Genetic inactivation of FAAP100 causes Fanconi anemia due to disruption of the monoubiquitin ligase core complex (original) (raw)
A variant in FAAP100 reveals an association with FA. We investigated the genetic causes of rare individuals with unclassified FA-like phenotypes. Seven DNAs from cell lines deficient in FANCD2 monoubiquitylation were included. SNP mapping in 1 DNA originating from a fetus (III-2, ID no. 1176), offspring of first cousins, revealed 15 autozygous regions of greater than 0.2 Mb in size (Figure 1A and Supplemental Table 1; supplemental material available online with this article; https://doi.org/10.1172/JCI187323DS1). Seven of them contained a total of 20 genes associated with DNA repair, including 3 regions with the FA and FA-associated genes FANCM, FANCV, MHF1/FAAP16, MHF2/FAAP10, and FAAP100 (Supplemental Table 1 and Figure 1B). After filtering and prioritization, whole-exome sequencing (WES) results identified a conspicuous change in FAAP100 (Supplemental Figure 1A). Sanger sequencing confirmed the homozygous sequence variant NM_025161.6(FAAP100):c.1624A>C, rs1598596062, chromosome position 17-81547458T>G (GRCh38) (Supplemental Figure 1B). It segregates in this family, consistent with an autosomal recessive trait (Figure 1C). No other DNA repair–related genes in the major autozygous regions were found to have disease-causing variants, including none in the 4 FA or FA-associated genes FANCM, FANCV, MHF1/FAAP16, or MHF2/FAAP10 that were excluded by sequence analysis in the WES. The WES also failed to detect homozygous pathogenic variants in DNA repair genes outside of these regions. The functions of 19 genes with homozygous variants in exons or at canonical splice sites that were predicted to be potentially deleterious were not related to nuclear DNA repair according to current knowledge (Supplemental Table 2). Meanwhile, the abnormalities in the fetus of interest, including growth retardation, radial ray defects, duodenal atresia, ventricular septal defect, and hydrocephalus (Supplemental Figure 2), as well as the cellular hypersensitivity to ICL induction by mitomycin C (MMC), as determined by the disproportionate accumulation of the amniocytes in G2 phase on flow cytometric cell-cycle analysis (Figure 1D and Supplemental Figure 1C), were consistent with characteristic manifestations of the FA spectrum. Because of the severe malformations, the pregnancy was discontinued.
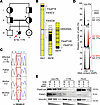
Origin, location, segregation, and implications of the FAAP100 variant. (A) Pedigree of a fetus with FA-suggestive phenotype (III-2, affected individual; solid and slashed circle, marked with a red arrow, ID no. 1176) and family. Squares denote males; circles denote females. Genotyped individuals of the core family are indicated by an asterisk, unassessed genotypes by a question mark, and proven or obligate heterozygotes by half-filled symbols. (B) Five FA (prefix FANC) and FA-associated (prefix FAAP) genes in major autozygous regions (black blocks) on 3 different chromosomes (Chr) in the affected III-2 (1176) are shown. (C) Sanger electropherograms identified a homozygous A>C variant sequence, heterozygous sequence, or WT sequence at FAAP100 position c.1624 (highlighted by a red frame), predicting p.(T542P), in relatives of the core family as indicated. (D) Cell-cycle analysis of cultured amniotic fibroblasts by flow cytometry. Exemplary individual measurements. Histograms of cells from a normal control (non-FA; top), a fetus with FA (positive control; middle), and fetus III-2 (1176; bottom). Black histograms are from untreated cultures and superimposed gray or gray/red histograms are from cultures exposed to MMC (10 ng/mL, 48 hours), shown individually in Supplemental Figure 1C. The percentage of cells in G2 phase is indicated. Red coloring and arrows denote an increased G2 compartment size. Variability and significance of G2 phase arrest in repeated measurements are shown in Supplemental Figure 4A. (E) Titration experiments show reduced levels of FAAP100 protein in _FAAP100T542P_-mutant 1176 cells from affected III-2 cells (approximately 3 steps of 1:3 dilution each lower than in non-FA cells). Reduced levels of FANCL are also suggested in 1176 cells (approximately 2 steps of 1:3 dilution lower than in non-FA cells). Wedges represent dilutions at the ratios indicated above the blots. FA-A, cells from an individual with FA, subtype A. Vinculin was used as a loading control.
