The mitochondrial origin
of postischemic arrhythmias (original) (raw)
mBzR antagonist stabilizes ΔΨm and the cellular AP. To demonstrate a mechanistic link between the mitochondrial energy state and electrical excitability, we used a previously described method for triggering whole-cell oscillations in ΔΨm by focal 2-photon laser excitation, in which laser-induced depolarization of a few mitochondria leads to a sustained autonomous oscillation in the entire mitochondrial network (8). ΔΨm, reported by tetramethylrhodamine methyl ester (TMRM) fluorescence, was imaged while APs were simultaneously recorded using the patch-clamp technique in current-clamp mode (Figure 1). The mBzR antagonist 4′-chlorodiazepam (4′-Cl-DZP) suppressed whole-cell oscillations in ΔΨm within minutes of application, and the effect was reversible, as ΔΨm oscillations returned within 10 minutes of washout of the compound (Figure 1A). Through its stabilizing effect on ΔΨm, 4′-Cl-DZP also eliminated oscillations in APD (Figure 1B). In the absence of the drug, APD90 decreased to an electrically inexcitable state within approximately 4 seconds (2–3 stimuli at 0.5 Hz) during each cycle of mitochondrial depolarization. Within approximately 2 minutes of drug application (64 μM; Figure 1, B and D), AP oscillations were suppressed and APD90 stabilized.

Blockade of mitochondrial oscillations and stabilization of the cellular AP by 4′-Cl-DZP. Freshly isolated cardiomyocytes were loaded with TMRM (100 nM) at 37°C, and APs were recorded under whole-cell current-clamp conditions on the stage of the microscope as described in Methods. (A) The reversible effect of acutely added 4′-Cl-DZP (32 μM) on mitochondrial ΔΨ m oscillations. (B) Mitochondrial oscillations in ΔΨ m and the sarcolemmal APD were triggered after a highly localized laser flash (3 minutes before the train of oscillating APDs shown in this panel). APs evoked by brief current injections were recorded during the oscillations. Previously, we showed that during a synchronized cell-wide depolarization-repolarization cycle, the AP shortens in synchrony with fast mitochondrial depolarization (4). (C) During the APD oscillations, the cell becomes inexcitable when ΔΨ m is fully depolarized (remaining upward spikes are from the stimulus only). (D) After addition of 64 μM 4′-Cl-DZP, a stable AP is restored and ΔΨ m oscillations are suppressed.
We have used the term mitochondrial criticality to refer to the state of the mitochondrial network just prior to cell-wide depolarization of ΔΨm, when the system becomes very sensitive to even small perturbations in conditions (20). Since an elevation of ROS and loss of ΔΨm also occurs during IR of the intact heart, we next tested whether mitochondrial criticality might contribute to alterations in the electrophysiological substrate leading to postischemic arrhythmias in the intact heart.
Baseline electrophysiological properties. In order to investigate whether mitochondrial ROS-induced activation of IMAC underlies the electrical dysfunction of hearts subjected to IR, we established a protocol for reproducibly inducing VF in Langendorff-perfused guinea pig hearts. To rule out direct effects of the compounds used in the study on sarcolemmal ion channels, we first determined whether 4′-Cl-DZP, CsA, N,N-Dihexyl-2-(4-fluorophenyl)indole-3-acetamide (FGIN-1-27), or glibenclamide (GLIBEN) altered intrinsic electrophysiological properties, including APD and conduction velocity (CV), during baseline perfusion. None of the compounds had a significant impact on baseline APD (186 ± 18, 174 ± 16, 168 ± 24, 188 ± 16 ms, 182 ± 16; control, FGIN-1-27, CsA, 4′Cl-DZP, GLIBEN, respectively) or CV (41 ± 4, 44 ± 6, 40 ± 4, 39 ± 5, 41 ± 5 cm/s) at the optimal concentration for each agent used in this study (4.6 μM FGIN-1-27, 0.2 μM CsA, 64 μM 4′Cl-DZP, and 10 μM GLIBEN). At a relatively high concentration (46 μM), the mBzR agonist FGIN-1-27, which we have previously shown to induce ΔΨm depolarization (8), produced significant APD shortening and membrane inexcitability that occurred within 15 minutes of drug delivery during normal perfusion (results not shown). The highest concentration (100 μM) of 4′Cl-DZP tested had no significant effect on APD or CV during baseline perfusion and was the most effective in blunting AP shortening during ischemia (see below); however, it was not as effective as the lower concentration of 64 μM in restoring the APD upon reperfusion and preventing arrhythmias. Hence the lower concentration was selected as the optimal concentration to be used in subsequent experiments.
Response to ischemia. We then investigated whether any of these pharmacological interventions modulated the electrophysiological response of hearts to no-flow global ischemia. As expected, ischemia resulted in progressive shortening of APD (Figure 2), resulting in complete loss of AP after 18.4 ± 3.3 minutes under control conditions (i.e., without pharmacological intervention prior to the ischemic episode). Also as expected, APD shortening in ischemia was accompanied by progressive reduction of the AP upstroke velocity (dF/dt), which occurred over a similar time frame (measured as a change in the rate of dye fluorescence).

