The Comparative RNA Web (CRW) Site: an online database of comparative sequence and structure information for ribosomal, intron, and other RNAs (original) (raw)
- Research article
- Published: 17 January 2002
- Sankar Subramanian1,2,
- Murray N Schnare3,
- James R Collett1,
- Lisa M D'Souza1,
- Yushi Du1,
- Brian Feng1,
- Nan Lin1,
- Lakshmi V Madabusi1,4,
- Kirsten M Müller1,5,
- Nupur Pande1,
- Zhidi Shang1,
- Nan Yu1 &
- …
- Robin R Gutell1
BMC Bioinformatics volume 3, Article number: 2 (2002)Cite this article
- 53k Accesses
- 917 Citations
- 16 Altmetric
- Metrics details
Abstract
Background
Comparative analysis of RNA sequences is the basis for the detailed and accurate predictions of RNA structure and the determination of phylogenetic relationships for organisms that span the entire phylogenetic tree. Underlying these accomplishments are very large, well-organized, and processed collections of RNA sequences. This data, starting with the sequences organized into a database management system and aligned to reveal their higher-order structure, and patterns of conservation and variation for organisms that span the phylogenetic tree, has been collected and analyzed. This type of information can be fundamental for and have an influence on the study of phylogenetic relationships, RNA structure, and the melding of these two fields.
Results
We have prepared a large web site that disseminates our comparative sequence and structure models and data. The four major types of comparative information and systems available for the three ribosomal RNAs (5S, 16S, and 23S rRNA), transfer RNA (tRNA), and two of the catalytic intron RNAs (group I and group II) are: (1) Current Comparative Structure Models; (2) Nucleotide Frequency and Conservation Information; (3) Sequence and Structure Data; and (4) Data Access Systems.
Conclusions
This online RNA sequence and structure information, the result of extensive analysis, interpretation, data collection, and computer program and web development, is accessible at our Comparative RNA Web (CRW) Site http://www.rna.icmb.utexas.edu. In the future, more data and information will be added to these existing categories, new categories will be developed, and additional RNAs will be studied and presented at the CRW Site.
Background
In the 1830's, Charles Darwin's investigation of the Galapagos finches led to an appreciation of the structural characteristics that varied and were conserved among the birds in this landmark comparative study. His analysis of the finches' structural features was the foundation for his theory on the origin and evolution of biological species [1]. Today, 150 years later, our understanding of cells from a molecular perspective, in parallel with the technological advances in nucleic acid sequencing and computer hardware and software, affords us the opportunity to determine and study the sequences for many genes from a comparative perspective, followed by the computational analysis, cataloging, and presentation of the resulting data on the World Wide Web.
In the 1970's, Woese and Fox revisited Darwinian evolution from a molecular sequence and structure perspective. Their two primary objectives were to determine phylogenetic relationships for all organisms, including those that can only be observed with a microscope, using a single molecular chronometer, the ribosomal RNA (rRNA), and to predict the correct structure for an RNA molecule, given that the number of possible structure models can be larger than the number of elemental particles in the universe. For the first objective, they rationalized that the origin of species and the related issue of the phylogenetic relationships for all organisms are encoded in the organism's rRNA, a molecule that encompasses two-thirds of the mass of the bacterial ribosome (ribosomal proteins comprise the other one-third). One of their first and most significant findings was the discovery of the third kingdom of life, the Archaebacteria (later renamed Archaea) [2–4]. Subsequently, the analysis of ribosomal RNA produced the first phylogenetic tree, based on the analysis of a single molecule, that included prokaryotes, protozoa, fungi, plants, and animals [4]. These accomplishments were the foundation for the subsequent revolution in rRNA-based phylogenetic analysis, which has resulted in the sequencing of more than 10,000 16S and 16S-like rRNA and 1,000 23S and 23S-like rRNA genes, from laboratories trying to resolve the phylogenetic relationships for organisms that occupy different sections of the big phylogenetic tree.
The prediction of tRNA structure with a comparative perspective in the 1960's [5–9] and subsequent validation with tRNA crystal structures [10, 11] established the foundation for Woese and Fox in the 1970's to begin predicting 5S rRNA structure from the analysis of multiple sequences. They realized that all sequences within the same functional RNA class (in this case, 5S rRNA) will form the same secondary and tertiary structure. Thus, for all of the possible RNA secondary and tertiary structures for any one RNA sequence, such as for Escherichia coli 5S rRNA, the correct structure for this sequence will be similar to the correct secondary structure for every other 5S rRNA sequence [12, 13].
While the first complete 16S rRNA sequence was determined for E. coli in 1978 [14], the first covariation-based structure models were not predicted until more 16S rRNA sequences were determined [15–17]. The first 23S rRNA sequence was determined for E. coli in 1980 [18]; the first covariation-based structure models were predicted the following year, once a few more complete 23S rRNA sequences were determined [19–21]. Both of these comparative structure models were improved as the number of sequences with different patterns of variation increased and the covariation algorithms were able to resolve different types and extents of covariation (see below). Initially, the alignments of 16S and 23S rRNA sequences were analyzed for the occurrence of G:C, A:U, or G:U base pairs that occur within potential helices in the 16S [15, 22] and 23S [19] rRNAs. The 16S and 23S rRNA covariation-based structure models have undergone numerous revisions [23–28]. Today, with a significantly larger number of sequences and more advanced covariation algorithms, we search for all positional covariations, regardless of the types of pairings and the proximity of those pairings with other paired and unpaired nucleotides. The net result is a highly refined secondary and tertiary covariation-based structure model for 16S and 23S rRNA. While the majority of these structure models contain standard G:C, A:U, and G:U base-pairings arranged into regular secondary structure helices, there were many novel base-pairing exchanges (e.g., U:U <-> C:C; A:A <-> G:G; G:U <-> A:C; etc.) and base pairs that form tertiary or tertiary-like structural elements. Thus, the comparative analysis of the rRNA sequences and structures has resulted in the prediction of structure and the identification of structural motifs [29].
Beyond the comparative structure analysis of the three ribosomal RNAs and transfer RNA, several other RNAs have been studied with this perspective. These include the group I [30–33] and II [34, 35] introns, RNase P [36–38], telomerase RNA [39, 40], tmRNA [41], U RNA [42], and the SRP RNA [43]. The comparative sequence analysis paradigm has been successful in determining structure over this wide range of RNA molecules.
Very recently, the authenticities of the ribosomal RNA comparative structure models have been determined [Gutell et al., manuscript in preparation]: 97–98% of the secondary and tertiary structure base pairs predicted with covariation analysis are present in the crystal structures for the 30S [44] and 50S [45] ribosomal subunits. Thus, the underlying premise for comparative analysis and our implementation of this method, including the algorithms, the sequence alignments, and the large collection of comparative structure models with different structural variations for each of the different RNA molecules (e.g., 16S and 23S rRNAs) have been validated.
The highly refined and accurate analysis of phylogenetic relationships and RNA structure with comparative analysis can require very large, phylogenetically and structurally diverse data sets that contain raw and analyzed data that is organized for further analysis and interpretation. With these requirements for our own analysis, and the utility of this comparative information for the greater scientific community, we have been assembling, organizing, analyzing, and disseminating this comparative information. Initially, a limited amount of sequence and comparative structure information was available online for our 16S (and 16S-like) [46, 47] and 23S (and 23S-like) ribosomal RNAs [48–52] and the group I introns [33]. In parallel, two other groups have been providing various forms of ribosomal RNA sequence and structure data (the RDP/RDP II [53, 54] and Belgium (5S/5.8S [55], small subunit [56, 57] and large subunit [58, 59]) groups). With significant increases in the amount of sequences available for the RNAs under study here, improved programs for the analysis of this data, and better web presentation software, we have established a new "Comparative RNA Web" (CRW) Site http://www.rna.icmb.utexas.edu/. This resource has been available to the public since January 2000.
Results and Discussion
The primary objectives and accomplishments for our Comparative RNA Web (CRW) Site are:
- I.
To study the following RNA molecules from a comparative perspective: - A.
Primary importance: 16S and 23S rRNA. - B.
Secondary importance: 5S rRNA, tRNA, group I and II introns. - II.
To provide the following comparative information for each of these RNA molecules: - A.
The newest comparative structure models for the primary RNA types. - B.
Nucleotide frequency tables for all individual positions, base pairs and base triples in the comparative structure models. This nucleotide frequency information is also mapped onto the complete NCBI phylogenetic tree [60, 61], revealing the type and extent of sequence and base pair conservation and variation at each position in the 16S and 23 S rRNAs at each node in the phylogenetic tree. - C.
A phylogenetic and structurally diverse set of secondary structure models (with diagrams and lists of positions that are base-paired) for each of the RNA types in this collection. - D.
Secondary structure diagrams revealing the extent of sequence and structure conservation for different phylogenetic groups at different levels in the phylogenetic tree. - E.
Basic information (organism name, RNA type, length, etc.) and NCBI GenBank [60] entries for each RNA sequence that is analyzed within the CRW Site. - F.
Sequence alignments created and maintained for comparative structure analysis. - III.
To catalog portions of this information in our relational database management system (RDBMS) and to dynamically retrieve it from our summary pages, full relational search, and phylogenetic tree-based search systems. - IV.
To present additional pages that: - A.
Reveal the evolution of the 16S and 23S rRNA structure models. - B.
Describe the comparative and covariation analysis techniques that we have utilized within the CRW Site. - C.
Formally define each of the primary RNA structure elements. - D.
Contain figures and data tables for our own publications detailing RNA structural motifs from a comparative perspective: - "Predicting U-turns in the ribosomal RNAs with comparative sequence analysis" [62].
- "A Story: unpaired adenosines in the ribosomal RNAs" [63].
- "AA.AG@helix.ends: AA and AG base-pairs at the ends of 16S and 23S rRNA helices" [64].
- E.
Contain figures and data tables for our own publications addressing RNA folding: - "A comparison of thermodynamic foldings with comparatively derived structures of 16S and 16S-like rRNAs"[65].
- "An Analysis of Large rRNA Sequences Folded by a Thermodynamic Method" [66].
- F.
Contain figures and data tables for our own publications that analyze RNA structure from a phylogenetic perspective: - "Phylogenetic Analysis of Molluscan Mitochondrial LSU rDNA Sequences and Secondary Structures" [67].
- "Accelerated Evolution of Functional Plastid rRNA and Elongation Factor Genes Due to Reduced Protein Synthetic Load After the Loss of Photosynthesis in the Chlorophyte Alga Polytoma" [68].
- "Group I Intron Lateral Transfer Between Red and Brown Algal Ribosomal RNA" [69].
The contents of our Comparative RNA Web (CRW) Site are outlined on its main page http://www.rna.icmb.utexas.edu/ (Figure 1). The detailed explanations of the data and their presentations in the first four sections of this site (1. Comparative Structure Models; 2. Nucleotide Frequency and Conservation Information; 3. Sequence and Structure Data; and 4. Data Access Systems) are presented here. To fully appreciate this description of the CRW Site, we encourage users to evaluate the pages at this web site while reading this manuscript; while a few of the pages and links at the CRW Site are shown as figures here, the reader is routinely referred to the actual web pages and the corresponding highlights on the "Table of Contents."
Figure 1
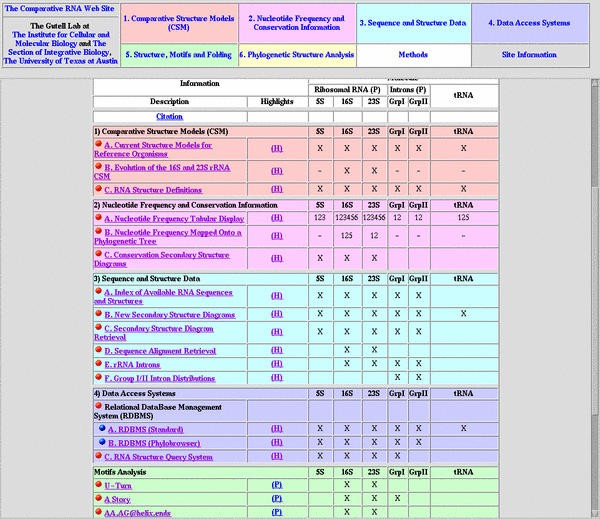
Introductory view of the CRW Site. The top frame divides the site into eight sections; the first four sections are the primary focus of this manuscript. The bottom frame contains the CRW Site's Table of Contents. Color-coding is used consistently throughout the CRW Site to help orient users.
1. Comparative structure models
1A. Current structure models for reference organisms
The first major category, Comparative Structure Models http://www.rna.icmb.utexas.edu/CSI/2STR/ contains our most recent 16S and 23S rRNA covariation-based structure models, which were adapted from the original Noller & Woese models (16S [15, 22] and 23S [19] rRNA), and the structure models for 5S rRNA [12], tRNA [5–9], and the group I [32] and group II [34] introns, as determined by others. This collection of RNA structure models was predicted with covariation analysis, as described at the CRW Site Methods Section http://www.rna.icmb.utexas.edu/METHODS/ and in several publications (see below).
Briefly, covariation analysis, a specific application of comparative analysis (as mentioned earlier), searches for helices and base pairs that are conserved in different sequences that form the same functionally equivalent molecule (e.g., tRNA sequences). It was determined very early in this methodology that the correct helix is the one that contains positions within a potential helix that vary in composition while maintaining G:C, A:U, and G:U base pairs. As more sequences for a given molecule were determined, we developed newer algorithms that searched for positions in an alignment of homologous sequences that had similar patterns of variation. This latter implementation of the covariation analysis helped us refine the secondary and tertiary structure models by eliminating previously proposed base pairs that are not underscored with positional covariation and identifying new secondary and tertiary structure base pairs that do have positional covariation [19, 70–72]. Our newest covariation analysis methods associate color-coded confidence ratings with each proposed base pair (see reference structure diagrams and Section 2A, "Nucleotide Frequency Tabular Display," for more details). One exception to this is the tRNA analysis, which was initially performed with the Mixy chi-square-based algorithm [71], and thus the color codes are based on that analysis.
When implemented properly, covariation analysis can predict RNA structure with extreme accuracy. All of the secondary structure base pairs and a few of the tertiary structure base pairs predicted with covariation analysis [5–9, 71–74] are present in the tRNA crystal structure [10, 11]. The analysis of fragments of 5S rRNA [75] and the group I intron [76] resulted in similar levels of success. Most recently, the high-resolution crystal structures for the 30S [44] and 50S [45] ribosomal subunits have given us the opportunity to evaluate our rRNA structure models. Approximately 97–98% of the 16S and 23S rRNA base pairs predicted with covariation analysis are in these crystal structures (Gutell et al., manuscript in preparation). This congruency between the comparative model and the crystal structure validates the comparative approach, the covariation algorithms, the accuracy of the juxtapositions of sequences in the alignments, and the accuracy of all of the comparative structure models presented herein and available at the CRW Site. However, while nearly all of the base pairs predicted with comparative analysis are present in the crystal structure solution, some interactions in the crystal structure, which are mostly tertiary interactions, do not have similar patterns of variation at the positions that interact (Gutell et al., manuscript in preparation). Thus, covariation analysis is unable to predict many of the tertiary base pairings in the crystal structure, although it does identify nearly all of the secondary structure base pairings.
Beyond the base pairs predicted with covariation analysis, comparative analysis has been used to predict some structural motifs that are conserved in structure although they do not necessarily have similar patterns of variation at the two paired positions. Our analyses of these motifs are available in the "Structure, Motifs, and Folding" section of our CRW Site.
While the secondary structure models for the 16S, 23S and 5S rRNAs, group I and II introns, and tRNA are available at the "Current Structure Models for Reference Organisms" page, our primary focus has been on the 16S and 23S rRNAs. Thus, some of our subsequent analysis and interpretation will emphasize only these two RNAs.