Expression and variant effect prediction in silico analyses suggest impaired stability and function of the FAAP100 variant. c_._1624 occurs in all FAAP100 transcript variants, and c.1624A>C is expressed in FAAP100 transcripts of the fibroblast line 1176 with the homozygous genomic variant, derived from the affected fetus (no. 1176) at 21+1 weeks of gestation (Supplemental Figure 1, D and E). The expression level of FAAP100 transcripts in 1176 cells was slightly lower than in a normal control fibroblast line (Supplemental Figure 1E). The presence of the mutation was confirmed in FAAP100 cDNA from 1176 cells (Supplemental Figure 1E). FAAP100 c.1624A>C results in the threonine-to-proline substitution p.(T542P) (NP_079437.5:p.(Thr542Pro)). T542P-mutant FAAP100 (FAAP100T542P) protein is present in 1176 cells at reduced levels compared with WT FAAP100 (FAAP100WT) in control and FA-A cells, which was determined by titration to be approximately 1/27 of normal (Figure 1E, and see also the corresponding blots below). FANCL protein also appears to be present at lower levels in FAAP100T542P-mutant 1176 fibroblasts at approximately one-ninth of normal (Figure 1E), whereas, conversely, FAAP100 protein levels are reduced in FANCL-deficient cells (23). Furthermore, lower levels of FAAP100WT are suggested in an FA-B line with the hemizygous pathogenic variant in FANCB c.832C>T predicting p.Q278* (25) compared with non-FA or FA-D2 cells (compare ratios on the corresponding blots below), suggesting destabilizing effects resulting from defects in the molecular interaction of FAAP100, FANCB, and FANCL. T542 is located in a region identified as belonging to a shared protein family named FANCAA (beyond amino acid position 448) (http://pfam.xfam.org/; https://www.ebi.ac.uk/interpro/; Supplemental Figure 1D and Supplemental Figure 3A), but not in a known functional motif or domain. T542 is highly conserved in vertebrates, but not in other organisms, and is surrounded in vertebrates by a block of also-conserved amino acids (Supplemental Figure 3B). p.T542P is unknown to most databases of normal or disease-associated human genetic diversity, including the gnomAD platform, version 4.1.0 (https://gnomad.broadinstitute.org/), ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), and other large-scale reference human genetic variation datasets (see Methods). However, it has received an entry in the NCBI dbSNP database (https://www.ncbi.nlm.nih.gov/snp/), reflecting its single observation in the Korean Genome Project (Korea1K) (26). In silico analyses using, among others (see Methods), Alamut Visual Plus, version 1.12 (https://www.sophiagenetics.com/sophia-ddm-for-genomics/alamut-visual-plus/), and AlphaMissense (https://alphamissense.hegelab.org/) classify the substitution as a variant of unknown significance or likely pathogenic, respectively. Structural modeling shows that the threonine at position 542 is part of a β-strand within an antiparallel β-sheet in a region of the protein with very high structural confidence (Supplemental Figure 3C). The exchange of threonine to proline redirects the dihedral angles Φ and Ψ away from the range favored for β-strand formation, shortening the β-strand (aa 541–549) by 2 amino acids (Supplemental Figure 3D). In addition, the polar contacts between T542 and F603 of the adjacent antiparallel β-strand are disrupted, destabilizing this β-sheet and potentially compromising β-strand mediated protein-protein interaction via an adjacent β-sheet of FANCB in the BLP100 module.
