FastGroup: A program to dereplicate libraries of 16S rDNA sequences (original) (raw)
Description of program and algorithms
Overview of FastGroup program
Figure 1 shows the FastGroup graphical user interface (GUI). The GUI reflects the order in which operations are carried out by the FastGroup program. First, sequences are loaded into the program from a directory of files (e.g., seq or txt files) or from a FASTA-formatted document. The program trims the sequences according to user-defined parameters and the trimmed sequences are matched against each other and grouped. In the Grouping step, the user can either define a percent sequence identity (PSI) that will be used to group the sequences together or a consecutive number of mismatches (MM) that will prevent sequences from grouping together (both algorithms are described below).
Figure 1
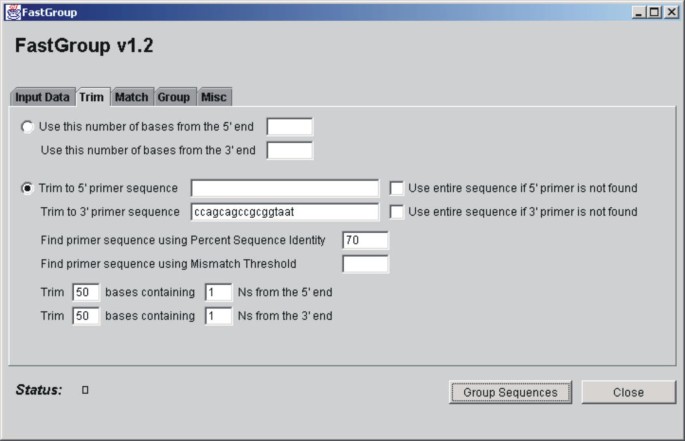
Graphical User Interface (GUI) for FastGroup.
Trimming sequences
Sections of the input sequences containing mismatched and/or ambiguous bases must be removed or they will prevent proper grouping. To make trimming as flexible as possible, FastGroup can trim sequences in three ways: 1) a user-specified number of bases from the 5' or 3' ends can be used (the rest of the sequence is discarded), 2) sequence 5' or 3' of a defined site can be removed, or 3) sequence with ambiguous bases (i.e., "Ns") can be removed from the ends. For the latter two methods, trimming criteria can be entered separately for the 5' and 3' ends. If a primer sequence is specified, the user may adjust the stringency of the match by varying the PSI or MM parameter (explained in detail below).
Matching
Both algorithms initiate grouping by first finding a window (i.e., a short sequence) that is shared between the two sequences being compared. Both the window size and direction of matching (e.g., 5' vs. 3') are specified by the user.
Overview of grouping step
When FastGroup is initiated, the first sequence in the library is trimmed and placed in a new group, g1. The second sequence in the library is then trimmed and compared against the sequence in g1. If the two sequences are determined to be similar, as defined by the user-derived matching and grouping criteria, both sequences are placed in group g1. If the sequences are not similar, the first sequence is placed into g1 and the second sequence is placed into a new group, g2. The next sequence in the input library is then retrieved, trimmed, and compared against the sequences in the groups. This process is repeated with every sequence in the library until all sequences belong to a group. New groups are created as necessary. Sequences in groups are Targets. A sequence being compared to the Targets is a Query sequence. It is important to note that the first sequence used to create a group is the sequence used for comparison against all subsequent sequences. The name for each group begins with "g#-", where the # is assigned sequentially as groups are found by the program. After the hyphen, the name of the first sequence put into the group is given.
Percent Sequence Identity (PSI) algorithm
The PSI algorithm starts at the first position after the matching window and compares each base in the Query sequence to that of the Target sequence. This is done in sequential order and at each position the algorithm records if the bases match. This process is repeated through the length of the smaller sequence. The PSI is calculated by dividing the number of bases found to be the same in both sequences by the number of bases in the smaller sequence. If two sequences have a percent sequence identity that is greater than or equal to the value entered by the user into the Percent Sequence Identity window, then the Query sequence is added to a Target sequence group.