Each RNA structure model presented here is based upon a single reference sequence, chosen as the most representative for that molecule (Table 1); for example, E. coli is the preferred choice as the reference sequence for rRNA (5S, 16S, and 23S), based on the early and continued research on the structure and functions of the ribosome [77, 78]. Each of the six structure models (5S, 16S and 23S rRNA, group I and II introns, and tRNA) in the "Current Structure Models for Reference Organisms" page http://www.rna.icmb.utexas.edu/CSI/2STR/ contains six or seven different diagrams for that molecule: Nucleotide, Tentative, Helix Numbering, Schematic, Histogram, Circular, and Matrix of All Possible Helices.
Table 1 Reference sequence and nucleotide frequency data available at the CRW Site. Nucleotide frequency data available in tabular form is indicated with "Y." Entries marked with "*" are also available mapped on the phylogenetic tree. L, Lousy; M, Model; T, Tentative.
Nucleotide: The standard format for the secondary structure diagrams with nucleotides (Figures 2A, 2B, and 2C) reveals our confidence for each base pair, as predicted by covariation analysis. Base pairs with a red identifier ("-" for G:C and A:U base pairs, small closed circles for G:U, large open circles for A:G, and large closed circles for any other base pair) have the greatest amount of covariation; thus, we have the most confidence in these predicted base pairs. Base pairs with a green, black, grey, or blue identifier have progressively lower covariation scores and are predicted due to the high percentages of A:U + G:C and/or G:U at these positions. The most current covariation-based E. coli 16S and 23S rRNA secondary structure models are shown in Figures 2A, 2B, and 2C. Note that the majority of the base pairs in the 16S and 23S rRNA have a red base pair symbol, our highest rating. These diagrams are the culmination of twenty years of comparative analysis. Approximately 8500 16S and 16S-like rRNA sequences and 1050 23S and 23S-like rRNA sequences were collected from all branches of the phylogenetic tree, as shown in Section 2, "Nucleotide Frequency and Conservation Information" and in Table 2. These sequences have been aligned and analyzed with several covariation algorithms, as described in more detail in the "Predicting RNA Structure with Comparative Methods" section of the CRW Site http://www.rna.icmb.utexas.edu/METHODS/ and in Section 2A. All of the secondary structure diagrams from the "Current Structure Models for Reference Organisms" page are available in three formats. The first two are standard printing formats, PostScript http://www.adobe.com/products/postscript/main.html and PDF http://www.adobe.com/products/acrobat/adobepdf.html. The third, named "bpseq," is a simple text format that contains the sequence, one nucleotide per line, its position number, and the position number of the pairing partner (or 0 if that nucleotide is unpaired in the covariation-based structure model).
Figure 2
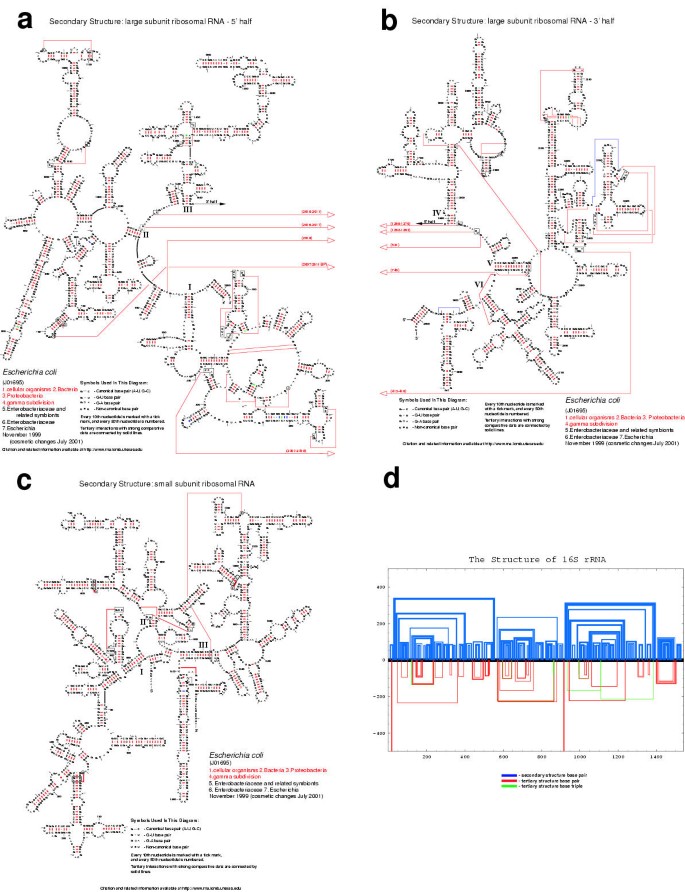
The most recent (November 1999) versions of the rRNA comparative structure models (see text for additional details). A. E. coli 23S rRNA, 5' half. B. E. coli 23S rRNA, 3' half. C. E. coli 16S rRNA. D. The "histogram" format for the E. coli 16S rRNA.
Table 2 Alignments available from the CRW Site. These alignments were used to generate conservation diagrams (rRNA only) and correspond to the alignments used in the nucleotide frequency tables.
Tentative: In addition to the 16S and 23S rRNA structure models, we have also identified some base pairs in the 16S and 23S rRNAs that have a lower, although significant, extent of covariation. These are considered 'tentative' and are shown on separate 16S and 23S rRNA secondary structure diagrams http://www.rna.icmb.utexas.edu/CSI/2STR/. These base pairs and base triples have fewer coordinated changes (or positional covariations) and/or a higher number of sequences that do not have the same pattern of variation present at the other paired position. Consequently, we have less confidence in these putative interactions, in contrast with the interactions predicted in our main structure models.
The Helix Numbering secondary structure diagrams illustrate our system for uniquely and unambiguously numbering each helix in a RNA molecule. Based upon the numbering of the reference sequence, each helix is named for the position number at the 5' end of the 5' half of the helix. For example, the first 16S rRNA helix, which spans E. coli positions 9–13/21–25, is named "9;" the helix at positions 939–943/1340–1344 is named "939." This numbering system is used in the Nucleotide Frequency Tabular Display tables (see below). The Schematic versions of the reference structure diagrams replace the nucleotides with a line traversing the RNA backbone.
The "Histogram" and "Circular" diagram formats http://www.rna.icmb.utexas.edu/CSI/2STR/ both abstract the global arrangement of the base pairs. For the histogram version (Figure 2D), the sequence is displayed as a line from left (5') to right (3'), with the secondary structure base pairs shown in blue above the sequence line; below this line, tertiary structure base pairs and base triples are shown in red and green, respectively. The distance from the baseline to the interaction line is proportional to the distance between the two interacting positions within the RNA sequence. In contrast, in the circular diagram, the sequence is drawn clockwise (5' to 3') in a circle, starting at the top. Secondary and tertiary base-base interactions are shown with lines traversing the circle, using the same coloring scheme as in the histogram diagram. The global arrangement and higher-order organization of the base pairs predicted with covariation analysis are revealed in part in these two alternative formats. The majority of the base pairs are clustered into regular secondary structure helices, and the majority of the helices are contained within the boundaries of another helix, forming large cooperative sets of nested helices. The remaining base pairs form tertiary interactions that either span two sets of nested helices, forming a pseudoknot, or are involved in base triple interactions.
In th*/e "Matrix of All Possible Helices" plot http://www.rna.icmb.utexas.edu/CSI/2STR/, the same RNA sequence is extended along the X- and Y-axes, with all potential helices that are comprised of at least four consecutive Watson-Crick (G:C and A:U) or G:U base pairs shown below the diagonal line. The helices in the present comparative structure model are shown above this line. The number of potential helices is larger than the actual number present in the biologically-active structure (see CRW Methods http://www.rna.icmb.utexas.edu/METHODS/). For example, the S. cerevisiae phenylalanine tRNA sequence, with a length of 76 nucleotides, has 37 possible helices (as defined above); only four of these are in the crystal structure. The E. coli 16S rRNA, with 1542 nucleotides (nt), has nearly 15,000 possible helices; only about 60 of these are in the crystal structure. For the E. coli 23S rRNA (2904 nt), there are more than 50,000 possible helices, with approximately 100 in the crystal structure. The number of possible secondary structure models is significantly larger than the number of possible helices, due to the exponential increase in the number of different combinations of these helices. The number of different tRNA secondary structure models is approximately 2.5 × 1019; there are approximately 10393 and 10740 possible structure models for 16S and 23S rRNA, respectively (see CRW Methods http://www.rna.icmb.utexas.edu/METHODS/). Covariation analysis accurately predicted the structures of the 16S and 23S rRNAs (see above) from this very large number of structure models.
1B. Evolution of the 16S and 23S rRNA comparative structure models
An analysis of the evolution of the Noller-Woese-Gutell comparative structure models for the 16S and 23S rRNAs is presented here http://www.rna.icmb.utexas.edu/CSI/EVOLUTION/ (H-1B.1). Our objective is to categorize the improvements in these covariation-based comparative structure models by tabulating the presence or absence of every proposed base pair in each version of the 16S and 23S rRNA structure models, starting with our first 16S [15] and 23S [19] rRNA models. Every base pair in each of the structure models was evaluated against the growing number and diversity of new rRNA sequences. Proposed base pairs were taken out of the structure model when the number of sequences without either a covariation or a G:C, A:U, or G:U base pair was greater than our allowed minimum threshold; the nucleotide frequencies for those base pairs are available from the "Lousy Base-Pair" tables that are discussed in the next section. New base pairs were proposed when a (new) significant covariation was identified with our newer and more sensitive algorithms that were applied to larger sequence alignments containing more inherent variation (see CRW Methods http://www.rna.icmb.utexas.edu/METHODS/ for more detail).
Although other comparative structure models and base pairs were predicted by other labs, those interactions are not included in this analysis of the improvements in our structure models. The four main structure models for 16S and 23S rRNA are very similar to one another. The Brimacombe [16, 20] and Strasburg [17, 21] structure models were determined independently of ours, while the De Wachter [58, 79] models were adapted from our earlier structure models and have incorporated some of the newer interactions proposed here.
This analysis produced two very large tables with 579 proposed 16S rRNA base pairs evaluated against six versions of the structure model and 1001 23S rRNA base pairs evaluated against five versions of the structure model. Some highlights from these detailed tables are captured in summary tables (Tables 3a and 3b, and http://www.rna.icmb.utexas.edu/CSI/EVOLUTION/) that compare the numbers of sequences and base pairs predicted correctly and incorrectly for each of the major versions of the 16S and 23S rRNA structure models. For this analysis, the current structure model is considered to be the correct structure; thus, values for comparisons are referenced to the numbers of sequences and base pairs in the current structure model (478 base pairs and approximately 7000 sequences for 16S rRNA, and 870 base pairs and approximately 1050 sequences for 23S rRNA). Three sets of 16S and 23S rRNA secondary structure diagrams were developed to reveal the improvements between the current model and earlier versions: 1) changes since the 1996 published structure models; 2) changes since 1983 (16S rRNA) or 1984 (23S rRNA); and 3) all previously proposed base pairs that are not in the most current structure models (H-1B.2).
Table 3a Summary of the Evolution of the Noller-Woese-Gutell 16S rRNA Comparative Structure Model. Categories marked with "*" are calculated compared to the 1999 version of the 16S rRNA model.
Table 3b Summary of the Evolution of the Noller-Woese-Gutell 23S rRNA Comparative Structure Model. Categories marked with "*" are calculated compared to the 1999 version of the 23S rRNA model.
An analysis of these tables reveals several major conclusions from the evolution of the 16S and 23S rRNA covariation-based structure models. First, approximately 60% of the 16S and nearly 80% of the 23S rRNA base pairs predicted in the initial structure models appear in the current structure models. The accuracy of these early models, produced from the analysis of only two well-chosen sequences, is remarkable. Second, the accuracy, number of secondary and tertiary structure interactions, and complexity of the structure models increase as the number and diversity of sequences increase and the covariation algorithms are improved. As well, some pairs predicted in the earlier structure models were removed from subsequent models due to the large number of exceptions to the positional covariation at the two paired positions. Third, the majority of the tertiary interactions were proposed in the last few versions of the structure models.
1C. RNA structure definitions
The RNA structure models presented here are composed of several different basic building blocks (or motifs) that are described and illustrated at our RNA Structure Definitions page http://www.rna.icmb.utexas.edu/CSI/DEFS/ (H-1C.1-2). The nucleotides in a comparative structure model can be either base paired or unpaired. Base paired nucleotides can be part of either a secondary structure helix (two or more consecutive, antiparallel and nested base pairs) or a tertiary interaction, which is a more heterogeneous collection of base pair interactions. These include any non-canonical base pair (not a G:C, A:U, or G:U; e.g., U:U), lone or single base pairs (when both positions in a base pair are not flanked by two nucleotides that are base paired to one another), base pairs in a pseudoknot arrangement, and base triples (a single nucleotide interacting with a base pair). Each of these base pair categories has a unique color code in the illustrations on the "RNA Structure Definitions" page, which provides multiple examples of each category from the 16S and 23S rRNA structure models. In contrast to the nucleotides that are base paired, nucleotides can also be unpaired in the comparative structure models. Within this category, they can be within a hairpin loop (nucleotides capping the end of a helix), internal loop (nucleotides within two helices), or in a multi-stem loop (nucleotides within three or more helices).
2. Nucleotide frequency and conservation information
Underpinning the comparative sequence analysis of RNA molecules are the realizations that every RNA has evolved to its present state and form, and that the same secondary and tertiary structure for an RNA can be derived from many different sequences that maintain the integrity and functionality of that structure. These evolutionary and structural dynamics have made it possible to predict RNA structure models with comparative analysis (as presented in the previous section). The tempo and mode of the evolution for every position in the RNA structure is defined by a complex and not-well-understood equation, with variables for global mutation rates and rates for specific branches on the phylogenetic tree, the allowed variance for each nucleotide and the structure with which it is associated, the coordination and dependence between nucleotides, and other constraints not yet defined. In an effort to begin to understand these dimensionalities associated with an RNA sequence and to catalogue the observed constraints in each of the RNA molecules maintained within our CRW Site, we have prepared online tables and figures that reveal the amount and type of conservation and variation for many of the RNAs available here.
The comparative information for a sequence is initially assembled in a sequence alignment (more information about alignments below at: "3. Sequence and Structure Data"). The extent and type of sequence and structure conservation and variation are presented in two general formats: (1) nucleotide frequency tables that contain the types of nucleotides and their frequencies for each position in the RNA molecule; and (2) secondary structure diagrams revealing the most conserved nucleotide at each position that is present in the vast majority of the sequences. The position numbers for the nucleotide frequency tables and conservation diagrams are based upon a reference sequence (see Table 1). While deletions relative to the reference sequence are shown in the tables with "-," insertions relative to the reference sequence are not shown. Conservation diagrams summarize the insertions and deletions relative to the reference sequence.
2A. Nucleotide frequency tabular display
The nucleotide frequency tables appear in two general presentation modes. In the traditional table, the nucleotide types are displayed in the columns, while their frequencies are shown for each alignment in the rows. The nucleotide frequencies were determined for single positions, base pairs, and base triples for a subset of the RNAs in the CRW Site collection (detailed in Table 1). Single nucleotide frequencies are available for all individual positions, based upon the reference sequence, for every RNA in this collection. Base pair frequencies are presented for a) all base pairs in the current covariation-based structure models, b) tentative base pairs predicted with covariation analysis, and c) base pairs previously proposed with comparative analysis that are not included in our current structure models due to a lack of comparative support from the analysis with our best covariation methods on our current alignments (named "Lousy" base pairs). Base triples are interactions between a base pair and a third unpaired nucleotide; base triple frequencies are provided for a) base triples in the current covariation-based structure models and b) tentative base triples predicted with covariation analysis.
For each of these frequency tables, the percentages of each of the nucleotides are determined for multiple alignments, where the most similar sequences are organized into the same alignment. For the three rRNAs, the alignments are partitioned by their phylogenetic relationships. There is an alignment for the nuclear-encoded rRNA for each of the three primary lines of descent ((1) Archaea, (2) Bacteria, and (3) Eucarya; [80]), each of the two Eucarya organelles (no alignments yet for the 5S rRNA; (4) Chloroplasts and (5) Mitochondria), and two larger alignments that include all of the (6) nuclear-encoded rRNA sequences for the Archaea, Bacteria, and Eucarya, and (7) these three phylogenetic groups and the two Eucarya organelles (Table 2).