FAAP100T542P-mutant 1176 cells are sensitive to ICL induction, which is complemented by FAAP100WT. To experimentally test the potential pathogenicity of the FAAP100T542P variant, cultured 1176 fibroblasts were transduced with FAAP100WT, FAAP100T542P, and empty vector pLVX as a sham control. The response of the cells to ICL-inducing chemical agents was assessed by various techniques. Chromosome breakage analysis revealed the accumulation of metaphases with high breakage rates (≥8 breaks per nucleus), including frequent radial figures, after exposure to MMC of nontransduced (parental) 1176 cells, 1176 cells that were transduced with FAAP100T542P, or mock-transduced 1176 cells (Figure 2, A and B). In contrast, 1176 cells ectopically expressing transduced FAAP100WT showed a break number distribution similar to non-FA control fibroblasts and very few metaphases with high break rates. In cell-cycle studies, the FA-typical G2 phase accumulation in parental 1176 cells in response to MMC exposure was restored to normal control levels in _FAAP100WT_-transduced 1176 cells, but not in _FAAP100_T542P transduced or mock-transduced 1176 cells (Figure 2C; for variability, compare Supplemental Figure 4A). Furthermore, survival after exposure to MMC or cisplatin (CDDP) was rescued in _FAAP100WT_-transduced 1176 cells, but not in parental, _FAAP100T542P_-transduced or mock-transduced 1176 cells, or in FA-B fibroblasts (Figure 2, D and E). A consequence of the homozygous c.1624A>C (FAAP100T542P) mutation in 1176 cells was defective FANCD2 monoubiquitylation (Supplemental Figure 5A), whereas the monoallelic c.1624A>C mutation status in cells from the unaffected heterozygous parents was associated with sufficient FANCD2 monoubiquitylation (Supplemental Figure 5B), suggesting that the FAAP100 mutation was nonfunctional in a recessive manner. Transduction of FAAP100WT complemented deficient FANCD2 monoubiquitylation in 1176 cells (Figure 3A), whereas transduction of FAAP100T542P did not (Figure 3B). Transduction of FAAP100WT or FAAP100T542P did not alter the deficient FANCD2 monoubiquitylation status of FA-B cells or the proficient FANCD2 monoubiquitylation of non-FA cells (Supplemental Figure 5, C and D). Furthermore, parental 1176 cells and, similarly, 1176 cells transduced with FAAP100T542P or empty vector were deficient in the formation of FANCD2 subnuclear foci, an accumulation of monoubiquitylated FANCD2 and other DNA repair proteins on chromatin (Supplemental Figure 6A). Transduction of FAAP100WT rescued the defect in 1176 cells (Supplemental Figure 6, A and B), whereas we observed no significant change in FANCD2 foci formation by the transduction of either type of FAAP100 in FA-B fibroblasts (deficient) or non-FA normal control fibroblasts (proficient) (Supplemental Figure 6C). Deficient FANCD2 monoubiquitylation was also the effect of FAAP100 silencing by depletion through transfection of HeLa cells with FAAP100 siRNA (Supplemental Figure 7, A and B).
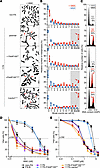
Ectopic expression of FAAP100WT or FAAP100T542P in FAAP100-deficient cells. (A) Metaphase micrographs of FAAP100T542P-mutant 1176 cells after exposure of the cultures to MMC (100 ng/mL, 48 hours). Parental 1176 cells and mock- or mutation-transduced (+vectorpLVX or +FAAP100T542P) 1176 cells show distinctly increased chromosome breakage, mostly of the chromatid type, whereas WT transduced 1176+FAAP100WT cells are rescued. Radials are marked with red arrows. (B) Box plots reflect the proportion of cells with the indicated number of chromosome breaks per metaphase; single value (♦), median (─), mean (□), IQR (─), minimum (×), and maximum (×) for the number of breaks from 3 independent experiments; blue symbols are from untreated cultures, and red are from cultures exposed to MMC (100 ng/mL, 48 hours). Light gray shading indicates high rates of 8 or more breaks per metaphase, and red arrows highlight pivotal rates of 10 or higher. Cell lines are the same as in A. Fifty metaphases were analyzed per experiment. (C) Cell-cycle analysis by flow cytometry. Exemplary individual measurements. Black histograms are from untreated cultures, and superimposed gray or gray/red are from cultures exposed to MMC (10 ng/mL, 48 hours). The percentage of cells in the G2 phase is indicated. Red coloring and arrows denote an increased G2 compartment size. Variability and significance of G2 phase arrest in repeated measurements are shown in Supplemental Figure 4A. Cell lines are the same as in A. (D and E) Dose-response (survival) curves of parental and mock FAAP100WT or FAAP100T542P transduced 1176 cells from cultures exposed to different concentrations of MMC (D) or CDDP (E) for 8 days. Data indicate the mean ± SD of 3 independent experiments. Cell lines are the same as in A and are identical in D and E. FA-B and non-FA are FA and normal control, respectively. LC50, 50% lethal concentration. Note that the transduction of FAAP100WT complements the repair defect in all assays, whereas FAAP100T542P does not.