Protection against ischemia-induced APD shortening by 4′-Cl-DZP. (A) Ischemia-induced APD shortening in control hearts and hearts treated with varying concentrations of 4′-Cl-DZP. (B) Representative APs from a control and a 100 μM 4′-Cl-DZP–treated heart recorded at various intervals during the ischemia protocol. At baseline, APD of control and 100 μM 4′-Cl-DZP–treated hearts were comparable.
Interestingly, treatment with the mBzR antagonist had a profound influence on the electrophysiological response to ischemia. 4′-Cl-DZP blunted APD shortening in a dose-dependent fashion during ischemia, consistent with its proposed role in preventing mitochondrial depolarization and the subsequent activation of sarcolemmal KATP currents (Figure 2A). Remarkably, at the highest concentration of 4′-Cl-DZP tested (100 μM), APs persisted even after 30 minutes of no-flow ischemia, a time when all of the untreated ischemic hearts became electrically inexcitable (Figure 2B).
Conversely, pretreatment of hearts with the mBzR agonist FGIN-1-27 accelerated APD shortening during ischemia and significantly reduced the time for the onset of inexcitability (12.7 ± 2.6 minutes) compared with control hearts (∼18 minutes, Figure 3A). In addition, FGIN-1-27 treatment was associated with disproportionate slowing of CV (see Supplemental Video 1, at 5 minutes; supplemental material available online with this article; doi:10.1172/JCI25371DS1) compared with untreated hearts (Supplemental Video 2, at 5 minutes). Such profound CV slowing in FGIN-1-27–treated hearts was shortly followed by the emergence of an area of functional conduction block within as little as 10 minutes of ischemia (Supplemental Video 1, at 10 minutes) while in control hearts, conduction was slowed but persisted beyond 10 minutes of ischemia (Supplemental Video 2, at 10 minutes).

Ischemia-induced APD shortening and representative APs recorded from hearts pretreated with 4.6 μM FGIN-1-27 (top panel), 0.2 μM CsA (middle panel), and 10 μM GLIBEN (bottom panel) compared with control untreated hearts.
Importantly, inhibition of the mitochondrial PTP with CsA did not have an impact on the response of hearts to ischemia, as APD shortening and onset of inexcitability were not significantly altered compared with control hearts (Figure 3B). In accord with the hypothesis that mitochondrial uncoupling was linked to sarcolemmal KATP channel activation, the KATP channel inhibitor GLIBEN caused a marked reduction in the rate of APD shortening with ischemia up to a point when the heart abruptly became inexcitable at approximately 18 minutes (Figure 3C). Interestingly, the time to inexcitability was similar to that of control hearts despite robust preservation of APD by GLIBEN at earlier time points (Figure 3C).
Ischemic elevation of extracellular K+ concentration, which may be mediated in part by KATP channel opening, might also be expected to partially depolarize the cellular resting membrane potential. This effect can be indirectly assessed by examining the AP amplitude (APA) and dF/dt, since these parameters are predominantly determined by the extent of inactivation of Na+ currents due to depolarization of the resting membrane potential. Therefore, in addition to measuring the marked changes in APD and the time to onset of inexcitability, we quantified ischemia-induced changes in the normalized APA (relative to the preischemia baseline level in each heart) and dF/dt in control hearts and hearts treated with FGIN-1-27, 4′Cl-DZP, GLIBEN, and a combination of FGIN-1-27 and GLIBEN. As expected, ischemia caused progressive reduction of normalized APA (Figure 4A) and dF/dt. Ischemia-induced reduction of APA and dF/dt was highly sensitive to treatment with FGIN-1-27, 4′Cl-DZP, and GLIBEN (Figure 4, B and C). While FGIN-1-27 accentuated the ischemia-induced reduction of APA and dF/dt, treatment with 4′-Cl-DZP and GLIBEN protected against such decrease in both parameters relative to control hearts (Figure 4, B and C). Finally, treatment with GLIBEN was also effective in abolishing the FGIN-1-27–induced reduction of APA and dF/dt when the heart was treated with both compounds.