For the tRNA and group I and II intron sequences, the most similar sequences are not necessarily from similar phylogenetic groups. Instead, the sequences that are most similar with one another are members of the same functional and/or structural class. The tRNA sequences are grouped according to the amino acids that are bound to the tRNA. Currently, only the type I tRNAs [81] are included here; the tRNAs are collected in 19 functional subgroup alignments and one total type I alignment. The group I and II intron alignments are based on the structural classifications determined by Michel (group I [32] and group II [34]) and Suh (group IE [82]). The group I introns are split into seven alignments: A, B, Cl-2, C3, D, E, and unknown. The group II introns are divided into the two major subgroups, IIA and IIB (Table 2).
For the standard nucleotide frequency tables (Highlight 2A (H-2A)), the left frame in the main frame window ("List Frame") contains the position numbers for the three types of tables: single bases, base pairs, and base triples. Clicking on a position, base pair, or base triple number will bring the detailed nucleotide occurrence and frequency information to the main window ("Data Frame;" H-2A.1). The collective scoring data (H-2A.2) used to predict the base pair is obtained, where available, by clicking the "Collective Score" link on the right-hand side of the base pair frequency table.
As discussed in Section 1A, we have established a confidence rating for the base pairs predicted with the covariation analysis; a detailed explanation of the covariation analysis methods and the confidence rating system will be available in the Methods section of the CRW Site http://www.rna.icmb.utexas.edu/METHODS/. The extent of base pair types and their mutual exchange pattern (e.g., A:U <-> G:C) is indicative of the covariation score. This value increases to the maximum score as the percentage and the amount of pure covariations (simultaneous changes at both positions) increase in parallel with a decrease in the number of single uncompensated changes, and the number of times these coordinated variations occur during the evolution of that RNA (for the rRNAs, the number of times this covariation occurs in the phylogenetic tree) increases. These scores are proportional to our confidence in the accuracy of the predicted base pair. Red, our highest confidence rating, denotes base pairs with the highest scores and with at least a few phylogenetic events (changes at both paired positions during the evolution of that base pair). The colors green, black, and grey denote base pairs with a G:C, A:U, and/or G:U in at least 80% of the sequences and within a potential helix that contains at least one red base pair. Base pairs with a green confidence rating have a good covariation score although not as high as (or with the confidence of) a red base pair. Black base pairs have a lower covariation score, while grey base pairs are invariant, or nearly so, in 98% of the sequences. Finally, blue base pairs do not satisfy these constraints; nevertheless, we are confident of their authenticity due to a significant number of covariations within the sequences in a subset of the phylogenetic tree or are an invariant G:C or A:U pairings in close proximity to the end of a helix.
The covariation score for each base pair is determined independently for each alignment (e.g., Three Domain/Two Organelle, Three Domain, Archaea, etc.). The collective score for each base pair is equivalent to the highest ranking score for any one of the alignments. For example, we have assigned our highest confidence rating to the 927:1390 base pair in 16S rRNA (Figure 2C; H-2A). Note that the entry for the 927:1390 base pair (H-2A) in the list of base pairs in the left frame is red in the C (or confidence) column. For this base pair, only the T (Three Phylogenetic Domains/Two Organelle) alignment has a significant covariation score (H-2A); thus, only the "T" alignment name is red. Of the nearly 6000 sequences in the T alignment, 69% of the sequences have a G:U base pair, A:U base pair at 16.2%, U:A at 6.9%, and less than 1% of the sequences have a G:C, C:G, U:U, or G:G base pair (H-2A.1). The collective scoring data (H-2A.2) reveals that there are 11 phylogenetic events (PE) for the T alignment, while the C1+C3 score is 1.00, greater than the minimum value for this RNA and this alignment (a more complete explanation of the collective scoring method is available at CRW Methods http://www.rna.icmb.utexas.edu/METHODS/). Note that the 928:1389 and 929:1388 base pairs are also both red. Here, six of the seven alignments have significant extents of covariation for both base pairs and are thus red. Each of the red alignments have at least two base pair types (e.g., G:C and A:U) that occur frequently, at least three phylogenetic events, and C1+C3 scores >= 1.5.
2B. Nucleotide frequency mapped onto a phylogenetic tree
The second presentation mode maps the same nucleotide frequency data in the previous section onto the NCBI phylogenetic tree http://www.ncbi.nlm.nih.gov/Taxonomy/taxonomyhome.html/[60, 61] (see Materials and Methods for details). This display allows the user to navigate through the phylogenetic tree and observe the nucleotide frequencies for any node and all of the branches off of that node. The number of nucleotide substitutions on each branch are displayed, with the number of mutual changes displayed for the base pairs and base triples. Currently, only the 16S and 23S rRNA nucleotide frequencies available in the first tabular presentation format are mapped onto the phylogenetic tree (see Table 1). As shown in CRW Section 2B (H-2B), the left frame in the main frame window contains the position numbers for the three types of data, single bases, base pairs, and base triples. Clicking on a position, base pair, or base triple number will initially reveal, in the larger section of the main frame, the root of the phylogenetic tree, with the frequencies for the selected single base, base pair, or base triple. The presentation for single bases (H-2B.1) reveals the nucleotides and their frequencies for all sequences at the root level, followed by the nucleotides and their frequencies for the Archaea, Bacteria, and Eukaryota (nuclear, mitochondrial, and chloroplast). Nucleotides that occur in less than 2%, 1.5%, 1%, 0.5%, 0.2%, and 0.1% of the sequences can be eliminated from the screen by changing the green "percentage limit" selection at the top of the main frame. The number of phylogenetic levels displayed on the screen can also be modulated with the yellow phylogenetic level button at the top of the main frame. Highlight 2B.1 displays only one level of the phylogenetic tree from the point of origin, which is the root level for this example. In contrast, Highlight 2B.2 displays four levels from the root. The number of single nucleotide changes on each branch of the phylogenetic tree is shown at the end of the row. For single bases, this number is in black. For base pairs, there are two numbers. The orange color refers to the number of changes at one of the two positions, while the pink color refers to the number of mutual changes (or covariations) that has occurred on that branch of the tree (H-2B.2). For example, for the 16S rRNA base pair 501:544, there are 65 mutual and 74 single changes in total for the Archaea, Bacteria, Eucarya nuclear, mitochondrial, and chloroplast. Within the Archaea, there are six mutual and five single changes. Five of these mutual changes are within the Euryarchaeota, and four of these are within the Halobacteriales (H-2B.2). The base pair types that result from a mutual change (or strict covariation) are marked with an asterisk ("*").
2C. Secondary structure conservation diagrams
Conservation secondary structure diagrams summarize nucleotide frequency data by revealing the nucleotides present at the most conserved positions and the positions that are present in nearly all sequences in the analyzed data set. The conservation information is overlaid on a secondary structure diagram from a sequence that is representative of the chosen group (e.g., E. coli for the gamma subdivision of the Proteobacteria, or S. cerevisiae for the Fungi; H-2C.1). All positions that are present in less than 95% of the sequences studied are considered variable, hidden from view, and replaced by arcs. These regions are labeled to show the minimum and maximum numbers of nucleotides present in that region in the group under study (e.g., [0–179] indicates that all sequences in the group contain a minimum of zero nucleotides but not more than 179 nucleotides in a particular variable region). The remaining positions, which are present in at least 95% of the sequences, are separated into four groups (H-2C.1): 1) those which are conserved in 98–100% of the sequences in the group (shown with red upper-case letters indicating the conserved nucleotide); 2) those which are conserved in 90–98% of the sequences in the group (shown with red lower-case letters indicating the conserved nucleotide); 3) those which are conserved in 80–90% of the sequences in the group (shown with large closed circles); and 4) those which are conserved in less than 80% of the sequences in the group (shown with small open circles).
Insertions relative to the reference sequence are identified with a blue line to the nucleotides between which the insertion occurs, and text in small blue font denoting the maximum number of nucleotides that are inserted and the percentage of the sequences with any length insertion at that place in the conservation secondary structure diagram (H-2C.1). All insertions greater than five nucleotides are tabulated, in addition to insertions of one to four nucleotides that occur in more than 10% of the sequences analyzed for that conservation diagram. Each diagram contains the full NCBI phylogenetic classification http://www.ncbi.nlm.nih.gov/Taxonomy/taxonomyhome.html/ for the group.
Currently, there are conservation diagrams for the 5S, 16S, and 23S rRNA for the broadest phylogenetic groups: (1) the three major phylogenetic groups and the two Eucarya organelles, chloroplasts and mitochondria; (2) the three major phylogenetic groups; (3) the Archaea; (4) the Bacteria; (5) the Eucarya (nuclear encoded); (6) the chloroplasts; and (7) the mitochondria. Longer term, our goal is to generate rRNA conservation diagrams for all branches of the phylogenetic tree that contain a significant number of sequences. Toward this end, we have generated 5S, 16S, and 23S rRNA conservation diagrams for many of the major phylogenetic groups within the Bacterial lineage (e.g., Firmicutes and Proteobacteria). We will also be generating conservation diagrams for the group I and II introns.
The CRW Site conservation diagram interface (H-2C.2) provides both the conservation diagrams (in PostScript and PDF formats) and useful auxiliary information. The display is sorted phylogenetically, with each row of the table containing all available conservation information for the rRNA sequences in that phylogenetic group. For each of the three rRNA molecules (5S, 16S, and 23S), three items are available: 1) the reference structure diagram, upon which the conservation information is overlaid; 2) the conservation diagram itself; and 3) the number of sequences summarized in the conservation diagram, which links to a web-formatted list of those sequences. The lists, for each sequence, contain: 1) organism name (NCBI scientific name); 2) GenBank accession number; 3) cell location; 4) RNA Type; 5) RNA Class; and 6) NCBI phylogeny. Users who want more information about a given sequence should consult the CRW RDBMS (see below). An equivalent presentation for intron conservation data is under development.
3. Sequence and structure data
Structure-based alignments and phylogenetic analysis of RNA structure
Analysis of the patterns of sequence conservation and variation present in RNA sequence alignments can reveal phylogenetic relationships and be utilized to predict RNA structure. The accuracy of the phylogenetic tree and the predicted RNA structure is directly dependent on the proper juxtapositioning of the sequences in the alignment. These alignments are an attempt to approximate the best juxtapositioning of sequences that represent similar placement of nucleotides in their three-dimensional structure. For sequences that are very similar, the proper juxtapositioning or alignment of sequences can be achieved simply by aligning the obviously similar or identical subsequences with one another. However, when there is a significant amount of variation between the sequences, it is not possible to align sequences accurately or with confidence based on sequence information alone. For these situations, we can juxtapose those sequences that form the same secondary and tertiary structure by aligning the positions that form the same components of the similar structure elements (e.g., align the positions that form the base of the helix, the hairpin loop, etc.). Given the accurate prediction of the 16S and 23S rRNA secondary structures from the analysis of the alignments we assembled, we are now even more confident in the accuracy of the positioning of the sequence positions in our alignments, and the process we utilize to build them.
Aligning new sequences
At this stage in our development of the sequence alignments, there are well-established and distinct patterns of sequence conservation and variation. From the base of the phylogenetic tree, we observe regions that are conserved in all of the rRNA sequences that span the three phylogenetic domains and the two eucaryotic organelles, the chloroplast and mitochondria. Other regions of the rRNA are conserved within the three phylogenetic domains although variable in the mitochondria. As we proceed into the phylogenetic tree, we observe positions that are conserved within one phylogenetic group and different at the same level in the other phylogenetic groups. For example, Bacterial rRNAs have positions that are conserved within all members of their group, but different from the Archaea and the Eucarya (nuclear-encoded). These types of patterns of conservation and variation transcend all levels of the phylogenetic tree and result in features in the rRNA sequences and structures that are characteristic for each of the phylogenetic groups at each level of the phylogenetic tree (e.g., level one: Bacterial, Archaea, Eucarya; level two: Crenarchaeota, Euryarchaeota in the Archaea; level three: gamma, alpha, beta, and delta/epsilon subdivisions in the Proteobacteria). Carl Woese likened the different rates of evolution at the positions in the rRNA to the hands on a clock [4]. The highly variable regions are associated with the second hand; these can change many times for each single change that occurs in the regions associated with the minute hand. Accordingly, the minute hand regions change many times for each single change in the hour hand regions of the rRNAs. In addition to the different rates of evolution, many of the positions in the rRNA are dependent on one another. The simplest of the dependencies, positional covariation, is the basis for the prediction of the same RNA structure from similar RNA sequences (see Section 1A, Covariation Analysis).
We utilize these underlying dynamics in the evolution and positional dependency of the RNA to facilitate the alignment and structural analysis of the RNA sequences. Our current RNA data sets contain a very large and diverse set of sequences that represent all sections of the major phylogenetic branches on the tree of life. This data collection also contains many structural variations, in addition to their conserved sequence and structure core. The majority of the new RNA sequences are very similar to at least one sequence that has already been aligned for maximum sequence and structure similarity; thus, these sequences are relatively simple to align. However, some of the new sequences contain subsequences that cannot be aligned with any of the previously aligned sequences, due to the excessive variation in these hypervariable regions. For these sequences, the majority of the sequence can be readily aligned with the more conserved elements, followed by a manual, visual analysis of the hypervariable regions. To align these hypervariable regions with more confidence, we usually need several more sequences with significant similarity in these regions that will allow us to identify positional covariation and subsequently to predict a new structural element. Thus, at this stage in the development of the alignments, the most conserved regions (i.e., hour hand regions) and semi-conserved regions (i.e., minute hand regions) have been aligned with high confidence. The second and sub-second (i.e., tenth and hundredth of a second) hand regions have been aligned for many of the sequences on the branches at the ends on the phylogenetic tree. However, regions of the sequences continue to challenge us. For example, the 545 and 1707 regions (E. coli numbering) contain an excessive amount of variation in the Eucarya nuclear-encoded 23S-like rRNAs. These two regions could not be well aligned and we could not predict a common structure with comparative analysis with ten Eucaryotic sequences in 1988 (see Figures 35–43 in [48]). However, once a larger number of related Eucaryotic 23S-like rRNA sequences was determined, we reanalyzed these two regions and were able to align those regions to other related organisms (e.g., S. cerevisiae with Schizosaccharomyces pombe, Cryptococcus neoformans, Pneumocystis carinii, Candida albicans, and Mucor racemosus) and predict a secondary structure that is common for all of these rRNAs (see Figures 3 and 6 in [52]). While the secondary structures for the fungal 23S-like rRNAs are determined in these regions, the animal rRNAs were only partially solved. We still need to determine a common secondary structure for the large variable-sized insertions in the animal rRNAs, and this will require even more animal 23S-like rRNA sequences from organisms that are very closely related to the organisms for which we currently have sequences.
Figure 3
RDBMS (Standard) search form.
A large sampling of secondary structure diagrams
We have generated secondary structure diagrams for sequences that represent the major phylogenetic groups, and for those sequences that reveal the major forms of sequence and structure conservation and variation. New secondary structure diagrams are templated from an existing secondary structure diagram and the alignment of these two sequences, the sequence for the new structure diagram and the sequence for the structure that has been templated. The nucleotides in the new sequence replace the templated sequence when they are in the same position in the alignment, while positions in the new sequence that are not juxtaposed with a nucleotide in the templated sequence are initially left unstructured. These nucleotides are then placed interactively into their correct location in the structure diagram with the program XRNA (Weiser & Noller, University of California, Santa Cruz) and base-paired when there is comparative support for that pairing in the alignment; otherwise, they are left unpaired.