Mismatching (MM) algorithm
The MM algorithm starts at the first position after the matching window and compares the bases in the Query sequence to the Target. If these two bases are the same, the program moves on to the next pair. If the bases are not equal, a one base pair gap is inserted into the Query sequence, effectively sliding the Query sequence relative to the Target sequence. The base in the Query sequence is then compared to the newly aligned Target base. If the bases match, the algorithm leaves the gap and moves to the next base for comparison. If the bases do not match, the gap in the Query sequence is removed and a gap is placed in the Target sequence. The newly aligned bases are then checked. If they are the same, the program moves to the next base in both sequences. However, if the gap in the Target sequence does not cause the bases to pair this is considered one mismatch. If the user-defined MM is <=1, the sequences will not be grouped. If a 2 base MM is assigned, the algorithm will also try using this size of the gap in both the Target and Query sequence, after initially using a 1 base gap. This algorithm is essentially the same as bounded diagonal band alignment [3].
Output files
Once all sequences in a data set have been analyzed by FastGroup, five output text files are produced. The fasta_groups.txt output file contains the group name and a representative sequence from each group in FASTA format. The fasta_groups.txt file is particularly useful for subsequent Clustal X (Clustal X Help http://www-igbmc.u-strasbg.fr/BioInfo/ClustalX/Top.html; [4, 5]) and BLAST analyses (BLAST http://www.ncbi.nlm.nih.gov/BLAST/; [6]). The second output file, group_seqs.txt, contains the group name and all sequences from the group. This file is most useful for visual confirmation of groupings. The third output file, group_files.txt, contains the group name, name of each sequence in the group, and the percent that each group makes of the total. The fourth output file, coverage.txt, shows how many sequences are in each group and calculates coverage by the method of Good [7]. Finally, the infile.txt file contains all the user specified parameters for a record of the analysis.
Testing of FastGroup
The library used to test FastGroup consisted of 94 bacterial 16S rDNA obtained from an environmental sample. The library was made by PCR amplifying with the bacterial-specific primers Bact27F and Bact1492R, cloning into a plasmid vector, and then sequencing the inserts using the Bact27F primer (Figure 2). All sequences were single-pass and unedited.
Figure 2

Schematic of bacterial 16S rDNA showing conserved and hypervariable regions. Detailed information about the primers and their superposition on the bacterial 16S rDNA can be found at http://rrna.uia.ac.be/primers/data/BS/sec_model_fw.html. Bact27F (5' AGA GTT TGA TCM TGG CTC AG 3') corresponds to positions 9–27 of the E. coli 16S rDNA and is similar to BSF8/20. Bact517 (5' ATT ACC GCG GCT GCT GG 3') corresponds to positions 517–534 of the E. coli 16S rDNA and is similar to BSF517/17. Bact1492R (5' TAC GGY TAC CTT GTT ACG ACT T 3') corresponds to positions 1492–1514 of the E. coli 16S rDNA. The approximate sites for hypervariable regions (V1-V3) are shown as shaded boxes.
A number of factors were considered when designing an approach for dereplicating 16S rDNA libraries. First, miscalled bases would prevent related sequences from grouping together. To remove these bases, it was assumed that: 1) miscalled and ambiguous bases occur together (i.e., the presence of N's could be used to differentiate "bad" sequence), and 2) as you move 3' of the sequencing primer miscalled and ambiguous bases become more prevalent, especially beyond ~600 bp. Therefore, trimming criteria that remove 3' sequence are necessary. A second factor influencing our trimming strategy arises from the fact that FastGroup must find a window in common between two sequences before it starts the grouping algorithm. Therefore, a conserved region at the matching end would be expected to increase analysis speed. For bacterial 16S rDNAs (Figure 2) sequenced using Bact27F, the Bact517 conserved site is ideal because: 1) the Bact517 site is highly conserved and should be easy to find in most bacterial 16S rDNA sequences 2) the site is ~500 bp away from the sequencing primer, therefore sequence 5' of this site should be good quality, and 3) the Bact517 site is just 3' of the V3 region. This final point is important because the V3 region is highly variable and usually contains information sufficient to differentiate between different bacterial species. Therefore, including the V3 region increases the resolving power of this approach for measuring bacterial diversity.
