Inflammatory breast cancer: Relationship between growth factor signaling and motility in aggressive cancers (original) (raw)
- Review
- Published: 01 June 2003
Breast Cancer Research volume 5, Article number: 174 (2003)Cite this article
- 11k Accesses
- 29 Citations
- Metrics details
Abstract
A variety of phenotypic characteristics are required for a cancer cell to successfully complete the metastatic cascade. Acquisition of a motile and invasive phenotype is one requirement for a cell to become metastatically competent. The Rho (Ras homology) GTPases are a subfamily of small GTP-binding proteins, which are related to the Ras oncogene. All aspects of cellular motility and invasion are controlled by the Rho GTPases and are closely linked to signals from the extracellular environment, particularly in response to growth factors. Dysregulation of Rho activation through aberrant growth-factor signaling, loss of function of key Rho-regulatory proteins or overexpression of Rho mRNA could result in increased Rho activity and cellular motility. Therefore, the importance of the Rho GTPases in the progression of aggressive cancers, is becoming more appreciated.
Introduction
Inflammatory breast cancer (IBC) is a highly aggressive form of locally advanced breast cancer that carries a guarded prognosis due to its propensity to disseminate via the dermal lymphatics and metastasize to distant organs [1]. Tumor recurrence or outgrowth of occult metastases are common in IBC and account for the lowest 5-year disease-free survival rate of all breast cancers (<45%, compared with an 86% 5-year disease-free survival rate for all breast cancers) [1, [2](/articles/10.1186/bcr598#ref-CR2 "Breast Cancer Facts and Figures 2001–2002. American Cancer Society, Atlanta, GA. 2002, [ http://www.cancer.org/downloads/STT/BrCaFF2001.pdf
]")\]. RhoC GTPase was found to be preferentially overexpressed in IBC tumors versus stage-matched, non-IBC tumors \[[3](/articles/10.1186/bcr598#ref-CR3 "van Golen KL, Davies S, Wu ZF, Wang Y, Bucana CD, Root H, Chandrasekharappa S, Strawderman M, Ethier SP, Merajver SD: A novel putative low-affinity insulin-like growth factor-binding protein, LIBC (lost in inflammatory breast cancer), and RhoC GTPase correlate with the inflammatory breast cancer phenotype. Clin Cancer Res. 1999, 5: 2511-2519.")\], and was found to be a breast-specific oncogene that could impart motile and invasive capabilities to breast epithelial cells akin to the IBC phenotype \[[4](/articles/10.1186/bcr598#ref-CR4 "van Golen KL, Wu ZF, Qiao XT, Bao LW, Mearajver SD: RhoC GTPase, a novel transforming oncogene for human mammary epithelial cells that partially recapitulates the inflammatory breast cancer phenotype. Cancer Res. 2000, 60: 5832-5838.")\]. The invasive and metastatic properties of pancreatic adenocarcinoma and melanoma have also been attributed to RhoC overexpression \[[5](/articles/10.1186/bcr598#ref-CR5 "Sahai E, Marshall CJ: Rho-GTPases and cancer. Nat Rev Cancer. 2002, 2: 133-142. 10.1038/nrc725.")\]. These examples of Rho protein expression and activation in aggressive cancers illustrate the importance of these molecules in understanding progression to a metastatic phenotype.Because of their similarity to Ras, a great deal of excitement has ensued since the discovery of the Rho GTPases in the mid-1980s as potential key oncogenes involved in cancer. Unlike Ras, no activating mutations have been demonstrated for Rho. Instead, overexpression and/or activation, in the absence of mutation, appears to be the rule in human cancers [6]. The importance of Rho proteins in conferring motile and invasive properties that contribute to the metastatic phenotype is becoming increasingly appreciated [4, 7]. The Rho GTPases control all aspects of cellular motility and invasion, including polarity, cytoskeletal organization, and transduction of signals from the extracellular environment, particularly from growth factors [4, 7, 8]. To be fully effective in achieving the invasive phenotype, the Rho proteins most likely need to complete a full GTPase cycle and continue to cycle, alternating between an inactive and active state [9–12]. Although one Rho protein may predominate in cancer cell, a reciprocal activation of different GTPases must occur during cycling (e.g. RhoC is active while Rac1 is inactive and vice versa), thus allowing cells to effectively reorganize the cytoskeleton and to form lamellipodia and filopodia. This is best illustrated when constitutively active RhoA is introduced into fibroblasts, leading to the formation of stable stress fibers and a nonmotile cell [7]. The need for reciprocal activation during cell motility is perhaps the reason why no activating mutations, such as those identified for Ras, have been found for Rho in human cancers.