The process of generating these secondary structure diagrams occurs in parallel with the development of the sequence alignments. In some cases, the generation of a structure diagram helps us identify problems with the sequence or its alignment. For example, anomalies in structural elements (in the new structure diagram) that had strong comparative support in the other sequences could be the result of a bad sequence or due to the misalignment of sequences in the helix region. In other cases, the new structure diagram reveals a possible helix in a variable region that was weakly predicted with comparative analysis. However, a re-inspection of a few related structure diagrams revealed another potential helix in this region that was then substantiated from an analysis of the corresponding region of the alignment. Thus, the process of generating additional secondary structure diagrams improves the sequence alignments and the predicted structures, in addition to the original purpose for these diagrams, to reveal the breadth of sequence conservation and variation for any one RNA type.
Our goals for the "Sequence and Structure Data" section of the CRW Site are to:
- A)
Align all rRNA, group I and II intron sequences that are greater than 90% complete and are available at GenBank; - B)
Generate rRNA and group I/II intron secondary structure diagrams for organisms that are representative of a phylogenetic group or representative of a type of RNA structural element. The generation of 5S, 16S, and 23S rRNAs secondary structures from genomic sequences generally has higher priority over other rRNA sequences. - C)
Enter pertinent information for each sequence and structure into our relational database management system. This computer system organizes all of our RNA sequence and structure entries, associates them with the organisms' complete NCBI phylogeny http://www.ncbi.nlm.nih.gov/Taxonomy/taxonomyhome.html/, and allows for the efficient retrieval of this data (see Section 4: Data Access Systems for more details).
Due in part to the technological improvements in the determination of nucleic acid sequence information, the number of ribosomal RNA and group I and II intron sequences has increased significantly within the past 10 years. As of December 2001, the approximate numbers of complete or nearly complete sequences and secondary structure diagrams for each of these RNAs for the major phylogenetic groups and structural categories are shown in Highlight 3A.1. At this time, the actual number of sequences that are both greater than 90% complete and available at GenBank is greater than the number in our CRW RDBMS.
The sequences, alignments, and secondary structure diagrams are available from several different web pages, which are described below in Sections 3A-3D and 4A-4B.
3A. Index of available sequences and structures
The top section of the "Index of Available Sequences and Structures" page (H-3A.1) reveals the numbers of available sequences for the Archaea, Bacteria, and Eucarya nuclear, mitochondrial, and chloroplast groups that are at least 90% complete and structure diagrams for the 5S, 16S, and 23S rRNAs and group I and group II introns. The remainder of the index page contains the numbers of sequences and structures for more expanded lists for each of those five phylogenetic/cell location groups. For example, the Archaea are expanded to the Crenarchaeota, Euryarchaeota, Korarchaeota, and unclassified Archaea. These counts are updated dynamically when the information in our relational database management system is revised. The numbers of sequences and structures are links that open the RDBMS "standard" output view (see below for details) for the selected target set. Secondary structure diagrams are available in PostScript, PDF, and BPSEQ (see above) formats from the structure links. The organism names in the output from these links are sorted alphabetically. The number of entries per output page is selectable (20, 50, 100, 200, or 400), with 20 set as a default. Entries not shown on the first page can be viewed by clicking on the "Next" button at the bottom left of the output page.
As of December 2001, our data collection contains 11,464 rRNA (5S, 16S, and 23S) and intron (group I, II, and other) sequences. The ribosomal RNAs comprise 80% of this total, and 16S rRNA represents 82% of the rRNA total; the remainder is split between the 23S and 5S rRNAs. Intron sequences comprise 20% of our total collection, with approximately twice as many group I introns than group II introns. Of the 406 secondary structure diagrams, the majority are for the 16S (71%) and 23S (20%) rRNAs. At this time, tRNA records are not maintained in our database system.
3B. New secondary structure diagrams
Secondary structure diagrams that have been created or modified recently are listed and available from their own page (H-3B.1). These diagrams are sorted into one of three categories: new or modified 1) in the past seven days (highlighted with red text); 2) in the past month (blue text); and 3) in the past three months (black text). Diagrams are listed alphabetically by organism name within each of the three time categories. The display also indicates the cell location and RNA Class (see below) for each diagram. The PostScript, PDF, and BPSEQ files can be viewed by clicking the appropriate radio button at the top of this page and then the links in the structure field.
3C. Secondary structure diagram retrieval
Multiple secondary structure diagrams can be downloaded from the Secondary Structure Retrieval Page (Highlight 3C.1). This system allows the user to select from organism names, phylogeny (general: Archaea, Bacteria, Eukaryota, and Virus), RNA Class (see Table 4), and cell location, as well as selecting for PostScript, PDF, or BPSEQ display formats. Once these selections are made, a list of the structure diagrams that fit those criteria appears. The user may select any or all of the diagrams to be downloaded. The 23S rRNA diagrams (which appear in two halves) are presented on one line as a single unit to ensure that both halves are downloaded. The system packages the secondary structure diagrams files into a compressed tar file, which can be uncompressed with appropriate software on Macintosh, Windows, and Unix computer platforms. (Note: due to a limitation in the web server software, it is currently not possible to reliably download more than 300 structures at one time. This limitation can be avoided by subdividing large queries.)
Table 4 RDBMS Fields and Short Descriptions.
3D. Sequence alignment retrieval
The Sequence Alignment Retrieval page (Highlight 3D.1) provides access to the sequence alignments used in the analyses presented at the CRW Site. Sequence alignments are available in GenBank and AE2 (Macke) formats (Table 2). These alignments will be updated periodically when the number of new sequences is significant. Newer alignments might also contain refinements in the alignments of the sequences. For each alignment, there is a corresponding list of sequences, their phylogenetic placement, and other information about the sequences (see conservation list of sequences for conservation diagrams). At present, only the rRNA alignments are available; the group I and group II intron alignments will be made available in June 2002.
3E. rRNA Introns
The introns that occur in 16S and 23S rRNAs are organized into four preconfigured online tables. These tables disseminate the intron information and emphasize the major dimensions inherent in this data: 1) intron position in the rRNA, 2) intron type, 3) phylogenetic distribution, and 4) number of introns per exon gene.
3E. rRNA Introns Table 1: Intron Position
The introns in rRNA Introns Table 1 are organized by their position numbers in the 16S and 23S rRNAs. The 16S and 23S rRNA position numbers are based on the E. coli rRNA reference sequence (J01695) (see Table 1). The intron occurs between the position number listed and the following position (e.g., the introns between position 516 and 517 are listed as 516). rRNA Introns Table 1 has four components.
The total number of introns and the number of positions with at least one intron in 16S and 23S rRNA are shown in rRNA Introns Table 1A (see highlights below and H-3E.1). The list of all publicly available rRNA introns, sorted by the numeric order of the intron positions, is contained in rRNA Introns Table 1B. This table has nine fields: 1) rRNA type (16S or 23S); 2) the intron position; 3) the number of documented introns occurring at that position; 4) the intron types (RNA classes) for each rRNA intron position; 5) the number of introns for each intron type for each rRNA position; 6) the length variation (minimum # – maximum #) for introns in each intron type; 7) the cell location for each intron type; 8) the number of phylogenetic groups for each intron type, (here, defined using the third column from rRNA Introns Table 3: Phylogenetic Distribution); and 9) the organism name and accession number.
These fields in rRNA Introns Table 1B (H-3E.1) allow for a natural dissemination of the introns that occur at each rRNA site. For example, of the 116 introns (as of December 2001) at position 516 in 16S rRNA, 55 of them are in the IC1 subgroup (H-3E.2); these introns range from 334–1789 nucleotides in length, all occur in the nucleus, and are distributed into four distinct phylogenetic groups. 54 of the introns at position 516 are in the IE subgroup, range from 190–622 nucleotides in length, all occur in the nucleus, and are also distributed into four distinct phylogenetic groups, etc.
Additional information is available in a new window for each of the values in rRNA Introns Table 1B (H-3E.3). This information is retrieved from the relational database management system (see section 4). The information for each intron entry in the new window are: 1) exon (16S or 23S rRNA); 2) intron position in the rRNA; 3) intron type (RNA class); 4) length of intron (in nucleotides); 5) cell location; 6) NCBI phylogeny; 7) organism name; 8) accession number; 9) link to structure diagram (if it is available); and 10) comment.
The number of intron types per intron position are tabulated in rRNA Introns Table 1C (H-3E.4), while the number of introns at each rRNA position are ranked in rRNA Introns Table 1D (H-3E.5). This latter table contains six fields of information for each rRNA: 1) number of introns per rRNA position; 2) number of positions with that number of introns; 3) the rRNA position numbers; 4) total number of introns (field #1 × field #2); 5) the Poisson probability (see rRNA Introns Table 1D for details); and 6) the expected number of introns for each of the observed number of introns per rRNA site.
The highlights from rRNA Introns Table 1 are: 1) As of December 2001, there are 1184 publicly available introns that occur in the rRNAs, with 900 in the 16S rRNA, and 284 in 23S rRNA. These introns are distributed over 152 different positions, 84 in the 16S rRNA and 68 in 23S rRNA. 2) Although 16S rRNA is approximately half the length of 23S rRNA, there are more than three times as many introns in 16S rRNA. However, this bias is due, at least in part, to the more prevalent sampling of 16S and 16S-like rRNAs for introns. 3) The sampling of introns at the intron positions is not evenly distributed (1184/152 = 7.79 introns per position for a random sampling). Instead, nearly 50% (71/152) of the intron positions contain a single intron and 89% (135/152) of the intron positions contain ten or less introns. In contrast, 59% (681/1163) of the introns are located at 9% of the intron positions and the three intron positions with the most introns (943, 516, and 1516 in 16S rRNA) contain 361, or 31% (361/1163), of the rRNA introns. 4) rRNA Introns Table 1D compares the observed distribution of rRNA introns with the Poisson distribution for the observed number of introns. The Poisson distribution, P(x) = e_-μ μ_x _x!_-1, where μ is the mean frequency of introns for positions in a particular exon and x is the target number of introns present at a particular position, allows the calculation of expected numbers of positions containing a particular number of introns. Based upon the observed raw numbers of introns in the 16S and 23S rRNAs, we expect to see no positions in 16S rRNA containing more than five introns and no positions in 23S rRNA containing more than three introns. However, thirty-five rRNA positions fall into one of those two categories. We also see both more positions without introns and fewer positions containing only one or two introns than expected. This observed distribution of rRNA introns among the available insertion positions is extremely unlikely to occur by chance. 5) While a single intron type occurs at the majority of the intron positions, several positions have more than one intron type. A few of the positions that deserve special attention have IC1 and IE introns at the same position (16S rRNA positions 516 and 1199, and 23S rRNA position 2563). The 16S rRNA position 788 has several examples each of IC1, IIB, and I introns.
3E. rRNA Introns Table 2: Intron Type
The introns are organized by intron type, as defined above, in rRNA Introns Table 2 (H-3E.6). The frequency of 16S and 23S rRNA exons, non-rRNA exons, number of intron positions in the 16S and 23S rRNA, cell locations, and number of phylogenetic groups for each intron type are tabulated. The highlights of this table are: 1) Of the 1184 known rRNA introns, 980 (83%) are group I, 21 (2%) are group II introns, and the remaining 183 (15%) are unclassified (see below). While only 2% of the rRNA introns are group II, 62% (728/1180) of the non-rRNA introns are group II. In addition to the group II introns, nearly all of the IC3 introns do not occur in rRNAs. 2) The majority of the rRNA group I introns (851/980 = 87%) fall into one of three subgroups: I (276 introns), IC1 (415 introns), and IE (160 introns). 3) As noted earlier, there are three times as many 16S rRNA group I introns than 23S rRNA group I introns (753 vs. 227). 4) Among the three cellular organelles in eucaryotes, 1010 introns (85%) occur in the nucleus, 133 (11%) in the mitochondria, and 41 (4%) in the chloroplasts. 5) The subgroups IC1, IC3 and IE are only present in the nucleus, while the IA, IB, IC2, ID, and II subgroups occur almost exclusively in chloroplasts and/or mitochondria.
The 183 introns described in rRNA Introns Table 2 as "Unclassified" merit special attention. All of these introns do not fall into either the group I and group II categories; however, two notable groups of introns are included within the "Unclassified" category. The first is a series of 43 introns occurring in Archaeal rRNAs (the Archaeal introns). Thirty-one of the known Archaeal introns are found in 16S rRNA and the remaining twelve are from 23S rRNA exons. The Archaeal introns range in length from 24 to 764 nucleotides, with an average length of 327 nucleotides. The second group contains 121 spliceosomal introns found in fungal rRNAs. 92 spliceosomal introns are from 16S rRNA and 29 are from 23S rRNA; the lengths of these introns range from 49 to 292 nucleotides. A future version of this database will include both of these groups as separate, distinct entries. Both the Archaeal and splicesomal introns occur only in nuclear rRNA genes and tend to occur at unique sites; the lone exception is the spliceosomal intron from Dibaeis baeomyces nuclear 23S rRNA position 787, a position where a group IIB intron occurs in mitochondrial Marchantia polymorpha rRNA. The Unclassified group contains 21 introns that do not fall into any of the four previously discussed categories (group I, group II, Archaeal, or spliceosomal), including all four mitochondrial introns in this group.
rRNA Introns Table 2 expands the presentation by providing links to twenty additional tables (H-3E.7), each of which provides expanded information about a specific intron type. The organism name, exon, intron position, cell location, and complete phylogeny are accessible for each intron from these tables. These online tables are dynamically updated daily as information about new introns is made available.
3E. rRNA Introns Table 3: Phylogenetic Distribution
The distribution of introns on the phylogenetic tree is tabulated in rRNA Introns Table 3A (H-3E.8) and 3B (H-3E.9). rRNA Introns Table 3A reveals the ratio of the number of rRNA introns per rRNA gene for the nuclear, chloroplast, and mitochondrial encoded RNAs for the major phylogenetic groups. The most noteworthy distributions are: 1) The majority (96%) of the rRNA introns occur in Eucarya, followed by the Archaea, and the Bacteria. 2) Only one rRNA intron has been documented in the Bacteria; due to the large number of rRNA gene sequences that have been determined, the ratio of rRNA introns per rRNA gene is essentially zero for the bacteria. 3) The frequency of introns in Archaea rRNAs is higher, with 43 examples documented as of December 2001. Within the Archaea, there is a higher ratio of rRNA introns in the Desulfurococcales and Thermoproteales subbranches in the Crenarchaeota branch. 4) For the three primary phylogenetic groups, the highest ratio of rRNA introns per rRNA gene is for the Eucarya, and for the phylogenetic groups within the Eucarya that have significant numbers of rRNA sequences, the ratio is highest in the fungi. Here, the ratios of rRNA introns per rRNA gene are similar between the nucleus and mitochondria (1.34 for the nucleus, 1.20 for the mitochondria). A significant number of rRNA introns occurs in the plants, with similar ratios of rRNA intron/rRNA gene for the nucleus, chloroplast, and mitochondria (0.36 for the nucleus, 0.38 for the chloroplast, and 0.34 for the mitochondria). In sharp contrast with the fungi and plants, only one intron has been documented in an animal rRNA, occurring within the Calliphora vicina nuclear-encoded 23S-like rRNA (GenBank accession number K02309).
Each of the two special "Unclassified" rRNA intron groups has a specific phylogenetic bias. Archaeal rRNA introns, which have unique sequence and structural characteristics [83], have not yet been observed within the Euryarchaeota or Korarchaeota; in fact, no non-Archaeal introns have been found in Archaea rRNAs to date. Splicesomal rRNA introns have only been reported in 31 different genera in the Ascomycota [84]. rRNA Introns Table 3A also presents the numbers of (complete or nearly so) rRNA sequences in the same phylogenetic groups in order to address the question of sampling bias. Two important caveats to this data must be considered. First, the numbers of rRNA sequences are an underestimate, since many rRNA introns are published with only short flanking exon sequences and do not meet the 90% completeness criterion for inclusion in this rRNA sequence count. The second caveat is that many rRNA sequences contain multiple introns (see rRNA Introns Table 4 and related discussion, below, for more information). Of the 51 phylogenetic group/cell location combinations shown in rRNA Introns Table 3 that may contain rRNA introns, 15 (29%) have a intron:rRNA sequence ratio greater than 1.0, indicating a bias toward introns within those groups. Introns are comparatively rare within the 26 (51%) groups that have a ratio below 0.3; ten of these 26 groups contain no known rRNA introns. Ten (20%) of the groups have intermediate ratios (between 0.3 and 1.0).