Analyses of trimming and matching parameters
Our analysis strategy necessitated that the Bact517 site be accurately identified in the 16S rDNA sequences. As shown in Table 1, if the Bact517 site must be perfectly matched to be identified (PSI for primer matching = 100%), 75 out of the 94 sequences (80%) were trimmed correctly. If the PSI parameter for matching to the Bact517 site was lowered to 70%, 82 out of the 94 sequences (87%) were correctly trimmed. However, lowering the detection stringency for the Bact517 site also increased the possibility that false positive sites would be detected, resulting in prematurely trimmed sequences. False sites did not appear to be a problem with PSI>=70%, but lowering the PSI for finding the Bact517 site to 60% did result in 9 false positives (Table 1). Therefore, for our data set, using a 70% PSI for finding the Bact517 site appeared optimal. We specifically chose a library of low quality sequences for the FastGroup analyses. Therefore, the bact517 position was not found in many of the test sequences because of sequencing errors.
Table 1 Effects of varying PSI on ability of FastGroup to correctly identify Bact517 site. To determine the number of times that the site was found by FastGroup, the sequences were displayed from the 3' direction and visually analyzed in the group_seqs.txt output file. The number of false positives were determined by looking for significantly truncated sequences (e.g., <400 bp) and then visually confirming that a false site was identified
As predicted, using the 3' conserved region for trimming and matching from the 3' end resulted in quicker FastGroup analysis (Table 2), presumably because the conserved region increases the chance that a window will be quickly found. Aligning from the 3' end also increased grouping frequency (Table 2), possibly because the conserved region increased the accuracy of the matching step. Because both algorithms require accurate matching for initiation, the added accuracy offered by the conserved regions as the matching sites increased the efficiency of grouping. Even when the trimming criteria did utilize the Bact517 site, the presence of this site in the sequence increased grouping efficiency. For an example of this phenomenon, compare the analyses where the sequence was trimmed by taking the first 500 bases and then was matched from the 5' versus the 3' ends (Table 2). The presence of the conserved sequence increased the grouping efficiency. Trimming to the Bact517 site also allowed smaller windows to be used, which dramatically increased grouping speed (Table 2). Trimming sections of sequence with ambiguous bases did not improve the sequence quality enough for accurate grouping (Table 2).
Table 2 Effects of matching direction and window size on grouping results and time to analyze data using the PSI algorithm.
Comparison of the PSI and MM algorithms
As shown in Table 3, the MM algorithm was much faster than PSI. The sequence composition of Groups obtained using a PSI value of 97% were roughly equivalent with those obtained using a MM = 2. The MM = 2 did result in some of the bigger groups being broken into two or more smaller groups. We believe that the PSI algorithm was more appropriate for analyses of 16S rDNA for a number of reasons. First, gaps in unedited sequences were not as big of a problem as we initially believed. We have analyzed one bacterial 16S rDNA library in which 96% of the sequences were grouped together using the PSI algorithm. This result would not have been obtained if gaps were a major problem. The second reason we prefer the PSI algorithm for analyses of 16S rDNA is that there are reasons to believe that bacteria with 16S rDNA >=97% identity belong to closely related bacteria [8].
Table 3 Comparison of PSI and MM Algorithms.
Analyzing partial sequences to increase speed of FastGroup analyses
With a large data set, it may be desirable to speed up the FastGroup analysis, possibly by using only part of the input sequence during grouping. This approach would only work if most of the information positions are not lost by the truncation. That is, if a sequence is 500 bp after trimming and only 80% of the sequence (i.e., 400 bp) is used in the Grouping step, how representative are the results? It was expected that, since the hypervariable region V3 is immediately 5' of the Bact517 site, grouping should be much faster and representative if matching was initiated from the 3' end. As shown in Table 4, using partial sequence does dramatically speed up FastGroup, but with a significant loss of resolution. The loss of resolution occurred even though the V3 region was included in the portion of the sequence analyzed. For this reason, we suggest using the longest sequence possible.