FANCD2 monoubiquitylation capacity of FAAP100-inactivated human and avian cell lines. (A) FAAP100T542P mutant 1176 cells (parental) lacked FANCD2 monoubiquitylation (◄D2) on immunoblots. FANCD2 monoubiquitylation was restored only in 1176+FAAP100WT transduced cells (◄D2-Ub), but not in 1176+FAAP100T542P or mock transduced cells. FA-L was a monoubiquitylation-deficient FA control. (B) FAAP100T542-mutant 1176 cells showed reduced levels of FAAP100 protein (see ratio to vinculin in parental and +[empty] vectorpLVX lanes) and lacked FANCD2 monoubiquitylation (◄D2) on immunoblots. 1176+FAAP100WT- and 1176+FAAP100T542P-transduced cells both overexpressed FAAP100, but FANCD2 monoubiquitylation was restored only in 1176+FAAP100WT cells (◄D2-Ub). FA-D2 was a control for absent FANCD2, and non-FA was a monoubiquitylation-proficient normal control. (C) FANCD2 monoubiquitylation was functional in HEK293T cells (not shown) and HEK293T cells mock transfected with the Cas9-containing vector PX459 (◄D2-Ub), but not in the FAAP100-inactivated HEK293T CRISPR/Cas9 clone Cr10 (parental) (◄D2), where it was rescued by transduction with FAAP100WT, resulting in overexpression of FAAP100 (◄D2-Ub), as shown by FANCD2 and FAAP100 immunoblots. (D) FANCD2 monoubiquitylation was functional in parental HAP1 cells (◄D2-Ub), but not in FAAP100-inactivated HAP1 FAAP100R149Vfs*2 cells (◄D2), where it was restored by transduction with FAAP100WT, resulting in overexpression of FAAP100 (◄D2-Ub), as shown by FANCD2 and FAAP100 immunoblots. (E) FANCD2 monoubiquitylation was functional in parental DT40 cells (◄D2-Ub), but absent in ΔFAAP100-DT40 cells (◄D2). In the latter, it was rescued by the expression of EGFP-FAAP100WT (◄D2-Ub), but not by EGFP-FAAP100T547P (◄D2), which contained the chicken equivalent T547P of human T542P, as shown by FANCD2 and EGFP immunoblots. Note that the strong overexpression of the FAAP100 protein by lentiviral transduction in B–D, where low- and high-exposure blots are shown, was in marked contrast to the transfection in E. Vinculin or RAD50 or tubulin was used as a loading control on all blots. The FAAP100/vinculin ratios were taken from the high-exposure FAAP100 blot. The FANCD2-Ub/FANCD2 ratios semiquantitatively estimated monoubiquitylation.
FAAP100-targeted established human and avian cell lines are sensitive to ICL induction and are complemented by FAAP100WT. We also studied the effects of FAAP100 deficiency in established cell lines genetically engineered to homozygously disrupt the FAAP100 gene. These lines included the CRISPR/Cas9 FAAP100 edited clones Cr10 and Cr12 derived from HEK293T cells, FAAP100R149Vfs* cells derived from HAP1 cells, and ΔFAAP100-DT40 cells derived from DT40 cells by chicken FAAP100 targeting (23). HEK293T Cr10 and HAP1-FAAP100R149Vfs* cells have a small duplication and deletion, respectively, resulting in frameshifts and truncation of the FAAP100 protein (Supplemental Table 3 and Supplemental Figure 8A). HEK293T Cr12 cells have an internal 27 bp in-frame FAAP100 deletion, and ΔFAAP100-DT40 cells have a large genomic FAAP100 deletion (Supplemental Table 3 and Supplemental Figure 8A). FAAP100 protein was not detected in HEK293T Cr10, HAP1-FAAP100R149Vfs*2 (Figure 3, C and D, and Supplemental Figure 8B), or ΔFAAP100-DT40 cells (23), in contrast to the presence of residual mutant FAAP100 protein in Cr12 cells (Supplemental Figure 8, B and C). HEK293T Cr12 cells also showed a residual mutant FAAP100 protein levels that were determined by titration to be approximately 1/27th of normal levels, similar to 1176 cells (Supplemental Figure 8C); a theoretical, small size difference compared with FAAP100WT could not be reliably detected (Supplemental Table 3 and Supplemental Figure 8B). HEK293T Cr10, HEK293T Cr12, and HAP1-FAAP100R149Vfs*2 cells overexpressed the full-length protein after transduction with FAAP100WT or FAAP100T542P, as did 1176 cells (Figure 3, B–D), whereas expression, but not overexpression, of FAAP100 was observed after transfection of ΔFAAP100-DT40 cells (Figure 3E). The FAAP100_-targeted cell lines exhibited MMC or CDDP sensitivity with FA-typical characteristics very similar to those of 1176 cells and, like 1176 cells, were rescued by the transduction with FAAP100WT but not FAAP100T542P or, in the case of ΔFAAP100-DT40 cells,_ by transfection of chFAAP100WT but not chFAAP100T547P, with the latter containing the chicken equivalent T547P of human T542P (Supplemental Figures 9–11; for G2 phase variability, compare Supplemental Figure 4, B and C). Transduction of FANCD2 monoubiquitylation–deficient HEK293T Cr10 or HAP1-FAAP100R149Vfs*2 cells with FAAP100WT rescued FANCD2 monoubiquitylation (Figure 3, C and D). Similarly, MMC-sensitive ΔFAAP100-DT40 cells were deficient in FANCD2 monoubiquitylation (Figure 3E). After stable transfection, they expressed EGFP- or FLAG-chFAAP100WT protein, or EGFP- or FLAG-chFAAP100T547P protein (Figure 3D). Only EGFP- or FLAG-chFAAP100WT, but not EGFP- or FLAG-chFAAP100T547P, rescued FANCD2 monoubiquitylation and survival (Supplemental Figure 11 and Figure 3E). These results underscore the idea that functional FAAP100 is an absolute requirement for the repair of ICL-like DNA defects and that FAAP100T542P or chFAAP100T547P are nonfunctional in this regard.