A more detailed phylogenetic distribution is available in rRNA Introns Table 3B (H-3E.10). The first three fields contain levels 2, 3, and 4 of the NCBI phylogeny, followed by fields for the genus of the organism, cell location, exon (16S or 23S rRNA), and intron type. Each of these classifications include a link to the complete details (organism name, phylogeny, cell location, exon, intron position, intron number, accession number, and structure diagram (when available)) for the intron sequences in that group.
3E. rRNA Introns Table 4: Number of Introns per Exon
rRNA Introns Table 4 presents the number of introns per rRNA gene (H-3E.11). While more than 80% of the documented rRNA genes do not have an intron, 646 16S and 182 23S rRNAs have at least one intron. Approximately 75% (623) of these genes have a single intron, 15% (127) have two introns, 0.5% (40) have three, 0.25% (20) have four, 0.1% (11) have five, two rRNA genes have 6, 7 or 8 introns, and one rRNA gene has 9 introns.
To determine the amount of bias in the distribution of introns among their exon sequences, the Poisson distribution (here, μ is the mean frequency of introns for a particular exon and x is the target number of introns per rRNA gene) has been used to calculate the number of rRNA sequences expected to contain a given number of introns (rRNA Introns Table 4). Based upon this data, no rRNA sequences are expected to contain four or more introns; in fact, we see 38 sequences that contain these large numbers of introns. The observed numbers of sequences exceed the expected values for all but one category: fewer rRNAs contain only one intron than expected.
The two molecules (16S and 23S rRNA) show a differing trend with respect to cell location for those sequences containing large numbers of introns. In 16S rRNA, only nuclear genes (ten) have been observed to contain five or more introns; indeed, of the 57 genes containing three or more introns, only two are not nuclear (both of these are mitochondrial). In 23S rRNA, the trend is both opposite and weaker; of the thirteen rRNA sequences containing four or more introns, five are nuclear (containing five introns), with four chloroplast and four mitochondrial genes comprising the remaining eight sequences.
rRNA Introns Table 4 provides access to seventeen additional tables (H-3E.12), which each present the complete information for every intron within a particular class (e.g., 16S rRNA genes containing two introns), grouped by their exons. As with the other online tables, this information will be updated daily to reflect new intron sequences that are added to this database.
The final components of the "rRNA Introns" page are 16S and 23S rRNA secondary structure diagrams that show the locations for all of the known rRNA introns (H-3E.13). The information collected here on the "rRNA Introns" page is the basis for two detailed analyses that will be published elsewhere: 1) the spatial distribution of introns on the three dimensional structure of the 16S and 23S rRNA (Jackson et al., manuscript in preparation); and 2) the statistical analysis of the distribution of introns on the rRNA (Bhattacharya et al., manuscript in preparation).
3F. Group 1/11 Intron distributions
For the CRW Site project, we collect group I and II introns and all other introns that occur in the ribosomal RNA. The "Intron Distribution Data" page contains three tables that compare intron types, phylogeny, exon, and cell location.
Intron Distribution Table 1 maps "Intron Type" vs. "Phylogeny" (and "Cell Location;" H-3F.1). Group I and II intron data are highlighted with yellow and blue backgrounds, respectively. The phylogenetic divisions are also split into the three possible cellular locations (nuclear, chloroplast, and mitochondria). A few of the highlights are:
- the Eukaryota contain the majority (2218 / 2349 = 94%) of the introns in the CRW RDBMS. 2) The Archaea have 42 introns that have unique characteristics and are called "Archaeal introns." 3) Group I introns are present in eukaryotes (nuclear-, chloroplast-, and mitochondrial-encoded genes) and in Bacteria. Group II introns have only been observed in Bacteria and in Eukaryotic chloroplast and mitochondrial genes.
Intron Distribution Table 2 shows "Intron Type" vs. "Exon" (and "Cell Location;" H-3F.2). Again, group I and II intron data are highlighted with yellow and blue backgrounds, respectively. In this table, the exon types are split into the three possible cellular locations (nuclear, chloroplast, and mitochondria). As of December 2001, the most obvious trend is that the exons with the most Group I introns are 16S rRNA (900), leucine tRNA (337), 23S rRNA (284), ribosomal protein S16 (214), and ribosomal protein L16 (152).
Intron Distribution Table 3 compartmentalizes the intron data by "Phylogeny" and "Exon" (and "Cell Location;" H-3F.3). In this table, color is used to highlight the three phylogenetic domains (Archaea in yellow, Bacteria in blue, and Eukaryota in green). As in Intron Distribution Table 2, the exon types are split into the three possible cellular locations (nuclear, chloroplast, and mitochondria).
Each of these three tables is dynamically created from a specific series of RDBMS queries on a daily basis. As of December 2001, links connecting to the specific RDBMS results are not available.
4. Data access systems
For our first generation of online comparative RNA structure databases (16S rRNA [46, 47], 23S rRNA [48–52], and group I Intron [33]), we organized the rRNA and group I intron secondary structures into a simple static set of manually-generated HTML pages. The structure diagrams were organized first by RNA type (for the rRNAs; e.g., all 16S rRNA diagrams were grouped together) or structural subtype (for group I introns; e.g., IC1) and then by the phylogenetic order of the organisms. This type of presentation is acceptable, although not ideal, for a small number of entries. However, it is grossly inadequate and inefficient for larger numbers of entries and more fields of information. Thus, with the anticipation that our database of comparative RNA information would grow significantly, the need to associate more fields of information with each entry, to automatically and dynamically generate the HTML output for all queries of the database, and the ability to search our database for entries with specific attributes in many fields and to sort those fields in the output with different priorities, we have developed a relational database management system (RDBMS) that is built on the MySQL database program (see Materials and Methods).
Our goal was to create a system that would allow for the following examples of dynamic searches of our CRW RDBMS. Find and output:
- A.
Homo sapiens 5S, 16S, and 23S rRNA entries. - B.
Enteric bacterial rRNA sequences and/or secondary structure diagrams. - C.
- Tunicate and 2) Coelacanth rRNA sequences.
- D.
All 23S rRNA sequences. Sort output by four methods: 1) organism name, alphabetically; 2) phylogenetic classification; 3) sequence length; and 4) first by cellular location, then by phylogenetic classification. - E.
Group I introns that occur: 1) in Saccharomyces cerevisiae, 2) in mitochondria, 3) in the exons 16S and 23S rRNA, 4) at position 516 in 16S rRNA, 5) in the IE subgroup, 6) in the IE subgroup at 16S rRNA position 516.
Each sequence and structure entry has the following fields or attributes: organism name, NCBI phylogeny, common name, cell location, RNA type, RNA class, sequence length, accession number, intron number, intron position, exon, open reading frame, link to secondary structure diagram (if it exists), and comment. An abbreviated explanation for each of these attributes is given in Table 5; a full explanation is available online at the RDBMS page.
The RDBMS and the data that it contains are accessed by several different graphical interfaces. One interface, the "Index of Available RNA Sequences and Structures," was described in Section 3, "Sequence and Structure Data." The SQL queries on this page were predetermined and restricted. The "Index" contains the number of sequences and structures for different molecules and phylogenetic groups. Clicking a link searches the current database for all entries that satisfy that specific query (e.g., bacterial 16S rRNA structures) and dynamically generates the output. The SQL queries for Sections 3E (rRNA Introns) and 3F (Group I/II Intron Distribution) are also preset. In contrast with the predetermined and restricted searches available on these pages, we have also developed two different graphical interactive interfaces for Section 4, "Data Access Systems," that allow the user to define and implement their own search of the same information in our relational database management system. The first one, called "Standard," is the least restrictive and allows the user to search for any values present in one or a combination of the attributes and to sort the output on any combination of attributes (see Section 4A below). The second one is semi-restrictive and allows the user to navigate through the phylogenetic tree to search for those entries that are within specific phylogenetic groups (see Section 4B below).
4A. RDBMS (Standard)
The "Standard" interface is the most fundamental of our interfaces to the CRW RDBMS information. While the restricted, specialized interface to the RDBMS information in Section 3A requires minimal instruction to use, the standard interface, with its ability to cull out all arrangements of information from the different fields with sophisticated search queries and output field sortings, requires a quick lesson for its operation. The selection process has three stages: 1) selection of attribute fields to display; 2) determination of values for the search; and 3) adjustment of the output field sort order.
A detailed explanation for each of the attributes is available from the links to the attribute names. This information is shown in the right frame. Additional examples of this system are available online.
Step 1. At the onset, the user selects the fields to be displayed on the screen and then clicks the "Go" button. While the user can select the individual fields (e.g., "Organism" or "Phylogeny"), for most applications the "QrRNA" (query rRNA), "Qintron" (query intron), or "All" options will automatically click the appropriate fields that are most important for searching for ribosomal RNA or group I and II intron entries.
Step 2. Select values for the fields or attributes. The acceptable values for the attributes in our RDBMS system are shown on the main frame of the query page (for list- and button-driven fields) or, for text input fields, can be determined with the "V" (values) button on the right side of the main frame; the results are displayed in the right frame (see Figure 3 and H-4A.1).
- The values for cellular location are Chl (chloroplast), Cya (cyanelle), Mit (mitochondria), Nuc (nuclear), and Vir (viral); each can be selected by simply checking the box to the left of its name.
- The values for the attributes RNA Type, ORF (open reading frame), Secondary Structures (entries with/without secondary structure diagrams), Results/Page, and Color Display are also displayed on the main frame, and can be selected by clicking the appropriate box or button.
The values for other attributes such as RNA Class, Sequence Length, and Exon can be determined by selecting one or more of the values in the scroll box. The values for these attributes can also be found by clicking the "V" button associated with each attribute. For example, clicking on the "Exon" "V" button will reveal, in the right frame, all of the exons that are contained in our database. The same exons are present in the scroll box.
- The values displayed for any one attribute are dependent on the settings of the other attributes. For example, when only rRNA is selected for the "RNA Type," then there are no values for "Exon." All of the possible exon values are displayed when "Intron" is the selected "RNA Type," while only a subset of the possible exon values are shown when Mit (mitochondria) is the selected "Cell Location." Note: no selection for an attribute signifies to this system that all of the values are possible.
The values present in our database for the attributes "Organism," "Phylogeny" (except for the first level – Archaea, Bacteria, and Eukaryota – that can be selected from the main frame), "Common Name" (except for the first level: "Animals," "Fungi&Plants," "Protists"), "Accession Number," "Intron Position," and "Comment" can only be observed in the right frame after clicking the "V" button.
- The values selected with the mouse in the right frame will appear in the appropriate attribute field.
- The values for each attribute are dependent on the settings for the other attributes. For example, if there are many values for the "Organism" field, selecting Archaea in the "Phylogeny" field will reduce the number of names in the "Organism" field to just those that are in this phylogenetic group.
- The number of possible values for an attribute can also be constrained by entering only part of a value in the field. For example, typing 'Esch' in the "Organism" field will output several organism names that contain 'Escherichia' when the "V" button is clicked. Typing "coli" in this field will list all organism names that contain "coli," as either part of a name or a complete word.
- Note that the system is case sensitive for all fields except "Common Name." The text 'esch' in the same "Organism" field will not output 'Escherichia' in the right frame.
The "Phylogeny" field with the values frame on the right was developed to allow the user to navigate through the phylogenetic tree. The information for the "Phylogeny" and "Common Name" fields is downloaded from the NCBI (see Materials and Methods; this information is downloaded daily to assure that we have the most current version of this data). There are two general modes of operation.
For mode one, you can systematically navigate through the phylogenetic tree to the selected goal point. For example, to get to the last phylogenetic group that contains Homo sapiens and gorillas, the user would click on the "Eukaryota" phylogeny button, then click on the "Fungi/Metazoa group" link in the right frame, followed by the "Metazoa," "Eumetazoa," "Bilateria," "Coelomata," "Deuterostomia," "Chordata," "Craniata," "Vertebrata," "Gnathostomata," "Teleostomi," "Euteleostomi," "Sarcopterygii," "Tetrapoda," "Amniota," "Mammalia," "Theria," "Eutheria," "Primates," "Catarrhini," and "Hominidae" links. The phylogenetic group Hominidae contains the genera Gorilla, Pan (chimpanzees), Pongo, and Homo (see Figure 3, H-4A.1, and H-4A.2). This type of navigation is useful when you know the links that will get you to the desired goal point; otherwise, mode two can help you jump to the appropriate node in the phylogenetic tree.
For the second mode, you type all or part of the name of an organism or phylogenetic group that is close to the phylogenetic node you want. For example, type "Homo sapiens" in the "Phylogeny" field and press the "V" button in the "Phylogeny" field. The right frame will display a few names; from these, select "Homo sapiens." The right frame now contains the entire phylogenetic path from the base of the tree to Humans (Figure 3 and H-4A.1).
The "Common Name" attribute can also help identify organism names in the CRW RDBMS. As with the phylogeny operation, two general modes for determining the values are available. For the first, the user would type the presumed common name in the "Common Name" field, and click the "V" button. A few general examples are: worm, fish, cat, dog, and human. More specific examples are: common earthworm (Lumbricus terrestris), European polecat (Mustela putorius), and duckbill platypus (Ornithorhynchus anatinus). These names must be in the "Common Name" database for the sequence entry to be identified with this method. In contrast, the second mode is intended to identify larger groups of organisms. The three buttons in the "Common Name" field ("Animals," "Fungi&Plants," "Protists;" H-4A.3) each reveal various low-level common names in the right frame that are arranged in a pseudo-phylogenetic structure. For example, a few of the lower animals (sponges, flatworms, etc.) are listed when the "Animals" button is pressed, in addition to the Protostomia, Deuterostomia, and organisms nested within these groups (Arthropoda, chordates, vertebrates, Mammals, etc.; H-4A.3). Accordingly, the "Fungi&Plants" and "Protists" buttons reveal the major groups of organisms within their respective groups. For the latter mode of operation, the user selects one of these common names, such as "Mammals." The phylogeny for this group then appears in the same right frame (cellular organisms, Eukaryota, Fungi/Metazoa group, Metazoa, Eumetazoa, Bilateria, Coelomata, Deuterostomia, Chordata, Craniata, Vertebrata, Gnathostomata, Teleostomi, Euteleostomi, Sarcopterygii, Tetrapoda, Amniota, Mammalia), along with the two phylogenetic groups within the Mammals (Mammalia), Prototheria and Theria. Another example is the common name "Mosses" in the Fungi&Plants. Selecting "Mosses" brings up the phylogeny for the Bryophyta. Note that these common names (i.e., "mammals" or "mosses") do not appear in the common name field in the output for the sequence entries that are within the Mammalian or Bryophyta phylogenetic groups. Thus, the common name field could be very useful to identify organisms and phylogenetically related organisms when you don't know their genus/species organism name or the phylogeny for that group of organisms.
Step 3. The last, critical step before submitting a query is to select the sort order for the attributes in the output. While a query will yield the same number of results with any sort order, the choice of sort order can make answering questions easier. Take, for example, a search for all Eucarya rRNA entries. By default, the entries are sorted alphabetically first by their phylogenetic classification, followed by organism name, cell location, and last by their RNA class. In contrast, the sort orders <phylogeny, organism name, cell location, and RNA class> and <organism name, RNA class, phylogeny, and cell location> produce significantly different orders and overall arrangements for the same set of entries (see online examples); the second sorting is more useful when searching for a particular organism, since its exact location on the phylogenetic tree may not be known to the user. The output page (H-4A.2) reveals the search strategy and attribute sort order at the bottom of the page. The default sort order for the attributes is shown on the "S" (or sort) buttons on the right side of the main frame (Figure 3 and H-4A.1). The sort order is changed by simply clicking the "S" buttons in the order the attributes are to be sorted. The resulting sort order for the attributes are shown in the small text box to the left of each attribute's S button; alternatively, you can type numbers into these boxes to set the sort order. The alphabetical/numerical order for any attribute can be reversed (z -> a, high number -> low number) by checking the box in the "R" (or reverse) column to the right of the Sort buttons. Finally, the sortings can be reset to the default values by clicking the "Sort Reset" button at the top of the query page.