Table 4 Effects of using only partial sequences during the Grouping step.
Comparison of FastGroup with ClustalX output
ClustalX [4] uses the ClustalW algorithm [5] to align sequences using a combination of progressive alignment and dynamic programming, making this algorithm sensitive to divergence between closely related sequences (<35% identity). The ClustalW algorithm was used to align the 94 test sequences using default parameters. A tree was generated from aligned sequences using ClustalX's Draw Neighbor Joining (NJ) Tree program. The resulting tree data were plotted (Figures 3) using NJPLOT, which was included as part of ClustalX software distribution. The average running time to produce an alignment from 94 sequences was one hour and 20 minutes plus an average of 5 minutes to generate tree data using Draw NJ tree.
Figure 3
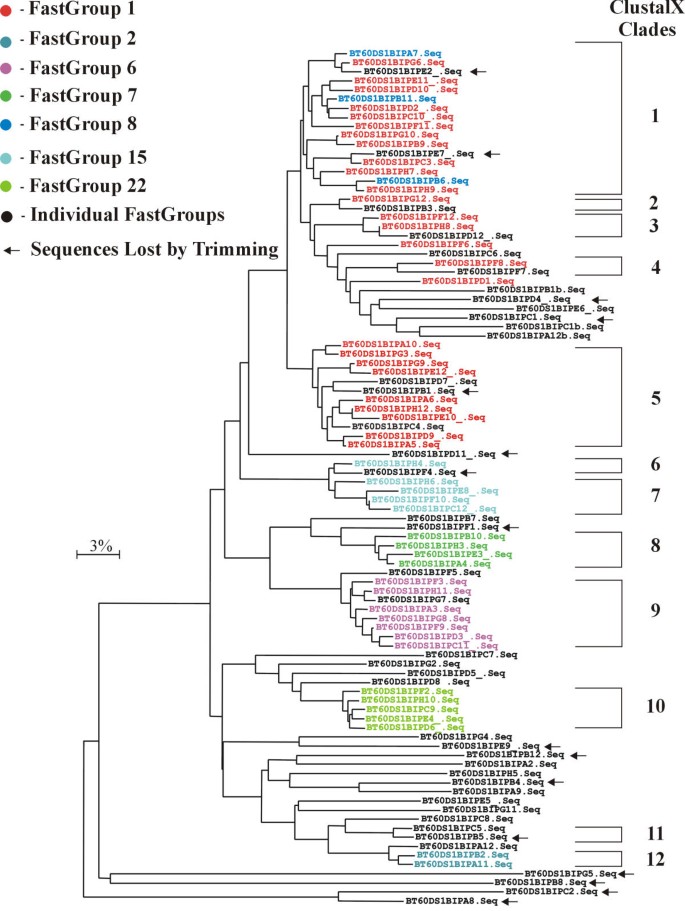
Comparison of ClustalX and FastGroup analyses. An alignment of the 16S rDNA library was performed using ClustalX and a NJ tree was constructed. The "ClustalX Clades" were made by grouping end nodes separated by approximately 3% divergence (i.e., the combined branch lengths). Sequences grouped together by FastGroup, using default trimming criteria and 97% PSI, were identified on this tree and color-coded.
FastGroup was used to group the same test sequences using the PSI algorithm. Sequences were trimmed at the 5' end for every N occurring within 50 bases, and at the 3' end to 70% of the Bact517 site. Trimmed sequences were grouped at 97% PSI. All other FastGroup parameters were left at default values. Run time was ~25 seconds.