Like Ras, Rho proteins are activated via extracellular signals that cause binding and hydrolyses of GTP and induction of downstream effector molecules, leading to a cellular response [8]. Although 21 members of the Rho-GTPase subfamily have been identified to date, the vast majority of data exists for RhoA, Rac1, and Cdc42 [7]. As suggested above, it is the activation state of these and other Rho proteins, and not the level of expression, that dictates the cellular effects. Therefore, attention is turning to potential mechanisms of aberrant Rho activation, such as alterations in the regulatory proteins or aberrant growth factor stimulation.
As outlined in Fig. 1, growth-factor-stimulated activation of Rho occurs through the interactions of several Rho regulatory proteins, such as the GTPase-activating proteins (GAPs), guanine nucleotide dissocation inhibitors (GDIs), GDI-dissociation factors (GDFs), and guanine nucleotide exchange factors (GEFs) [12–15]. The entire cycle is balanced by GDIs, which prevent guanosine diphosphate (GDP) dissociation and sequester Rho in the cytoplasm. The GDP-bound, inactive Rho is liberated from the GDI by the GDF and is localized to the inner plasma membrane. Activation of the GEFs by tyrosine kinase growth factor receptors leads to exchange of GDP for guanosine triphosphate (GTP), thus activating Rho [14, 15]. Simultaneously, growth-factor-receptor-induced phosphorylation of p120 RasGAP leads to heterodimer binding and inactivation of p190 RhoGAP, allowing the GTPase to remain active [13]. GTP binding produces a conformational change in Rho leading to interaction with and activation of downstream effector proteins, such as Rho kinase (ROCK) for RhoA and RhoC, or p21 activated kinase (Pak) for Rac and Cdc42 [16–18]. Binding affinities for the effector proteins vary with the GTPases. For example, RhoC has a greater binding affinity for ROCK than RhoA [16]. These differences in binding may be key in producing an invasive phenotype in cells preferentially expressing a GTPase such as RhoC [16, 17]. Through unknown mechanisms, the RasGAP/RhoGAP heterodimer dissociates, and the now active GAPs catalyze hydrolysis of remaining GTP to GDP, inactivating Rho and closing the cycle [13].
Figure 1
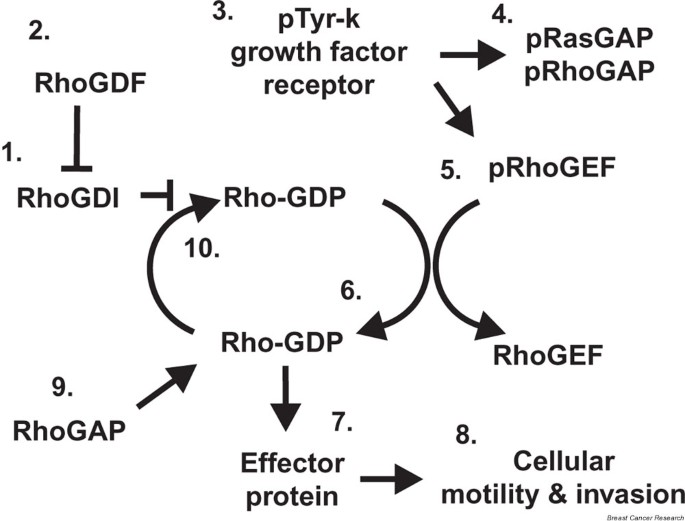
The GTPase cycle. RhoGDIs (1) sequester Rho GTPases in the cytoplasm and prevent GDP dissociation until released by RhoGDFs (2). The Rho protein attaches to the inner cytoplasmic membrane by prenylation of the protein. When a protein tyrosine kinase growth factor receptor (3), such as EGFR, is activated, p120 RasGAP is phosphorylated and forms a heterodimer with p190 RhoGAP, leading to inactivation of those proteins. The RhoGEF proteins (4) are also phosphorylated, causing exchange of GDP for GTP (6) on the Rho proteins, leading to Rho activation. Active, GTP-bound Rho proteins activate downstream Rho effector proteins (7), which stimulate cellular motility and invasion (8). RhoGAP (9) is released and activated, catalyzing the hydrolysis of GTP to GDP (10) and inactivating the Rho protein. EGFR = EGF receptor; GAP = GTPase activating factor; GDF = GDI-dissociation factor; GDI = guanine nucleotide dissociation inhibitor; GDP = guanosine diphosphate ; GEF = guanine nucleotide exchange factor; GTP = guanosine triphosphate.