Before submitting the query, a few attributes deserve more attention.
- Secondary Structures: a comparative secondary structure model has been developed for more than 400 of the sequence entries (see Section 3). The 'secondary structure' attribute near the bottom of the query page is an option to output all sequence and structure entries, only those entries with a secondary structure, or entries without a secondary structure diagram.
- Results/Page: the number of entries per output page can be modulated. While the system defaults to 50 entries per page, the maximum number of entries per output page can be set to 20, 100, 200, and 400. The user can scroll to those entries that do not appear on the first page by selecting the "Next" button on the left bottom frame in the output window and use the "Previous" button in the same frame to move toward the first page, as necessary.
- Color Display: to help distinguish the organism names on the output pages, the entries have the same color when the organism names are the same. The colors (pink and white) alternate for changes in the organism names in the output entries.
- Group ID and Group Class: these two attributes are currently not fully functional; thus, we do not encourage their use at this time.
- RNA Type/Class: currently, we do not have data entries for the following RNA Types and Classes: mRNA, tRNA, SnRNA, and Other.
After clicking the submit button at the top or bottom of the query page, a new window will open. This window distributes the results into three frames (H-4A.2). The main frame contains the sequence and structure entries that satisfy the search query. The frame in the lower left indicates the number of entries shown in the window and the entry numbers currently shown, and, if necessary, contains buttons to scroll to the next or previous set of entries. The third frame at the bottom middle-right displays the total number of entries that satisfy the query, the search strategy and the sort order for this query.
The three formats for the secondary structure diagrams, PostScript, PDF, and BPSEQ (see Section 1A and the online help from the "Secondary Structure" and "StrDiags" links on the RDBMS query and results pages) can be retrieved from the results window. The system defaults to PostScript when the secondary structure link is clicked; PDF or BPSEQ files can be obtained instead from the structure link by selecting the corresponding radio button at the top left section of the main frame. An explanation of the structure link names (d.5, d.l6, d.235, d.233, b.Il, and a.I2) and the longer names that are associated with the downloaded structure files is also available online.
The GenBank accession number for each entry is a link to a new window that retrieves the specified entry from NCBI. Sequence entries with more than one GenBank number contain a "m" to the right of the accession number. Clicking the "m" link opens a new window with all of the GenBank numbers associated with this sequence.
Each entry is associated with a NCBI phylogeny listing that can be retrieved in a new window by clicking the "m" button in the Phylogeny column. This listing also contains the known common names associated with each level of the phylogenetic tree (H-4A.4). The phylogeny for all of the entries in the results window is available in a new window when the "M" button in the header line of the phylogeny field is clicked.
4B. RDBMS (PhyloBrowser)
The PhyloBrowser interface to the CRW RDBMS was developed to facilitate the identification and retrieval of sequence and structure entries that are associated with specific phylogenetic groups. While the Standard interface will reveal all sequence entries for any one phylogenetic group, it does not show the phylogenetic groups that do not have the requested sequences; the PhyloBrowser interface displays the entire phylogenetic tree, including those branches that do not have corresponding entries. This interface is based on the Taxonomy Browser developed by NCBI http://www.ncbi.nlm.nih.gov/Taxonomy/taxonomyhome.html/ and uses the NCBI taxonomy database [60, 61]. Here, we describe the PhyloBrowser interface, ways to navigate through the phylogenetic data, and how to retrieve RNA information using this system.
The PhyloBrowser uses three frames (Figure 4 and H-4B.1). At the bottom of the page is the Results Frame (white background), which displays the selected portion of the phylogenetic tree and any RNA information. In the upper left is the Selection Frame (pink background), where the user can select the phylogenetic and RNA information shown in the Results Frame. Help is provided in the Help Frame, at the upper right (blue background).
Figure 4
RDBMS (PhyloBrowser) basic phylogenetic search screen, showing two additional levels of phylogeny.
Starting at the root, the entire phylogenetic tree can be navigated with this system. The base phylogenetic level name is shown in green. The number of phylogenetic levels displayed (below the base level) can be modulated from one (the default) to five levels using the "Display Phylogenetic Levels" control in the Selection Frame. The phylogenetic level number for each group is shown in red preceding the phylogenetic group name, and common name information, where available, is shown in black text in parentheses after the group name. Each phylogenetic group name is a link that reveals additional phylogenetic levels (Figure 4 and H-4B.1), allowing the user to navigate onto the branches of the phylogenetic tree.
In addition to this mode of transversing the phylogenetic tree, starting at the root and knowing the pathway to the desired end point, this system has the facility to jump to specific places in the phylogenetic tree. The user can enter a partial or complete scientific or common name in the white text field in the lower, purple-colored panel of the Selection Frame (e.g., "human;" see H-4B.2). Once the appropriate scientific or common name radio button is set, different names that satisfy the user-entered text can be viewed in the Results Frame by checking the "View" box. Clicking the appropriate name in the Results Frame will enter that name into the text field; unchecking the "View" check box and clicking "Submit" will reveal the phylogenetic branch for this organism (H-4B.3).
To navigate toward the root of the phylogenetic tree, click the "Parents" button in the Selection Frame. This will open a new window with the complete NCBI phylogeny from the root to the level of the organism of interest. This window (H-4B.4) also reveals the phylogenetic level number and common names. Simply clicking on a node name in this window (e.g., the "Eutheria" node in H-4B.4) will reveal this section of the phylogenetic tree in the Results Frame.
RNA information can be mapped onto the phylogenetic tree in the Results Frame at any time. In the white panel in the Selection Frame, the user can choose to view six RNA types (5S, 16S, and 23S rRNA; group I, II and other introns) from five cellular locations (chloroplast, cyanelle, mitochondria, nucleus, and viral) by checking the boxes to the left of the desired selections. After clicking the white "Submit" button, all entries that satisfy the RNA type and cell location selections are mapped onto the phylogenetic tree in the Results Frame (H-4B.3). There, the numbers of sequences and structure diagrams available in our CRW RDBMS are shown adjacent to each phylogenetic group name at all levels of the phylogenetic tree and enclosed in brackets; the format of this information for each individual RNA type is: [cell location, # sequences/# structures, cell location, # sequences/# structures, ...]. The RNA types are indicated in different colors (rRNA: 5S, green; 16S, red; 23S, blue; introns: group I, black; II, brown; other intron types, magenta) and the cell locations are abbreviated (N, nucleus; M, mitochondria; C, chloroplast; Y, cyanelle; V, viral). These values in brackets link to the Standard RDBMS results page, as described in the previous section, and allow the user to view the available sequence and structure information. The PhyloBrowser page (H-4B.3) reveals the "Homo sapiens" phylogenetic group with the number of sequences and structures available in our CRW RDBMS for RNA types (e.g., 16S and group I introns) that are present in the selected cell locations (e.g., Chl, Mit, Nuc).
Additional documentation for the use of this page is available from the PhyloBrowser page. A short description is displayed in the top-right frame by placing the mouse over each of the attributes ("Molecule," "Cell Location," "Phylogenetic Levels," "Go to Parents," "Query," and "Acknowledgement"). Additional information for each of these attributes is then displayed in a new window by clicking on either the attribute link or the additional information link in the top-right frame (Figure 4 and H-4B.1).
4C. RNA Structure Query System
Currently, we are unable to reliably and accurately predict an RNA structure from its underlying sequence due in part to the lack of more fundamental RNA structure rules that relate families of RNA sequences with specific RNA structural elements. Given this limitation, we have utilized comparative analysis to determine that RNA structure that is common to a set of functionally and structurally equivalent sequences. This analysis, as mentioned earlier, is very accurate: nearly 98% of the basepairings in our 16S and 23S rRNA comparative structure models are present in the high-resolution crystal structures for the 30S [44] and 50S [45] ribosomal subunits. In the process of predicting these comparative structure models, we have determined a large number of 5S, 16S, and 23S rRNA and group I intron comparative structure models from sequences that are representative of all types of structural variations and conservation. Thus, with the correct rRNA structure models and a large sampling of structurally diverse structure models, we now want to decipher more relationships between RNA sequences and RNA structural elements. Toward this end, we developed a system for the identification of biases in short sequences associated with simple structural elements in our set of comparative structure models. The first set of examples reveals a sampling of structure-based sequence biases. Recently, we utilized this system to identify and quantitate the following biases for adenosines in the Bacterial 16S and 23S rRNA covariation-based structure models [63]: 1) approximately 2/3 of the adenosines are unpaired; 2) more than 50% of the 3' ends of loops in the 16S and 23S rRNA have an A; 3) there is a bias for adenosines to be adjacent to other adenosines (66% of these are at two unpaired positions, and 15% of these are at paired/unpaired junctions); and 4) the majority of the As at the 3' end of loops are adjacent to a paired G. These results were discerned with this system and are shown in part in Figure 5 and H-4C.
Figure 5
Analysis of the Bacterial 16S and 23S rRNA structure models using the "RNA Structure Query System." The entire system (selection frame and results) is shown with the results for the distribution of single nucleotides, sorted in order of decreasing prevalence in unpaired regions.
This RNA sequence/structure query system has three primary fields of input to be selected by the user: the RNA type, phylogenetic group/cell location, and the nucleotide/structural element. The options for each of these fields are listed in Table 5. The system currently supports four RNA types (5S, 16S, and 23S rRNAs, and group I introns) and five phylogenetic groups/cell locations (Bacteria, Archaea, Eucarya nuclear-encoded, mitochondrial, and chloroplast). Any combination and number of RNA types and phylogenetic/cell location groups can be selected, although at least one RNA type and one phylogenetic/cell location group must be selected. The bacterial 16S and 23S rRNAs were selected for the examples in Figure 5 and H-4C. Five nucleotide categories are searchable: single nucleotides, (two) adjacent nucleotides, base pairs, three nucleotides, and four nucleotides. Each category can be searched against a defined set of structural elements, as outlined in Table 5. The structural elements for these nucleotide categories are based on 1) positions that are paired and unpaired and 2) positions at the center or 5' and 3' ends of helices and loops.
Table 5 Attributes for the "RNA Structure Query System." The 5' and 3' ends of helices and loops are based on the global orientation determined from the 5' and 3' ends of the entire RNA molecule.
The sorting function dynamically ranks the nucleotide patterns. The resulting output reveals, for any of the selected structural elements, the most frequent nucleotide pattern, followed by other patterns in descending order to the least frequent nucleotide pattern. For the "A Story" example mentioned earlier, adenosine is the most frequent nucleotide at unpaired positions (42.64%), followed by G (23.6%), U (21.27%), and C (12.49%) (Figure 5 and H-4C.1). These values are contained in the orange columns, and reveal the percentages for each of the nucleotides within each of the structural elements listed (i.e., paired, unpaired, etc.). This same figure reveals that 53.5% of the 3' end of loops contain an A. The unpaired to paired ratio is shown in yellow in Figure 5 and H-4C.1; this ratio is greatest for adenosines, where the value is nearly two (i.e., there are two unpaired adenosines for every A that is paired), and lowest for C, where less than three out of ten cytosines are unpaired. In contrast with the percentage values in the orange boxes that reveal the percentage of nucleotides within each structural element, the percentages in the green boxes reveal the distribution of nucleotides in different structural elements for each nucleotide. For example, 33.76% of the adenosines are paired, while 66.24% are unpaired. In contrast, 77.71% of the C's are paired and only 22.29% of the C's are unpaired.
The most common adjacent nucleotides in any structural environment in the Bacterial 16S and 23S rRNAs are GG (9.86%; H-4C.2), while in loops the most common dinucleotides are AA (19.2%; H-4C.3), followed by GA (13.35%), UA (9.821%), AU (6.703%), etc. The most frequent adjacent nucleotides at the 3'loop-5'helix junction are AG (24.99%; H-4C.4), followed by AC (13.28%), GG (8.28%), etc. For the adjacent AA sequences, nearly 75% occur in loops, while approximately 12% of the AA sequences occur in helices, another 12% occur at the 3'loop-5'helix junction, and less than 5% occur in 3'helix-5'loop junctions. Thus, these analyses of single and adjacent nucleotides reveal several strong biases in the distribution of nucleotides in different structural environments.
The top section of the output page (Figure 5 and H-4C.1) displays the types of data (RNA molecules and phylogenetic/cell location groups) that were selected and analyzed. This section also reveals the number of structure models that were analyzed; 175 16S and 71 23S rRNA structure models were analyzed in Figure 5 and H-4C.
A few of the other biases in the distribution of nucleotide patterns that were determined with this sequence/structure query system of our comparative structure models are displayed in Table 6. A more detailed accounting of this information is available online.
Table 6 Significant values from the "RNA Structure Query System." The 5' and 3' ends of helices and loops are based on the global orientation determined from the 5' and 3' ends of the entire RNA molecule. Values are for the Bacterial 16S and 23S rRNA comparative structure models.
Auxiliary components of the CRW site
In addition to the sections described above, the CRW Site also includes online appendices to work published elsewhere. The "Structure, Motifs, and Folding" section presently contains three RNA motif projects ("U-Tum" [62], "A Story" [63], and "AA.AG@helix.ends" [64]) and two RNA folding projects ("16S rRNA Folding" [65] and "23S rRNA Folding" [66]). In the "Phylogenetic Structure Analysis" section, additional information for three publications is available: "Mollusk Mitochondria" [67], "Polytoma Leucoplasts" [68], and "Algal Introns" [69].
Conclusions
Nearly 10 years ago, our initial goals for our RNA web page was to disseminate some of the comparative information we collected and analyzed for our prediction of 16S and 23S rRNA structure with comparative analysis. With dramatic increases in the number of ribosomal RNA sequences, we developed a relational database system to organize basic information about each sequence and structure entry to maintain an inventory of our collection, and to retrieve any one or set of entries that satisfy the conditions of the search. In parallel, with the significant advancements in computational and networking hardware and software, our need for more detailed and quantitative comparative information for each RNA molecule under study, and our interest in studying more RNA molecules beyond 16S and 23S rRNA, we have greatly expanded our web site, and named it the "Comparative RNA Web" (CRW) Site.
The major types of information available for each RNA molecule are:
- the current comparative RNA structure model;
- nucleotide and base pair frequency tables for all positions in the reference structure;
- secondary structure conservation diagrams that reveal the extent of conservation in the RNA sequence and structure;
- representative secondary structure diagrams for organisms from phylogenetic groups that span the phylogenetic tree and reveal the major forms of structural variation;
- a semi-complete/partial collection of publicly available sequences that are 90% or more complete; and
- sequence alignments.
At this time, we maintain the most current comparative sequence and structure information about the 16S and 23S rRNA. The other RNA molecules we maintain (5S rRNA, tRNA, and group I and II introns) are not as advanced at the time of this writing.
Our future aims for the CRW Site are to: 1) maintain a complete collection of sequences in our database management system for each of the RNAs under study; 2) once or twice a year, release new sequence alignments that contain A) improvements (if necessary) in the positioning of the sequences that are associated with similar structural elements, and B) increases in the number of aligned sequences; 3) generate more secondary structure diagrams for sequences that span the phylogenetic tree and reveal all forms of structural variation; 4) generate more secondary structure conservation diagrams and nucleotide and base pair frequency tables for more phylogenetic groups (e.g. Fungi: Basidiomycota, Ascomycota, and Zygomycota); 5) update the structure models when warranted by the analysis; 6) update current nucleotide and base pair frequency tables when the alignments they are derived from have been updated, and generate more frequency tables for more phylogenetic groups (see "4)" above); 7) add new types of comparative RNA sequence/structure information and new modes of presenting the data; and 8) analyze more types of RNA molecules from a comparative perspective, and present this data in the same formats utilized for the RNA molecules currently supported.