The NJ tree from the ClustalX analysis is shown in Figure 3. The groups from FastGroup are color coded on the Tree (Figure 3). In general, the ClustalX Clades and FastGroups are identical. The main exception were the FastGroups 1 and 8, which corresponds to ClustalX Clades 1–5. If the PSI is raised to 99% (i.e., 1 bp change per 100 bp) in FastGroup, then the two major ClustalX Groups become apparent (e.g., Group 1 includes Clades 1–4 and Group 2 is equivalent to Clade 5). The FastGroup 8 contain sequences that differ from FastGroup 1 by a one bp gap, which explains the reason that ClustalX placed these sequences in Clade 1. Because this gap occurred in all four FastGroup 8 sequences, and not in any of the FastGroup 1 sequences, these two groups probably represent different 16S rDNAs and possibly two bacterial species.
What other options exist for dereplicating large libraries of 16S rDNA besides FastGroup? One possibility is to align the sequences with Clustal X and then use the alignments to determine which sequences are the same. This approach is time consuming because it requires that: 1) the sequences be trimmed individually before the alignment, and 2) duplicate sequences be manually removed from the original library after the alignment. Advantages of the Clustal X approach is that visual confirmation of grouping is easy. However, results can also be visualized in FastGroup by having the program display the sequences from the 3' end and looking at the group_seqs.txt output file. FastGroup can also speed up alignment analysis by rapidly trimming, dereplicating, and outputting sequences to the fasta_groups.txt file, which is ideal for Clustal X alignments. A second possible approach to library dereplication is to compare sequences against each other using BLAST2 http://www.ncbi.nlm.nih.gov/blast/bl2seq/bl2.html and then delete duplicates. This approach works well but is too time consuming for libraries over a couple of hundred sequences. A third way that large libraries are often dereplicated requires submitting the sequences as batch files to a database (either local or remote), then searching the same sequences against the updated database using BLAST or Sequence Similarity http://www.cme.msu.edu/RDP/docs/sim_matrix_issues.html. Again, this method works well for a small number of sequences but is very time intensive with large data sets.
Due to technological advances, it is now possible to cheaply sequence thousands of 16S rDNA per day. This change in sequencing power necessitates a reassessment of how microbial diversity and biogeography is studied. Many of the techniques commonly used for these sorts of studies were designed to minimize efforts and cost in the pre-genomics era [9, 10]. However, these techniques suffer from a number of limitations. In the case of denaturing gradient gel electrophoresis (DGGE) it is essentially impossible to compare samples from one gel to another. Because the DGGE banding patterns can not be standardized, DGGE data does not represent a permanent record of microbial diversity or biogeography. In fact, to get a permanent record of what microbe each band on the DGGE represents it is necessary to clone and sequence the band. This is costly both in time and reagents. Terminal-restriction fragment length polymorphism (T-RFLP) banding patterns can be standardized. Therefore, T-RFLP data represents a permanent record of microbial diversity. T-RFLP resolution is, however, limited (e.g., it is dependent on the different restriction sites being present) and it is hard to link the T-RFLP pattern to a specific microbial species. To make this connection, it is necessary to analyze clones both by T-RFLP and by sequencing. In contrast, 16S rDNA sequences allow bacteria to be placed in taxonomical groups. Ribosomal 16S DNA sequences also allow the occurrence of a specific phylotype to be documented in an unequivocal manner. This, in turn, will allow databases of microbial biogeography to be constructed.
Sequencing large 16S rDNA libraries as we have outlined here offers the advantages of sequence data, while minimizing cost (i.e., 1 sequencing reaction per clone). The disadvantages of this approach include: 1) an underestimation of diversity because only part of the 16S rDNA locus is used, 2) the smaller sequences (~500 bp) are not ideal for taxonomical identification, and 3) dirty data (i.e., sequences with mistakes). A conscious effort should be made to save these libraries. That way, if the "cleaner" data or larger sequences are needed in the future, the libraries can be resequenced. Another concern with this approach is that it will cost more money than alternative methods. High-throughput sequencing is becoming very cheap. For example, the Joint Genome Institute estimates that each sequencing reaction costs $1.00–1.50 (Paul Predki, personal communication). When compared to the cost of people-power, extra reagents, and impermanence of the data of the other approaches, sequencing of 16S rDNA libraries is probably already a bargain, and it is only getting cheaper.