Effects on ischemia-induced AP amplitude and upstroke velocity. (A) Progressive reduction in the APA during the first 10 minutes of ischemia in control, FGIN-1-27–, and 4′-Cl-DZP–treated hearts. FGIN-1-27–treated hearts exhibited a more enhanced reduction of APA compared with control and 4′Cl-DZP–treated hearts. (B) Comparison of normalized APA and dF/dt after 10 minutes of ischemia compared with preischemic baseline perfusion in untreated control hearts and hearts treated with FGIN-1-27, 4′-Cl-DZP, GLIBEN, or a combination of FGIN-1-27 and GLIBEN. *P < 0.05 vs. control; †P < 0.05 vs. FGIN-1-27. (C) Representative raw traces of dF/dt in control hearts and hearts treated with FGIN-1-27, 4′-Cl-DZP, GLIBEN, or a combination of FGIN-1-27 and GLIBEN.
There was a more pronounced reduction in membrane excitability during ischemia in hearts treated with FGIN-1-27. Figure 5 shows a sequence of isopotential contour maps recorded every 1.2 ms that demonstrates the sequential spread of the AP wavefront (depolarized myocardium shown in red and resting myocardium shown in blue) across the epicardial surface of a representative heart treated with FGIN-1-27 after 11 minutes of ischemia. Also shown (Figure 5B) are representative AP traces recorded at 11 minutes of ischemia (left) and following 10 minutes of reperfusion (right). Clearly, 11 minutes of ischemia resulted in conduction block (upper left corner of isopotential contour maps), as the depolarization wavefront failed to propagate into this electrically silent area. Importantly, these areas of electrical silence were also present at the same location in the heart during reperfusion and likely participated in the formation of sustained arrhythmias (Figure 5B, right). A similar pattern of conduction block upon reperfusion is shown dynamically in another heart treated with FGIN-1-27 (Supplemental Video 3).

Metabolic sink/block as a mechanism of conduction failure and arrhythmias. (A) Sequential isopotential contour maps recorded every 1.2 ms that display the level of membrane potential (color coded) across 464 epicardial sites simultaneously. Red indicates depolarized membrane potential, and blue indicates resting membrane potential. These maps demonstrate the sequential spread of activation across the epicardium of a representative guinea pig heart treated with FGIN-1-27 at 11 minutes of ischemia. Under these conditions, the wavefront fails to propagate across the entire mapping field (i.e., metabolic sink/block). (B) Representative AP traces (A to B, location marked on first contour map) recorded at 11 minutes of ischemia (left) and 10 minutes of reperfusion (right) indicating presence of conduction block (left) and arrhythmias with electrical silence at the same sites of conduction block (right).
Response to reperfusion. After characterizing the ischemia-induced electrophysiological changes, we next investigated the response of hearts to reperfusion following the 30-minute ischemic episode with and without pretreatment with 4′-Cl-DZP, FGIN-1-27, CsA, GLIBEN, or a combination of FGIN-1-27 and GLIBEN. Reperfusion of untreated control hearts was associated with sustained VF in 89% of hearts (Figure 6).
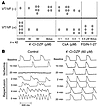
Effects of drugs on post-ischemic arrhythmias. (A) Incidence of reperfusion-related arrhythmias in all groups. (B) Representative AP traces recorded in a control heart (left) and a heart pretreated with 4′-Cl-DZP (right) at various time points during IR, demonstrating the protection of 4′-Cl-DZP against reperfusion arrhythmias.
In addition to preventing ischemia-induced APD shortening, treatment of hearts with 4′-Cl-DZP promoted the rapid recovery of AP morphology and APD upon reperfusion (Figures 6B and 7A) and markedly decreased the incidence of postischemic arrhythmias (10 of 12 hearts exhibited no sustained arrhythmias; Figure 6A). Prevention of reperfusion-related VF was evident both when 4′-Cl-DZP (32-64 μM) was delivered continuously prior to ischemia (6 of 8) and when it was given in a high dose (320 μM) bolus (4 of 4) 5 minutes prior to the onset of reperfusion (Figure 6A).

Post-ischemic AP recovery and arrhythmias. (A) Representative APs during recovery upon reperfusion in control, 4′-Cl-DZP–, CsA-, and FGIN-1-27–treated hearts. (B) Recovery of APD after 5 minutes of reperfusion as a percentage of baseline APD in hearts treated with 64 μM 4′-Cl-DZP and various concentrations of CsA, indicating that the optimal concentration was 0.2 μM. (C) Plot of the recovery of APD upon reperfusion normalized to the baseline APD before ischemia in 4′-Cl-DZP– and CsA-treated hearts.
In contrast, FGIN-1-27 treatment resulted in a prolonged period of electrical silence upon reperfusion, followed by ventricular tachycardia (VT) in all (6 of 6) treated hearts (Figures 6A and 7A). Compared with control hearts, VT presented with a significantly longer cycle length in FGIN-1-27–treated hearts compared with polymorphic VT/VF in normal hearts (not shown).
Treatment with 0.2 μM CsA was also associated with a markedly (P < 0.01) longer period of electrical inexcitability upon reperfusion (8 minutes for CsA compared with less than 2 minutes for 4′-Cl-DZP), followed by a slower, partial recovery of the AP as compared with that in 4′-Cl-DZP–pretreated hearts (Figure 7, A and C). This concentration of CsA was chosen based on an earlier report that demonstrated that it was optimal for inhibiting PTP in the reperfused heart (14). This was confirmed by further experiments using higher (0.4, 1 μM) or lower (0.1 μM) CsA concentrations, which resulted in diminished AP recovery (Figure 7B) and a higher incidence of VT/VF (Figure 6A).