Materials and Methods
Sequence collection
The majority of the sequence alignments presented at the CRW Site were assembled in the Gutell laboratory. The alignments that were based on another laboratory's initial effort and enlarged and refined for the CRW project are: 1) the prokaryotic (Archaea and Bacteria) alignments for 16S rRNA [85]; 2) the 5S rRNA alignments [55]; and 3) the tRNA alignments [81]. The group I and II intron alignments were originally based upon sequences collected by Michel [32, 34].
New rRNA and intron sequences were found by searching the nucleic acid sequence database at GenBank using the NCBI Entrez system http://www.ncbi.nlm.nih.gov/Entrez/ at least once per week with appropriate search criteria (e.g., "rrna" [Feature key] and "intron" [Feature key] to find introns that occur in rRNA). While the majority of the RNA sequences of importance to this database are available online at GenBank, a few sequences are only available in the literature (e.g., the Urospora penicilliformis intron [86]) or in a thesis; these sequences were manually entered into the appropriate sequence alignment. A few sequences were found in GenBank with the sequence similarity searching program BLAST [87]. At this time, we are only trying to identify all sequences that are more than 90% complete since all sequences that are less than 90% complete are not currently retrieved with the CRW RDBMS.
Deviations in GenBank entries
The majority of GenBank entries contain accurate annotations of the RNAs. However, some GenBank entries deviate from this norm in a variety of ways. In some entries, the presence of the rRNA was not annotated and the rRNA was found by searching for short sequences that are characteristic of that rRNA (a few examples). Sometimes, intron sequences are not annotated and were discovered during the alignment of the corresponding rRNA exons (e.g., the unannotated intron in the uncultured archaeon SAGMA-B 16S rRNA (AB050206) and many Fungi, including AF401965 [88]). Other GenBank entries contain incorrect annotations for the RNAs; the boundaries may be misidentified by a small or large number of nucleotides.
RNA sequence alignment and classification of intron sequences
Alignment and determination of intron-exon boundaries
The sequence alignments used for this analysis are maintained by us at the University of Texas; these alignments, containing all publicly available sequences used in the analysis, are or will be available from the CRW Site http://www.rna.icmb.utexas.edu (Table 2). rRNA, Type 1 tRNA, and intron sequences were manually aligned to maximize sequence and structural identity using the alignment editor AE2 (T. Macke, Scripps Clinic, San Diego, CA). The rRNA alignments are sorted by phylogeny and cell location, the intron alignments are sorted by subgroup, exon, insertion point (for rRNA introns), and phylogeny, and the tRNA alignments are sorted by aminoacyl type and phylogeny. Alignment of the rRNA exons (when available) between closely-related sequences provided an independent evaluation of the intron-exon borders for each intron-containing rRNA sequence; the large number of rRNA sequences in our collection and the high level of sequence conservation at intron insertion points provide great confidence in this evaluation.
Classification of introns
Group I and II intron sequences were classified into one of the structural subgroups defined by Michel [32, 34] or the more recently determined subgroup IE [82] based upon sequence and structural homology to previously-aligned sequences. Uncertainties in these assignments come from two main sources. First, some introns are referred to in rRNA GenBank entries without the intron sequence being provided; in these cases, we represent the intron as having length "NSEQ" (No SEQuence information) and accept the authors' major intron classification (e.g., group I or group II) but not the specific intron type (e.g., if an author classified an intron as IA1 and did not publish the sequence, our system designates its type as "I"). In the second case, we do have sequence information but cannot fully classify the intron with confidence; here, we provide the most plausible classification. The classifications "I" and "II," respectively, are group I and II introns of undefined subtype. An intron described as "IB' has the characteristic features of the IB subgroup but cannot be subclassified as IB1, IB2, IB3, or IB4. Those introns that do not belong to either group I or group II are generally classified as "Unknown" in the "RNA Class" field (see Section 4A and Table 5); included in this category are the Archaeal and spliceosomal introns. At present, the Archaeal and spliceosomal introns are identified with the phrases "Archaeal" and "spliceosomal," respectively, in the Comment field of the RDBMS; a standard designation for these introns will be added to a future version of the system. Although the introns in our collection have been judiciously placed into one of the intron subgroups and are roughly correct, these intron placements will be reanalyzed to assure the accurate assignment of subgroups.
Identification of unannotated or misannotated introns, with examples
Some examples of introns that were identified or clarified by the alignment process are: 1) Aureoumbra lagunensis (U40258; the intron was annotated as an insertion); 2) Exophiala dermatitidis (X78481; the intron was not annotated); and 3) Chara sp. Qiu 96222 (AF191800; the intron annotations were shifted approximately 15 positions toward the 5' end of the rRNA sequence).
About TBD and NSEQ
Information that could not be determined either from the GenBank entries or by using these methods is represented in the RDBMS system as TBD (To Be Determined). When a sequence is known but not available (for example, when an intron is inferred from a rRNA GenBank entry), the sequence length and percent completeness are instead represented as NSEQ (No SEQuence), to show that the sequence itself is not available.
Database System
Contents of the RDBMS (general and intron-specific)
The relational database management system (RDBMS) available from the Comparative RNA Web Site http://www.rna.icmb.utexas.edu described in this work utilizes the MySQL engine http://www.mysql.com/. The system contains vital statistics for each sequence (Table 4). The primary fields are: 1) organism name; 2) complete phylogeny; 3) cell location; 4) RNA type (general category; e.g., rRNA or intron); 5) RNA class (more detailed identification; e.g., 16S or IC1); 6) GenBank Accession Number (linked to GenBank); and 7) secondary structure diagrams for selected sequences. Intron-specific data stored in the system are the exon, intron number (index for multiple introns from a single exon), intron position (for rRNA introns only: the E. coli (GenBank Accession Number J01695) equivalent position number immediately before the intron), and open reading frame presence. Note that only sequences that are at least 90% complete are made available through this system. The majority of this data is manually entered into the database system; one exception is the complete NCBI phylogeny database [60, 61], which is automatically downloaded and incorporated into this system daily so that all RDBMS entries appear using the current NCBI scientific name for a given organism. Changes to the RDBMS phylogeny data are identified automatically during the incorporation process and then updated manually. Any changes made to the data become available to the public on the next day.
Secondary Structure and Conservation Diagrams
Secondary structure and conservation diagrams were developed entirely or in part with the interactive graphics program XRNA (Weiser & Noller, University of California, Santa Cruz). The PostScript files output by XRNA were converted into PDF using ghostscript (version 7.00; http://www.cs.wisc.edu/~ghost/index.htm).
Computer details
Hardware and software used
The Comparative RNA Web Site http://www.rna.icmb.utexas.edu is hosted on a Sun Microsystems Enterprise 250 dual-processor server. Apache web server version 1.3.20, from the Apache Software Foundation http://www.apache.org/, provides the site's connectivity interface. The MySQL database (version 3.23.29; http://www.mysql.com/) provides the RDBMS functions. Web site statistics are collected using webalizer (version 2.01; http://www.mrunix.net/webalizer/).
Authentication system
The Comparative RNA Web Site has instituted an authorization system for its users. Information is collected to assist in web server administration and error tracking. On their initial visits, users will select a username, provide a current email address (for verificiation purposes), and review the terms and conditions for use of the CRW Site. An email will be sent to the provided email address containing a validation URL for that account. At this URL, the user may provide additional information; the system will then email an initial password to the user at the selected email account. The user then has the two pieces of information (username and password) necessary to log in and use the CRW Site. Once logged in, the user may change the password and update the user information at any time.
URL rewriting
We strongly encourage all users to access the Comparative RNA Web Site http://www.rna.icmb.utexas.edu using its main address, http://www.rna.icmb.utexas.edu/, rather than through specific URLs. As the site grows, specific pages may be moved, changed, or deleted. As well, use of more specific URLs may not include the navigation system for the site, providing the user with a suboptimal operating experience of the entire site. Therefore, the system is configured to route an initial request for a more specific URL to an introductory page, which will offer users access to the main page and a selection of specific URLs.
Abbreviations
CRW =:
Comparative RNA Web
NCBI =:
National Center for Biotechnology Information.
nt =:
nucleotide
RDBMS =:
Relational Database Management System.
URL =:
Uniform Resource Locator
References
- Darwin C: Origin of Species by Means of Natural Selection, or the Preservation of Favoured Races in the Struggle for Life. First edition, 1859; second edition, 1860; third edition, 1861; fourth edition, 1866; fifth edition, 1869; sixth and final edition, 1872. Amherst NY, Prometheus Books.
- Woese CR, Fox GE: Phylogenetic structure of the prokaryotic domain: the primary kingdoms. Proc Natl Acad Sci USA. 1977, 74: 5088–5090.
Article PubMed Central CAS PubMed Google Scholar - Woese CR, Magrum LJ, Fox GE: Archaebacteria. J Mol Evol 1978, 11: 245–251.
Article CAS PubMed Google Scholar - Woese CR: Bacterial evolution. Microbiol Rev. 1987, 51: 221–271.
PubMed Central CAS PubMed Google Scholar - Holley RW, Apgar J, Everett GA, Madison JT, Marquisee M, Merrill SH, Penswick JR, Zamir A: Structure of a ribonucleic acid. Science 1965, 147: 1462–1465.
Article CAS PubMed Google Scholar - RajBhandary UL, Stuart A, Faulkner RD, Chang SH, Khorana HG: Nucleotide sequence studies on yeast phenylalanine sRNA. Cold Spring Harb Symp Quant Biol 1966, 31: 425–434.
Article CAS PubMed Google Scholar - Madison JT, Everett GA, Kung HK: On the nucleotide sequence of yeast tyrosine transfer RNA. Cold Spring Harb Symp Quant Biol 1966, 31: 409–416.
Article CAS PubMed Google Scholar - Zachau HG, Dutting D, Feldman H, Melchers F, Karau W: Serine specific transfer ribonucleic acids. XIV. Comparison of nucleotide sequences and secondary structure models. Cold Spring Harb Symp Quant Biol 1966, 31: 417–424.
Article CAS PubMed Google Scholar - Levitt M: Detailed molecular model for transfer ribonucleic acid. Nature 1969, 224: 759–763.
Article CAS PubMed Google Scholar - Kim SH, Suddath FL, Quigley GJ, McPherson A, Sussman JL, Wang AH, Seeman NC, Rich A: Three-dimensional tertiary structure of yeast phenylalanine transfer RNA. Science 1974, 185: 435–440.
Article CAS PubMed Google Scholar - Robertus JD, Ladner JE, Finch JT, Rhodes D, Brown RS, Clark BF, Klug A: Structure of yeast phenylalanine tRNA at 3Å resolution. Nature 1974, 250: 546–551.
Article CAS PubMed Google Scholar - Fox GE, Woese CR: 5S RNA secondary structure. Nature 1975, 256: 505–507.
Article CAS PubMed Google Scholar - Fox GE, Woese CR: The architecture of 5S rRNA and its relation to function. J Mol Evol 1975, 6: 61–76.
Article CAS PubMed Google Scholar - Brosius J, Palmer ML, Kennedy PJ, Noller HF: Complete nucleotide sequence of a 16S ribosomal RNA gene from Escherichia coli. Proc Natl Acad Sci USA. 1978, 75: 4801–4805.
Article PubMed Central CAS PubMed Google Scholar - Woese CR, Magrum LJ, Gupta R, Siegel RB, Stahl DA, Kop J, Crawford N, Brosius J, Gutell R, Hogan JJ, Noller HF: Secondary structure model for bacterial 16S ribosomal RNA: phylogenetic, enzymatic and chemical evidence. Nucl Acids Res 1980, 8: 2275–2293.
Article PubMed Central CAS PubMed Google Scholar - Zwieb C, Glotz C, Brimacombe R: Secondary structure comparisons between small subunit ribosomal RNA molecules from six different species. Nucl Acids Res 1981, 9: 3621–3640.
Article PubMed Central CAS PubMed Google Scholar - Stiegler P, Carbon P, Zuker M, Ebel JP, Ehresmann C: [Secondary and topographic structure of ribosomal RNA 16S of Escherichia coli]. C R Seances Acad Sci D. 1980, 291: 937–940.
CAS PubMed Google Scholar - Brosius J, Dull TJ, Noller HF: Complete nucleotide sequence of a 23S ribosomal RNA gene from Escherichia coli. Proc Natl Acad Sci USA. 1980, 77: 201–204.
Article PubMed Central CAS PubMed Google Scholar - Noller HF, Kop J, Wheaton V, Brosius J, Gutell RR, Kopylov AM, Dohme F, Herr W, Stahl DA, Gupta R, Woese CR: Secondary structure model for 23S ribosomal RNA. Nucl Acids Res 1981, 9: 6167–6189.
Article PubMed Central CAS PubMed Google Scholar - Glotz C, Zwieb C, Brimacombe R, Edwards K, Kossel H: Secondary structure of the large subunit ribosomal RNA from Escherichia coli, Zea mays chloroplast, and human and mouse mitochondrial ribosomes. Nucl Acids Res 1981, 9: 3287–3306.
Article PubMed Central CAS PubMed Google Scholar - Branlant C, Krol A, Machatt MA, Pouyet J, Ebel JP, Edwards K, Kossel H: Primary and secondary structures of Escherichia coli MRE 600 23S ribosomal RNA. Comparison with models of secondary structure for maize chloroplast 23S rRNA and for large portions of mouse and human 16S mitochondrial rRNAs. Nucl Acids Res 1981, 9: 4303–4324.
Article PubMed Central CAS PubMed Google Scholar - Noller HF, Woese CR: Secondary Structure of 16S Ribosomal RNA. Science 1981, 212: 403–411.
Article CAS PubMed Google Scholar - Woese CR, Gutell R, Gupta R, Noller HF: Detailed analysis of the higher-order structure of 16S-like ribosomal ribonucleic acids. Microbiol Rev 1983, 47: 621–669.
PubMed Central CAS PubMed Google Scholar - Noller HF: Structure of ribosomal RNA. Annu Rev Biochem 1984, 53: 119–162. 10.1146/annurev.bi.53.070184.001003
Article CAS PubMed Google Scholar - Haselman T, Camp DG, Fox GE: Phylogenetic evidence for tertiary interactions in 16S-like ribosomal RNA. Nucl Acids Res 1989, 17: 2215–2221.
Article PubMed Central CAS PubMed Google Scholar - Haselman T, Gutell RR, Jurka J, Fox GE: Additional Watson-Crick interactions suggest a structural core in large subunit ribosomal RNA. J Biomol Struct Dyn 1989, 7: 181–186.
CAS PubMed Google Scholar - Larsen N: Higher order interactions in 23s rRNA. Proc Natl Acad Sci U S A 1992, 89: 5044–5048.
Article PubMed Central CAS PubMed Google Scholar - Gutell RR, Larsen N, Woese CR: Lessons from an evolving rRNA: 16S and 23S rRNA structures from a comparative perspective. Microbiol Rev. 1994, 58: 10–26.
PubMed Central CAS PubMed Google Scholar - Gutell RR: Comparative sequence analysis and the structure of 16 S and 23 S rRNA. In: Ribosomal RNA: Structure, Evolution, Processing, and Function in Protein Biosynthesis (Edited by: Zimmerman RA, Dahlberg AE). Boca Raton, CRC Press 1996, 111–128.
Google Scholar - Michel F, Jacquier A, Dujon B: Comparison of fungal mitochondrial introns reveals extensive homologies in RNA secondary structure. Biochimie 1982, 64: 867–881.
Article CAS PubMed Google Scholar - Cech TR: Conserved sequences and structures of group I introns: building an active site for RNA catalysis-a review. Gene 1988, 73: 259–271. 10.1016/0378-1119(88)90492-1
Article CAS PubMed Google Scholar - Michel F, Westhof E: Modelling of the Three-dimensional Architecture of Group I Catalytic Introns Based on Comparative Sequence Analysis. J Mol Biol 1990, 216: 585–610.
Article CAS PubMed Google Scholar - Damberger SH, Gutell RR: A comparative database of group I intron structures. Nucl Acids Res 1994, 22: 3508–3510.