faap100 KO in zebrafish results in FA phenotypes. We further investigated the consequences of faap100 deficiency in a previously generated zebrafish _faap100_-KO (ENSDARG00000079457) model using CRISPR/Cas9-mediated gene editing (27). Here,we report the functional characterization of primary cell cultures established from caudal fins of faap100–/– fish homozygous for the faap100 c.1133_1136delACCC, Δ4 (hg72) deletion (Figure 4A). We performed cell growth studies and chromosome breakage, cell-cycle, and survival analyses. faap100–/– cells in culture showed significantly reduced growth compared with WT cultures (Figure 4B). The doubling time of faap100–/– cultures was almost 3 times slower than that of WT controls (5.13 vs. 1.81 days). Chromosome breakage analysis of faap100–/– cells after exposure to MMC revealed the accumulation of metaphases with high breakage rates (≥8 breaks per nucleus), including multiple radial figures (Figure 4, C and D). Cell-cycle analysis showed the FA-typical disproportionately increased G2-phase arrest following exposure to MMC (Figure 4E). In addition, decreased faap100–/– cell survival was observed after exposure to MMC (Figure 4F). These results demonstrate that zebrafish cells required faap100 to prevent MMC-induced chromosomal and cell-cycle aberrations and cell death. Furthermore, homozygous mutants of 17 zebrafish fanc genes had displayed a female-to-male sex reversal phenotype (27), highlighting the role of FA pathway genes in meiotic oocyte survival. Here, sex determination of adult fish revealed that faap100–/– zebrafish also had complete female-to-male sex reversal (Figure 4G). We tested the fertility of male homozygous KOs by outcrossing them with WT fish and found no fertility defects (data not shown), which is consistent with the observations for most (15 of 17) of the zebrafish fanc gene KOs (27). In summary, our zebrafish data show that disruption of faap100 resulted in functional cellular phenotypes consistent with fanc gene KOs.
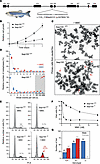
Cellular FA phenotype of _faap100_-KO zebrafish. (A) Schematic of the CRISPR/Cas9-mediated faap100 gene–KO (si:dkey-57h18.1). (B) Growth curves show slower proliferation of primary cell cultures from faap100–/– fish (circles) than from faap100+/+ fish (squares). The mean ± SD for multiples of the initial cell number of 3 independent subcultures is shown for each time point, with day 0 counts set to 1. ***P < 0.001 (t test) at day 4. Data were exponentially fitted. (C) Metaphase micrographs after exposure of faap100+/+ or faap100–/– cell cultures to MMC (2.5 ng/mL, 24 hours). faap100–/– cells showed markedly increased chromosome breakage, mostly of the chromatid type. Red arrows indicate radials and black arrows other types of breakage. Original magnification, ×1,000. (D) Box plots reflect the proportion of faap100+/+ (top) or faap100–/– (bottom) cells with the indicated number of chromosome breaks per metaphase. Single value (♦), median (─), mean (□), IQR (─), minimum (x), and maximum (x) for the number of breaks from 2 independent experiments; blue symbols represent data from untreated cultures, and red symbols represent data from cultures exposed to MMC (2.5 ng/mL, 24 hours). Light gray shading indicates high rates of 8 or more breaks per metaphase, and the red arrow highlights a pivotal rate of 10 or higher. A total of 31–53 metaphases were analyzed per experiment. (E) Flow cytometric cell-cycle analysis of faap100+/+ (top) and faap100–/– (bottom) cell cultures without MMC (– MMC) or after exposure to MMC (+ MMC) (5 ng/mL, 48 hours). Exemplary individual measurements are shown. The percentages of cells in G2 are shown. Red coloring and arrow indicate an increased G2 compartment size. (F) Dose-response (survival) curves of faap100+/+ (top) and faap100–/– (bottom) cells from cultures exposed to different concentrations of MMC. The mean ± SD of triplicates is shown. (G) Homozygous KO of faap100 resulted in complete sex reversal from female to male. The numbers above the bars represent the number of fish in each sex and genotype category.
