and assessed for surface markers and intracellular Foxp3 by flow cytometry. (G–I) Naive CD4+ T cells and Tregs were sorted from 11-day-old CD4-NIKtg and littermate WT control spleens and LNs. Naive T cells were labeled with CFSE and cultured with WT APCs and soluble anti-CD3 at various ratios with Tregs. Gates in G indicate proportion of T cells that underwent at least 1 cell division. nd, not done. Data (± SD) are representative of 2 independent experiments. *P < 0.05.
Despite in vitro resistance to Treg-mediated suppression, CD4-NIKtg Tconvs were suppressible in vivo, as assessed by BM chimeras. Lethally irradiated CD45.1 recipients of a mixture of CD4-NIKtg and congenically marked WT BM remained healthy for at least 10 months and did not show signs of inflammation such as enlarged LNs. The same was true for recipients of CD4-NIKtg BM alone, due to radioresistant Tregs that reconstitute the host after irradiation and have previously been shown to prevent disease after transplantation of Foxp3scurfy BM (32). The mixed BM chimeras demonstrated that the CD4-NIKtg Tconv and Treg phenotypes are intrinsic rather than secondary to inflammation, because although the mice remained healthy, (a) the CD4-NIKtg Tconvs had an expanded population of CD44hi cells; (b) WT Tregs were expanded relative to CD4-NIKtg Tregs; and (c) NIKtg Tregs showed decreased CD44 expression, a phenotype consistent with poor regulatory capacity (Supplemental Figure 9).
These chimeras also allowed us to obtain CD4-NIKtg Tregs from healthy mice to stringently test the Treg-intrinsic role of NIK. The decreased suppressive capacity of CD4-NIKtg Tregs from 11-day-old CD4-NIKtg mice could have been secondary to an inflammatory environment in these prediseased mice, rather than an intrinsic effect of NIK in Tregs. We sorted CD4-NIKtg and WT Tregs and Tconvs from healthy BM chimeras and performed in vitro Treg suppression assays using various combinations of Treg and Tconv cell populations. The results of this experiment were similar to those obtained using 11-day-old CD4-NIKtg mice: CD4-NIKtg Tregs from healthy BM chimeras were poorer suppressors of WT Tconvs than were WT Tregs, and CD4-NIKtg Tconvs were more resistant to suppression by WT Tregs than were WT Tconvs (Figure 7). However, CD4-NIKtg Tregs from the healthy mice were only modestly impaired in suppressive capacity, which suggests that both Treg-intrinsic and inflammatory phenomena contribute to the demise of Treg function in CD4-NIKtg mice.

NIK intrinsically inhibits Treg-suppressive capacity and increases Tconv resistance to suppression. CD4-NIKtg and littermate WT control Tregs and naive T cells were sorted from spleen and LN of healthy BM chimeras and cultured with CD45.1 Tregs or naive T cells plus WT APCs and soluble anti-CD3. (A) Naive T cells were labeled with CellTrace Violet to assess division over the course of 3 days. Gates indicate proportion of T cells that underwent at least 1 cell division. (B and C) Data (± SD) are representative of 2 independent experiments.
These in vitro data suggested that overexpression of NIK in T cells causes aggressive lethal autoimmunity via intrinsic effects on both Tconvs and Tregs; combined, these effects lead to hyperactive effector T cells. Furthermore, the observation that lethally irradiated CD45.1 WT recipients of CD4-NIKtg BM (which harbor radioresistant WT host Tregs) remained healthy, but Rag1–/– recipients (which lack host Tregs) succumbed to disease (data not shown), suggested that WT Tregs can regulate CD4-NIKtg effector T cells and, by extension, that CD4-NIKtg Tregs cannot. To formally test this, we made mixed BM chimeras in which 1:1 mixtures of CD4-NIKtg plus Foxp3scurfy BM or CD4-NIKtg plus WT (FoxP3scurfy littermate) BM were used to reconstitute Rag1–/– recipients. In this experiment, the sole difference between the groups was the absence or presence of WT Foxp3+ Tregs. We found that 80% of the CD4-NIKtg plus WT BM recipients survived, but nearly all of the CD4-NIKtg plus FoxP3scurfy BM recipients succumbed to disease within 8 weeks (Supplemental Figure 10). We thus concluded that CD4-NIKtg Tregs have poor regulatory function in vivo.
In our genetic systems of NIK induction, T cells overexpressed NIK from the double-positive thymocyte stage. We wondered whether acute NIK induction in mature Tregs would have the same effect, so we developed a system to induce NIK expression in vitro using TAT-Cre fusion protein transduction, in which we attained approximately 50%–70% transduction efficiency. We treated purified NIKtg or control CD4+ T cells with TAT-Cre, cultured under Treg-inducing conditions, and then sorted iTregs from control cultures and GFP+ iTregs from NIKtg cultures. NIK overexpression did not interfere with iTreg differentiation in these conditions, as assessed by Foxp3 and CD25 expression (Figure 8A and data not shown). However, when we tested these sorted iTregs for suppressive function against WT Tconvs, we found that iTregs in which NIK was acutely expressed were much poorer suppressors than TAT-Cre–treated control iTregs (Figure 8, B and C). This showed that NIK overexpression affected Treg function independently of developmental effects that could occur during selection in the thymus.
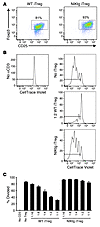
Acute overexpression of NIK in mature T cells inhibits iTreg suppressive capacity. CD4+ T cells were magnetically purified from NIKtg and littermate WT control spleen and LN, treated with TAT-Cre, and cultured under iTreg-inducing conditions for 3 days. Tregs were then sorted on the basis of CD4, CD25, and GFP, and cultured with CD45.1 naive T cells plus WT APCs and soluble anti-CD3 for an additional 3 days. (A) After iTreg induction and sorting, WT and NIKtg iTregs expressed equivalent Foxp3 and CD25. Plots are gated on live T cells. Numbers reflect percent Foxp3+ T cells. (B) Naive CD45.1 CD4+ T cells were labeled with CellTrace Violet to assess division over the course of 3 days. (C) Data (± SD) are representative of 2 independent experiments.