Growth-factor-induced Rho activation and cancer cell motility
Studies have suggested that dysregulation of the GTPase cycle is due to altered expression of the key Rho regulatory proteins described above. The best examples of this are the GEF proteins, which were originally identified as oncogenes, and p190-A RhoGAP, which was first described as a tumor suppressor gene [15, 18, 19].
Several other studies have focused on the role of growth factor receptors and their ligands in the activation of Rho GTPases and the induction of cancer cell motility. Oncogenic versions of the IGF-I receptor (IGF-IR) or the Her family of growth factor receptors activate RhoA, Rac1, Cdc42, and possibly RhoC [20, 21]. During cancer progression, early dysregulation of growth factor signaling, such as EGFR amplification or Her2/neu expression, may lead to a mitogenic effect required for tumor cell growth and survival. However, later in the progression of the cancer, these same signals may activate overexpressed Rho GTPases, driving metastasis [22]. Therefore, overexpression of Rho by itself is not sufficient to achieve a metastatic phenotype. However, when coupled with aberrant growth factor signaling, the active form of the GTPase drives metastasis.
The insulin-like growth factors (IGFs)
The process of motility is similar for both normal migrating cells and invasive or metastatic cancer cells, requiring dynamic reorganization of the actin cytoskeleton in response to external stimuli. Normal epithelial cells grow and undergo lateral migration in tight sheets in which cells are connected to one another by cell–cell adhesions (i.e. cadherin-catenin complexes) and to the extracellular matrix (ECM) by cell–matrix adhesions [23]. The ability of an epithelial cell to migrate individually is a hallmark of tumorigenic cells that requires cellular depolarization and turnover of cell-ECM contact sites known as focal adhesions [23, 24]. Focal adhesions contain integrins, cytoskeletal and signaling proteins such as α-actinin and focal adhesion kinase (FAK). Focal adhesion turnover and cellular motility is decreased in fibroblasts derived from FAK -/- mice [23]. Reexpression of FAK leads to transient inhibition of RhoA and RhoC activity, ROCK activation, restoring focal adhesion turnover, and cell migration [23, 25], suggesting that completion of the GTPase cycle by RhoA and RhoC is required for focal adhesion turnover and cell motility. Loss of FAK activity results in constitutive activation of Rho/ROCK and decreased migration.
The observations made in fibroblasts provide a framework for studying growth factor induced motility in epithelial derived cancers. Stimulation of IGF-IR overexpressing MCF-7 breast cancer cells with IGF-I leads to actin filament disassembly, cellular depolarization, and the formation of fascin microspikes associated with lamellipodia [24, 26]. Signaling from IGF-IR to the actin cytoskelton during depolarization and cell separation appears to be mediated by the actin-binding protein α-actinin [27]. These cellular changes involve the activation of the phosphotidylinositol 3-kinase (PI3K) pathway and transient phosphotyrosine-phosphotase-mediated dephosphorylation of FAK, and two other focal adhesion-associated molecules, p130-Crk-associated substrate (Cas) and paxillin [26]. However, increased activation of the mitogen-activated protein kinase (MAPK) pathway is needed for complete cellular migration [27]. Transient dephosphorylation of FAK leads to Rho activation, actin depolymerization, focal adhesion turnover, and loss of cellular polarity. Phosphorylation of FAK inhibits Rho activity, promoting stablization of focal adhesions and activation of Rac and Cdc42, both of which are required for fascin microspike formation and lamellipodial formation [28, 29, 7], demonstrating that the Rho GTPases must work in concert to produce a motile cell.
Contrary to these studies, it is suggested that decreased IGF-IR levels lead to increased motility and invasiveness in MCF-7 cells by increasing Rac and Cdc42 activity and decreasing Rho activity and E-cadherin expression [30]. These data imply that activation of IGF-IR stabilizes the E-cadherin–β-catenin complex and potentially activates RhoA, promoting cellular aggregation.