A customized Faap100-KO mouse recapitulates the phenotypes observed in other FA mouse models. As another animal model of FAAP100 deficiency, we generated and studied an _Faap100_-KO mouse. Cre-induced deletion of exon 3 resulted in the mutation c.285_1240del with the deduced effect p.(Ser96*) (Supplemental Figure 12A). Male goGermline mice were bred with C57BL/6 females to produce heterozygous Faap100+/– germline offspring on a C57BL/6 background (28). Gene dosage studies identified Faap100+/+, Faap100+/–, and Faap100–/– mice (Supplemental Figure 12B). Real-time quantitative PCR (RT-qPCR) analysis of normalized relative Faap100 mRNA expression in Faap100+/+ and Faap100–/– mice revealed virtually no amplification of a transcript region bridging exons 3–4 in Faap100–/– mice (Supplemental Figure 12C). This confirmed the genomic deletion of exon 3 at the mRNA level. Notably, Faap100 mRNA spanning exons 6–8 was present in Faap100–/– mice at approximately half the level of Faap100+/+ mice (Supplemental Figure 12D). This indicates the expression of Faap100 transcripts that, however, lacked the sequence associated with exon 3. This skipping shifted the downstream exons out of the Faap100 reading frame and rendered these transcripts nonfunctional. Faap100–/– mice were viable but were born at a sub-Mendelian rate of 6.1% offspring from heterozygous mating, suggesting embryonic and/or fetal lethality (Figure 5A). Faap100–/– pups had a significantly reduced birth weight and nose-to-tail length (Figure 5, B and C). Faap100–/– mice also showed a small but significant impairment in weight gain and nose-to-tail growth (Figure 5, D and E). Histological examination of Faap100–/– mice revealed testicular and ovarian atrophy (Figure 5F). In Faap100–/– males, the testes showed atrophic seminiferous tubules. Most appeared empty, with little evidence of any active cell division. Spermatozoa were absent from the epididymis. Female Faap100–/– mice had ovarian hypoplasia with small, malformed ovaries and very immature and dysfunctional ovarian tissue, with a predominance of stromal and luteal cells but inactive epithelium. No follicular differentiation or development was observed. The oviducts were relatively well differentiated. These findings suggest that sexual differentiation was largely intact in Faap100–/– mice, but germ cell formation was abolished. Faap100–/– mice were consistently sterile in various test mating constellations. Anophthalmia, hydrocephalus, and limb malformations were observed in 6 of 62 Faap100–/– and Faap100+/– mice (Supplemental Table 4). The rate of malformations was slightly higher than that published for basic anomalies in newborn, inbred C57BL/6 mice (29). In addition, 23 Faap100+/+ mice maintained under the same conditions showed no malformations. Pancytopenia or bone marrow failure, as assessed by peripheral blood counts, was not present in Faap100–/– mice in a 6-month pilot study (Supplemental Table 5). Faap100–/– mouse embryonic fibroblasts (MEFs) were obtained from embryonic tissue cultures and confirmed for the absence or heterozygous or homozygous presence of the Faap100 exon 3 deletion (Supplemental Figure 12E). Faap100–/– MEFs exhibited decreased survival after exposure to CDDP (Figure 5G). Karyotypes showed frequent radial-like chromatid exchanges, increased breakage predominantly of the chromatid type, and an accumulation of metaphases with a high number of breaks (≥8 breaks per nucleus) after exposure to MMC (Figure 5, H and I). Faap100–/– MEFs also showed increased MMC-induced G2-phase arrest (Figure 5J; for G2 phase variability, compare Supplemental Figure 4D). Taken together, the organismic and cellular characteristics of Faap100–/– mice appeared compatible with an FA phenotype.