Article PubMed Central CAS PubMed Google Scholar - Michel F, Umesono K, Ozeki H: Comparative and functional anatomy of group II catalytic introns – a review. Gene 1989, 82: 5–30. 10.1016/0378-1119(89)90026-7
Article CAS PubMed Google Scholar - Yu N: Comparative Sequence Analysis of Group II Intron and tmRNA and Database. M.A. thesis, University of Texas at Austin, 2000.
Google Scholar - James BD, Olsen GJ, Liu JS, Pace NR: The secondary structure of ribonuclease P RNA, the catalytic element of a ribonucleoprotein enzyme. Cell 1988, 52: 19–26.
Article CAS PubMed Google Scholar - Brown JW, Haas ES, James BD, Hunt DA, Liu JS, Pace NR: Phylogenetic analysis and evolution of RNase P RNA in proteobacteria. J Bacteriol 1991, 173: 3855–3863.
PubMed Central CAS PubMed Google Scholar - Harris JK, Haas ES, Williams D, Frank DN, Brown JW: New insight into RNase P RNA structure from comparative analysis of the archaeal RNA. RNA 2001, 7: 220–232. 10.1017/S1355838201001777
Article PubMed Central CAS PubMed Google Scholar - Romero DP, Blackburn EH: A conserved secondary structure for telomerase RNA. Cell 1991, 67: 343–353.
Article CAS PubMed Google Scholar - Chen JL, Blasco MA, Greider CW: Secondary structure of vertebrate telomerase RNA. Cell 2000, 100: 503–514.
Article CAS PubMed Google Scholar - Williams KP, Bartel DP: Phylogenetic analysis of tmRNA secondary structure. RNA 1996, 2: 1306–1310.
PubMed Central CAS PubMed Google Scholar - Guthrie C, Patterson B: Spliceosomal snRNAs. Annu Rev Genet 1988, 22: 387–419. 10.1146/annurev.ge.22.120188.002131
Article CAS PubMed Google Scholar - Zwieb C: Structure and function of signal recognition particle RNA. Prog Nucleic Acid Res Mol Biol 1989, 37: 207–234.
Article CAS PubMed Google Scholar - Wimberly BT, Brodersen DE, Clemons WM Jr, Morgan-Warren RJ, Carter AP, Vonhein C, Hartsch T, Ramakrishnan V: Structure of the 30S ribosomal subunit. Nature 2000, 407: 327–339. 10.1016/S0168-9002(98)00044-8
Article CAS PubMed Google Scholar - Ban N, Nissen P, Hansen J, Moore PB, Steitz TA: The complete atomic structure of the large ribosomal subunit at 2.4 A resolution. Science 2000, 289: 905–920. 10.1006/jmbi.1999.2786
Article CAS PubMed Google Scholar - Gutell RR: Collection of Small Subunit (16S- and 16S-like) ribosomal RNA structures. Nucl Acids Res 1993, 21: 3051–3054.
Article PubMed Central CAS PubMed Google Scholar - Gutell RR: Collection of Small Subunit (16S- and 16S-like) ribosomal RNA structures: 1994. Nucl Acids Res 1994, 22: 3502–3507.
Article PubMed Central CAS PubMed Google Scholar - Gutell RR, Fox GE: A compilation of large subunit RNA sequences presented in a structural format. Nucl Acids Res 1988, 16 Suppl: rl75-r269.
Google Scholar - Gutell RR, Schnare MN, Gray MW: A compilation of large subunit (23S-like) ribosomal RNA sequences presented in a secondary structure format. Nucl Acids Res 1990, 18 Suppl: 2319–2330.
Article CAS PubMed Google Scholar - Gutell RR, Schnare MN, Gray MW: A compilation of large subunit (23S- and 23S-like) ribosomal RNA structures. Nucl Acids Res 1992, 20 Suppl: 2095–2109.
Article CAS PubMed Google Scholar - Gutell RR, Gray MW, Schnare MN: A compilation of large subunit (23S- and 23S-like) ribosomal RNA structures:1993. Nucl Acids Res 1993, 21: 3055–3074.
Article PubMed Central CAS PubMed Google Scholar - Schnare MN, Damberger SH, Gray MW, Gutell RR: Comprehensive Comparison of Structural Characteristics in Eukaryotic Cytoplasmic Large Subunit (23S-like) Ribosomal RNA. J Mol Biol 1996, 256: 701–719. 10.1006/jmbi.1996.0119
Article CAS PubMed Google Scholar - Olsen GJ, Overbeek R, Larsen N, Marsh TL, McCaughey MJ, Maciukenas MA, Kuan WM, Macke TJ, Xing Y, Woese CR: The Ribosomal Database Project. Nucl Acids Res 1992, 20 Suppl: 2199–2200.
Article CAS PubMed Google Scholar - Maidak BL, Cole JR, Lilbum TG, Parker CT Jr, Saxman PR, Farris RJ, Garrity GM, Olsen GJ, Schmidt TM, Tiedje JM: The RDP-II (Ribosomal Database Project). Nucl Acids Res 2001, 29: 173–174. 10.1093/nar/29.1.173
Article PubMed Central CAS PubMed Google Scholar - Erdmann VA, Huysmans E, Vandenberghe A, De Wachter R: Collection of published 5S and 5.8S ribosomal RNA sequences. Nucl Acids Res 1983, 11: rl05-rl33.
Article Google Scholar - Huysmans E, De Wachter R: Compilation of small ribosomal subunit RNA sequences. Nucleic Acids Res. 1986, 14 Suppl: r73–118.
Article CAS PubMed Google Scholar - Van de Peer Y, De Rijk P, Wuyts J, Winkelmans T, De Wachter R: The European small subunit ribosomal RNA database. Nucl Acids Res 2000, 28: 175–176. 10.1093/nar/28.1.175
Article PubMed Central CAS PubMed Google Scholar - De Rijk P, Van de Peer Y, Chapelle S, De Wachter R: Database on the structure of large ribosomal subunit RNA. Nucl Acids Res 1994, 22: 3495–3501.
Article PubMed Central CAS PubMed Google Scholar - Wuyts J, De Rijk P, Van de Peer Y, Winkelmans T, De Wachter R: The European Large Subunit Ribosomal RNA Database. Nucl Acids Res 2001, 29: 175–177. 10.1093/nar/29.1.175
Article PubMed Central CAS PubMed Google Scholar - Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, Rapp BA, Wheeler DL: GenBank. Nucl Acids Res 2000, 28: 15–18. 10.1093/nar/28.1.15
Article PubMed Central CAS PubMed Google Scholar - Wheeler DL, Chappey C, Lash AE, Leipe DD, Madden TL, Schuler GD, Tatusova TA, Rapp BA: Database resources of the National Center for Biotechnology Information. Nucl Acids Res 2000, 28: 10–14. 10.1093/nar/28.1.10
Article PubMed Central CAS PubMed Google Scholar - Gutell RR, Cannone JJ, Konings D, Gautheret D: Predicting U-turns in Ribosomal RNA with Comparative Sequence Analysis. J Mol Biol 2000, 300: 791–803. 10.1006/jmbi.2000.3900
Article CAS PubMed Google Scholar - Gutell RR, Cannone JJ, Shang Z, Du Y, Serra M: A Story: Unpaired Adenosines in Ribosomal RNAs. J Mol Biol 2000, 304: 335–354. 10.1006/jmbi.2000.4172
Article CAS PubMed Google Scholar - Elgavish T, Cannone JJ, Lee JC, Harvey SC, Gutell RR: AA.AG@Helix.Ends: A:A and A:G Base-pairs at the Ends of 16 S and 23 S rRNA Helices. J Mol Biol 2001, 310: 735–753. 10.1006/jmbi.2001.4807
Article CAS PubMed Google Scholar - Konings DAM, Gutell RR: A comparison of thermodynamic foldings with comparatively derived structures of l6S and 16S-like rRNAs. RNA 1995, 1: 559–574.
PubMed Central CAS PubMed Google Scholar - Fields DS, Gutell RR: An Analysis of Large rRNA Sequences Folded by a Thermodynamic Method. FoldDes 1996, 1: 419–430.
Article CAS Google Scholar - Lydeard C, Holznagel WE, Schnare MN, Gutell RR: Phylogenetic Analysis of Molluscan Mitochondrial LSU rDNA Sequences and Secondary Structures. Mol Phylogenet Evol 2000, 15: 83–102. 10.1006/mpev.1999.0719
Article CAS PubMed Google Scholar - Vernon D, Gutell RR, Cannone JJ, Rumpf RW, Birky CW Jr: Accelerated Evolution of Functional Plastid rRNA and Elongation Factor Genes Due to Reduced Protein Synthetic Load After the Loss of Photosynthesis in the Chlorophyte Alga Polytoma. Mol Biol Evol 2001, 18: 1810–1822.
Article CAS PubMed Google Scholar - Bhattacharya D, Cannone JJ, Gutell RR: Group I Intron Lateral Transfer Between Red and Brown Algal Ribosomal RNA. Curr Genet 2001, 40: 82–90. 10.1007/s002940100227
Article CAS Google Scholar - Gutell RR, Weiser B, Woese CR, Noller HF: Comparative Anatomy of 16-S-like Ribosomal RNA. Prog Nucleic Acid Res Mol Biol 1985, 32: 155–216.
Article CAS PubMed Google Scholar - Gutell RR, Power A, Hertz GZ, Putz EJ, Stormo GD: Identifying constraints on the higher-order structure of RNA: continued development and application of comparative sequence analysis methods. Nucl Acids Res 1992, 20: 5785–5795.
Article PubMed Central CAS PubMed Google Scholar - Gautheret D, Damberger SH, Gutell RR: Identification of base-triples in RNA using comparative sequence analysis. J Mol Biol 1995, 248: 27–43. 10.1006/jmbi.1995.0200
Article CAS PubMed Google Scholar - Olsen GJ: Comparative analysis of nucleotide sequence data. Ph.D. thesis, University of Colorado Health Sciences Center, 1983.
Google Scholar - Chiu DK, Kolodziejczak T: Inferring consensus structure from nucleic acid sequences. Comput Appl Biosci 1991, 7: 347–352.
CAS PubMed Google Scholar - Correll CC, Freeborn B, Moore PB, Steitz TA: Metals, motifs, and recognition in the crystal structure of a 5S rRNA domain. Cell 1997, 91: 705–712.
Article CAS PubMed Google Scholar - Cate JH, Gooding AR, Podell E, Zhou K, Golden BL, Kundrot CE, Cech TR, Doudna JA: Crystal structure of a group I ribozyme domain: principles of RNA packing. Science 1996, 273: 1678–1685.
Article CAS PubMed Google Scholar - Hill WE, Dahlberg AE, Garrett RA, Moore PB, Schlessinger D, Warner JR, editors: The Ribosome: Structure, Function, and Evolution. Washington DC, American Society for Microbiology 1990.
- Zimmerman RA, Dahlberg AE, editors: Ribosomal RNA: Structure, Evolution, Processing, and Function in Protein Biosynthesis. BocaRaton, CRC Press 1996.
- Neefs JM, Van de Peer Y, Hendriks L, De Wachter R: Compilation of small ribosomal subunit RNA sequences. Nucl Acids Res. 1990, 18 Suppl: 2237–2317.
Article CAS PubMed Google Scholar - Woese CR, Kandler O, Wheelis ML: Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc Nati Acad Sci USA 1990, 87(12):4576–4579.
Article CAS Google Scholar - Sprinzl M, Dank N, Nock S, Schon A: Compilation of tRNA sequences and sequences of tRNA genes. Nucl Acids Res 1991, 19 Suppl: 2127–2171.
Article CAS PubMed Google Scholar - Suh SO, Jones KG, Blackwell M: A Group I Intron in the Nuclear Small Subunit rRNA Gene of Cryptendoxyla hypophloia, an Ascomycetous Fungus: Evidence for a New Major Class of Group I Introns. J Mol Evol 1999, 48: 493–500.
Article CAS PubMed Google Scholar - Kjems J, Garrett RA: Ribosomal RNA introns in archaea and evidence for RNA conformational changes associated with splicing. Proc Natl Acad Sci U S A 1991, 88: 439–443.
Article PubMed Central CAS PubMed Google Scholar - Bhattacharya D, Lutzoni F, Reeb V, Simon D, Nason J, Fernandez F: Widespread occurrence of spliceosomal introns in the rDNA genes of ascomycetes. Mol Biol Evol 2000, 17: 1971–1984.
Article CAS PubMed Google Scholar - Maidak BL, Olsen GJ, Larsen N, Overbeek R, McCaughey MJ, Woese CR: The RDP (Ribosomal Database Project). Nucl Acids Res 1997, 25: 109–111. 10.1093/nar/25.1.109
Article PubMed Central CAS PubMed Google Scholar - Van Oppen MJH, Olsen JL, Stam WT: Evidence for Independent Acquisition of Group I Introns in Green Algae. Mol Biol Evol 1993, 10: 1317–1326.
CAS PubMed Google Scholar - Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ: Basic local alignment search tool. J Mol Biol. 1990, 215: 403–410. 10.1006/jmbi.1990.9999
Article CAS PubMed Google Scholar - Lutzoni F, Pagel M, Reeb V: Major fungal lineages are derived from lichen symbiotic ancestors. Nature 2001, 411: 937–940. 10.1038/35082053
Article CAS PubMed Google Scholar
Acknowledgements
This work was supported by the National Institutes of Health (GM48207), the Welch Foundation (F-1427), startup funds from the Institute for Cellular and Molecular Biology at the University of Texas at Austin (awarded to RRG), and funding from the Ibis Therapeutics division of Isis Pharmaceuticals. J. Collett was supported from NSF IGERT grant DGE-0114387.
We thank John Eargle, Daniella Konings Viloya Schweiker, Chris Simmons, Bryn Weiser, and Ping Ye for their contributions to this project.
Author information
Authors and Affiliations
- Institute for Cellular and Molecular Biology, Section of Integrative Biology, University of Texas at Austin, 2500 Speedway, Austin, TX, 78712-1095, USA
Jamie J Cannone, Sankar Subramanian, James R Collett, Lisa M D'Souza, Yushi Du, Brian Feng, Nan Lin, Lakshmi V Madabusi, Kirsten M Müller, Nupur Pande, Zhidi Shang, Nan Yu & Robin R Gutell - Department of Biology, Arizona State University, Tempe, AZ, 85287-1501, USA
Sankar Subramanian - Department of Biochemistry and Molecular Biology, Dalhousie University, Halifax, Nova Scotia, B3H 4H7, Canada
Murray N Schnare - Ambion, Inc., Austin, TX, 78744-1832, USA
Lakshmi V Madabusi - Department of Biology, University of Waterloo, Waterloo, Ontario, N2L 3G1, Canada
Kirsten M Müller
Authors
- Jamie J Cannone
You can also search for this author inPubMed Google Scholar - Sankar Subramanian
You can also search for this author inPubMed Google Scholar - Murray N Schnare
You can also search for this author inPubMed Google Scholar - James R Collett
You can also search for this author inPubMed Google Scholar - Lisa M D'Souza
You can also search for this author inPubMed Google Scholar - Yushi Du
You can also search for this author inPubMed Google Scholar - Brian Feng
You can also search for this author inPubMed Google Scholar - Nan Lin
You can also search for this author inPubMed Google Scholar - Lakshmi V Madabusi
You can also search for this author inPubMed Google Scholar - Kirsten M Müller
You can also search for this author inPubMed Google Scholar - Nupur Pande
You can also search for this author inPubMed Google Scholar - Zhidi Shang
You can also search for this author inPubMed Google Scholar - Nan Yu
You can also search for this author inPubMed Google Scholar - Robin R Gutell
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toRobin R Gutell.
Authors’ original submitted files for images
Rights and permissions
About this article
Cite this article
Cannone, J.J., Subramanian, S., Schnare, M.N. et al. The Comparative RNA Web (CRW) Site: an online database of comparative sequence and structure information for ribosomal, intron, and other RNAs.BMC Bioinformatics 3, 2 (2002). https://doi.org/10.1186/1471-2105-3-2
- Received: 07 December 2001
- Accepted: 17 January 2002
- Published: 17 January 2002
- DOI: https://doi.org/10.1186/1471-2105-3-2