Studies demonstrating the effect of IGF-I on neuroblastoma cell motility suggest a peripheral role for RhoA or RhoC. These studies are based on observations made of IGF-I-stimulated neuroglial Schwann cell motility [31]. IGF-I stimulation of SH-SY5Y and SHEP human neuroblastoma cells leads to Rac1 mediated lamellipodial extension through the PI3K and MAPK pathways [31]. Blocking SH-SY5Y IGF-IR with αIR-3 antibody inhibits cellular motility. Furthermore, when PTEN was expressed in SH-SY5Y cells, motility was also inhibited. The presence of PTEN prevents phosphorylation and activation of xxAkt by PI3K suggesting activation of Akt is integral in neuroblastoma motility.
A positive feedback loop of growth factor signaling has been described in Src transformed human pancreatic cancer cells. Activation of the PI3K pathway leads to phosphorylation of Akt and upregulation of IGF-IR [32]. IGF-IR expression is diminished when PTEN is expressed, indicating that Src transformation leads to an increase in PI3K activation, phosphorylation of Akt, and upregulation of IGF-IR. IGF-IR is activated by IGF-I, increasing the level of PI3K activation, increasing Akt phosphorylation, sustaining IGF-IR expression, and increasing cellular motility and metastasis. Since pancreatic cancer cells have not been shown to produce IGF-I, it is thought that the pancreatic stroma, a hallmark of pancreatic tumors and a rich source of IGF, is the main source of IGF in this system.
In the RIP1–Tag2 murine model of pancreatic tumorigenesis, IGF-IR is elevated in invasive tumors [33]. In this model, IGF-II activates IGF-IR in hyperproliferative, oncogene-expressing β cells and is uniformly expressed in invasive tumors. As a consequence, overexpression of IGF-IR, activated by IGF-II in an autocrine manner, accelerates tumor progression and enhances invasive and metastatic capabilities of the cells, presumably mediated by Rac and Rho.
A comparison of two human neuroblastoma cell lines, SH-SY5Y- and IGF-IR-transfected SHEP (SHEP/IGF-IR), shows that the two cell lines are equally stimulated to migrate in response to IGF-I stimulation via Rac activation [31]. Interestingly, SHEP/IGF-IR and SH-SY5Y utilize different isoforms of insulin receptor substrate (IRS-1 and IRS-2, respectively) to transduce signals from IGF-IR. Treatment with either a PI3K inhibitor (LY294002) or a MEK1 inhibitor (PD98059), alone or in combination, decreases cellular motility equally in the two cell types. Regardless of which IRS is used to transduce the signal from IGF-IR, both PI3K and MAPK are utilized in Rac1-mediated neuroblastoma motility.
IGF-I treatment of the MDA-MB-231-BO breast cancer cell line, a metastatic variant derived from an osseous metastasis of MDA-MB-231 parent cells, stimulates motility in the variant but not the parental cell lines [34]. In contrast to the neuroblastoma studies, inhibition of ROCK with Y27632 implicates activation of RhoA and RhoC in conferring MDA-MB-231 BO motility. IRS-2 is preferentially phosphorylated in the metastatic variant, leading to increased activation of the PI3K and MAPK pathways. In addition to motility, signaling through IGF-IR and IRS-2 leads to an increase in anchorage-independent growth and cell adhesion. These data demonstrate that a variety of aspects of the breast cancer metastatic phenotype are specifically regulated through IRS-2. Taken together, the observations made in MCF-7, MDA-231 BO, and neuroblastoma cells suggest that activation of the GTPases may be dependent on the adapter molecules that transduce the signal from IGF-IR.
To add to the complexity, both IGF-I and IGF-II can interact with IGF-IR, the insulin receptor (IR), or a heterodimer of IGF-IR/IR, thus eliciting numerous cellular responses [35]. These responses can be further modulated by the IGF-binding proteins (IGFBPs) and the IGFBP-related proteins (IGFBP-rPs), which can limit binding of ligand to receptor or elicit their own cellular signals. To date, several IGFBPs and IGFBP-rPs have been identified and play an active role in IGF signaling in normal and transformed cells. Limiting access of IGF to its receptor can further regulate activation of IGF-IR and breast cancer cell motility by IGFBPs. This is specifically illustrated in studies showing that IGFBP-1 reduces MDA-MB-231-BO cell motility by limiting IGF-I effects on the cell [35].
In the same study that identified RhoC GTPase as being expressed in IBC, another gene, LIBC/Wisp3, a bona fide tumor suppressor gene, was cloned and identified as being lost in a significant number of IBC tumors [3, 36]. The tumor suppressor activity of LIBC/Wisp3 reduces IBC anchorage-independent growth, invasiveness, and angiogenic potential, and decreases tumor formation when implanted orthotopically in nude mice [36]. LIBC/Wisp3 is a low-affinity IGFBP, or more specifically an IGFBP-rP, denoted IGFBP-rP9. Data suggest that loss of IGFBP-rP9 in IBC may lead to an increase in IGF signaling, which potentially activates the Rho GTPases, specifically RhoC.