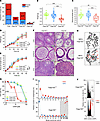
Characteristics of the Faap100–/– mouse. (A) Proportions and numbers of female (red) and male (blue) Faap100+/+, Faap100+/–, and Faap100–/– mouse offspring from heterozygous mating. (B) Faap100–/– mice show significantly lower birth weight. Box-and-whisker plots: single value (■, ●, ▲), median (─), mean (□), IQR (─), whiskers (–), and range (x). n = 24 (Faap100+/+), n = 73 (Faap100+/–), and n = 18 (Faap100–/–). ***P < 0.001, by 1-way, repeated-measures ANOVA with Tukey’s test (B and C). (C) Shortened nose-to-tail length in Faap100–/– neonatal mice. n = 23 (Faap100+/+) , n = 68 (Faap100+/–), and n = 15 (Faap100–/–). **P 0.01 and ***P < 0.001. (D) Reduced postnatal weight gain in Faap100–/– mice. n = 9–30 (Faap100+/+), n = 10–83 (Faap100+/–), and n = 6–11 (Faap100–/–). *P < 0.05 and **P < 0.01, by 2_-_way, repeated-measures ANOVA with post hoc Tukey’s test (D and E). (E) Slower growth in body length of Faap100–/– mice. n = 9 (Faap100+/+), n = 11–42 (Faap100+/–), and n = 9 (Faap100–/–). ***P < 0.001. (F) Gonads in Faap100–/– mice appear dysplastic, unlike normal gonads in Faap100+/+ mice. Scale bars: 50 μm. (G) Dose-response survival curves of MEFs exposed to CDDP (8 days) show reduced survival of Faap100–/– cells. Data are the mean ± SD of triplicates. LC50, 50% lethal concentration. (H) Metaphase micrographs after MMC exposure (100 ng/mL, 48 hours) show increased radials (red arrows) and chromatid breaks in Faap100–/– MEFs. Original micrographs have been magnified approximately ×1,000. (I) Box plots of breaks per metaphase from 2 independent experiments. Red symbols: MMC-treated; blue: untreated. Gray zone = 8 or more breaks; red arrow = 10 or more breaks. n = 50 metaphases/experiment. (J) Cell-cycle analysis reveals G2-phase arrest in Faap100–/– MEFs after MMC exposure (10 ng/mL, 48 hours). Exemplary individual measurements. Black histograms: untreated; gray or gray/red overlaid histograms: MMC-treated. The percentage of G2 cells is indicated. Red coloring and arrow: increased G2 compartment size. Variability and significance of G2-phase arrest in repeated measurements are shown in Supplemental Figure 4D.
The FAAP100T542P-mutant protein disrupts FA core complex functions biochemically and mechanistically. To determine whether FAAP100T542P affects FANCD2 monoubiquitylation in a biochemically defined system, we reconstituted the FANCD2 monoubiquitylation reaction in vitro using purified human FANCD2-FANCI (30), FAAP100WT (Supplemental Figure 13A), FAAP100T542P, FANCB, and FANCL as indicated in Figure 6A, in the presence of purified ubiquitin (UB), ubiquitin-activating enzyme E1 (UBE1), and ubiquitin-conjugating enzyme E2 T (UBE2T) (31). With FANCL alone, only limited monoubiquitylation of FANCD2 occurred (Figure 6B, lane 6). As described previously (31), FAAP100WT dramatically enhanced FANCL-mediated monoubiquitylation of FANCD2 (Figure 6B, compare lanes 2 and 4 with lane 6). In contrast, FAAP100T542P was defective in stimulating the FANCD2 monoubiquitylation reaction in the presence of FANCB (Figure 6B, compare lane 7 with lane 2, and Figure 6C). Similarly, the FAAP100T542P mutant was also significantly less efficient than FAAP100WT in the absence of FANCB (Figure 6B, compare lane 8 with lane 4, and Figure 6C). In conclusion, FAAP100T542P was defective in stimulating FANCL-mediated monoubiquitylation of FANCD2. A titration experiment, performed to exclude potential inhibitory or dominant-negative effects, further confirmed the stimulatory defect of FAAP100T542P (Figure 6, C and D). In support of this observation, we also found that, unlike FAAP100WT, FAAP100T542P was unable to form a complex with FANCB and FANCL in insect cells. As shown in Supplemental Figure 13B, cotransfection of FANCB, FANCL, and FAAP100WT baculoviruses in insect cells resulted in the formation of the BLP100 complex, which could be purified as a trimeric entity (Supplemental Figure 13B, lanes 2, 4, and 6). However, purification of a putative complex after coexpression of FANCB, FANCL, and FAAP100T542P by baculovirus in insect cells yielded only a purified FANCB protein without FANCL and FAAP100 (Supplemental Figure 13B, lanes 3, 5, and 7). To further investigate the interaction of FAAP100T542P with FANCB and FANCL in a different cellular background, we performed mammalian 2- and 3-hybrid (M2/3H) assays in HEK293T cells. M2H studies revealed no interaction between FAAP100T542P and FANCB (Figure 6E). Consequently, M3H experiments showed no interaction between FAAP100T542P and FANCL in the presence of FANCB (Figure 6F). Co-IP studies in 1176 cells using FANCB for pull-down confirmed the absence of FAAP100T542P binding to FANCB (Figure 6G). A similar result was obtained in HEK293T Cr12 cells expressing the FAAP100L543_S551del CRISPR mutant (Figure 6H). The BLP100 subcomplex is normally formed in the cytosol and imported into the nucleus in a FANCA- and FANCM-dependent manner (13, 23). Our in silico analysis confirmed that FAAP100 did not possess a core nuclear localization signal (NLS). Subcellular protein fractionation of 1176 and HEK293T Cr12 cells revealed that, in contrast to FAAP100WT, FAAP100T542P or FAAP100L543_S551del was present only in the cytoplasm and was unable to enter the nucleus and access chromatin (Figure 6I). In summary, the mutagenic effect of FAAP100T542P was manifested at several levels: it failed to support the E3 ubiquitin ligase activity of FANCL toward FANCD2 because it abolished the normal interaction of FAAP100 with FANCB so that no BLP100 complex was formed, and because of the lack of indirect interaction of FAAP100 with FANCA, no nuclear translocation occurred.

Ligase activity, interaction, and subcellular localization studies with FAAP100T542P. (A) Reconstitution of FANCD2 monoubiquitylation using purified proteins, including HA-ubiquitin, UBE1, UBE2T, and FANCD2-FANCI complex and, as indicated, FAAP100WT or FAAP100T542P, FANCB, and/or FANCL. FANCD2 immunoblot: ◄D2-Ub, monoubiquitylated; ◄D2, nonubiquitylated. (B) Quantitation of results from A. FANCD2-Ub divided by total FANCD2 (percentage) indicates the ubiquitylation efficiency. Lane numbers are identical to those in A. Box plots: single value (♦), median (─), mean (□), IQR (─), minimum (x), and maximum (x) from 3 independent experiments. ***P < 0.001, by 1-way, repeated-measures ANOVA with post hoc Tukey’s test. (C) Titration of FAAP100WT (blue) and FAAP100T542P (red). For reaction mixtures, see A, including FAAP100WT or FAAP100T542P, FANCB, and FANCL. (D) Quantitation of results from C (see B and C for details). Data indicate the mean ± SD of 3 independent experiments. (E) In mammalian 2-hybrid assays, FAAP100WT, but not FAAP100T542P, interacted with FANCB. FAAP100 fused to the activation domain (orange) or the DNA-binding domain (red). Neither direction of FAAP100 fusion directly interacted with FANCL. Box plots: single value (♦), median (─), mean (□), IQR (─), minimum (x), and maximum (x) of 3 independent experiments. Induction factor, multiples of negative control. *P < 0.05 and **P < 0.01, by 1-way, repeated-measures ANOVA with post hoc Tukey’s test. (F) In mammalian 3-hybrid assays, FAAP100WT, but not FAAP100T542P, interacts with FANCL in both fusion directions in the presence of stably overexpressed FANCB. Controls, calculations, and statistical tests are the same as in E. *P < 0.05 and **P < 0.01, by 1-way, repeated-measures ANOVA with post hoc Tukey’s test. (G and H) Co-IPs from cells transfected with FANCB vector and exposed to MMC (40 ng/mL,16 hours). FAAP100T542P in 1176 cells and FAAP100L543_S551del in HEK293T clone Cr12 did not interact with FANCB. Transduced FAAP100WT rescued the pull-down of FAAP100 by FANCB. Vinculin was used as a loading control. (I) On subcellular protein fractionation, FAAP100T542P in 1176 fibroblasts and FAAP100L543_S551del in the HEK293T clone Cr12 were not detected in nuclear extracts (NE) or on chromatin (CB). Transduced FAAP100WT rescuedFAAP100 relocalization. CE, cytoplasmic extracts. Tubulin, YY1, and histone H3 were used as loading controls. Cells were exposed to MMC (40 ng/mL, 16 hours).