Epidermal growth factor (EGF)
The EGF receptor (EGFR) and its oncogenic relative Her2/neu are commonly expressed in breast cancer, including up to 60% of IBC tumors [1]. EGFR overexpression is also said to occur in as many as 90% of pancreatic tumors [37]. EGF-induced cellular motility and invasion by the Rho is well documented. Small breast tumors (<1 cm) with a propensity to metastasize, particularly IBC tumors, concordantly overexpress RhoC GTPase and Her2/neu [38].
In vivo invasive and metastatic properties of the MTLn3 rat mammary carcinoma cell line are significantly diminished when transfected with dominant negative forms of RhoA, Rac1, and Cdc42 [39]. MTLn3 cells harboring dominant negative forms of the GTPases are also refractory to EGF-induced growth and invasion in vitro. Similarly, a dose-dependent increase of RhoA-mediated in vitro cellular invasion is seen for the Panc-1 human pancreatic cancer cell line stimulated with EGF [40]. Treatment of the cells with statins, inhibitors of 3-hydroxy-3-methyl-coenzyme A, abrogates EGF-induced invasion, presumably by preventing RhoA from localizing in the inner plasma membrane, where it can be activated by RhoGEFs [40].
EGF activation of the Rho GTPases can occur through activation of the RhoGEFs and inactivation of the RhoGAPs. The Vav family of RhoGEFs is an example of a group of GEFs that are activated in response to EGF stimulation of EGFR [15, 18]. The Vav family consists of three members that interact with and activate RhoA, Rac1, and Cdc42 [15, 18]. Vav1 is found primarily in hematopoietic cells, while Vav2 and Vav3 are expressed ubiquitously in tissues [15, 18]. All three Vav proteins have been implicated in breast cancer metastasis and may prove to be very important in the aberrant activation of Rho GTPases during the metastatic cascade.
p190 RhoGAP was first identified as a phosphorylated protein associated with p120 RasGAP in _v-src_-transformed, EGF stimulated cells [13, 41]. EGF stimulation induces tyrosine phosphorylation of p190 RhoGAP, leading to heterodimer formation with p120 RasGAP and a loss in GTP-binding, decreased GTP hydrolysis, and actin reorganization [13]. At the time of EGF activation, the p190 and p120 proteins are located at the inner plasma membrane [41]. Upon heterodimer formation, the p190/p120 complex becomes associated with EGFR in an endocytic compartment and is internalized [41]. Reasonably, during receptor recycling the GAPs are released and can inactivate remaining GTP-bound Rho. The p190-A isoform was first identified as a tumor suppressor gene, thus suggesting that loss or inactivation of GAP proteins can lead to dysregulation and subsequent activation of Rho GTPases [19].
Conclusion
A great deal of work performed over the past five years has given insight into mechanisms of growth-factor-induced motility. Clearly, the Rho GTPases occupy a central and pivotal role as a molecular switch, controlling aspects of the cystoskeletal rearrangement during motility and invasion. As more studies focus on the role of the Rho GTPases in metastasis, a complex web of molecules involved in regulating activation of the GTPases and crosstalk between different Rho subfamily members will be elucidated. Given the central role occupied by these molecules, understanding their biology is essential in understanding a key element of the metastatic phenotype.
Note
This article is the second in a review series on Inflammatory breast cancer, edited by Sofia Merajver.
Other articles in the series can be found at http://breast-cancer-research.com/articles/series.asp?rqs=merajver
Abbreviations
EGF:
= epidermal growth factor
EGFR:
= EGF receptor
FAK:
= focal adhesion kinase
GAP:
= GTPase activating factor
GDF:
= GDI-dissociation factor
GDI:
= guanine nucleotide dissociation inhibitor
GDP:
= guanosine diphosphate
GEF:
= guanine nucleotide exchange factor
GTP:
= guanosine triphosphate
IBC:
= inflammatory breast cancer
IGF:
= insulin-like growth factor
IGFBP:
= IGF-binding protein
IGFBP-rP:
= IGFBP-related protein
IGF-IR:
= IGF-I receptor
IR:
= insulin receptor
IRS:
= insulin receptor substrate
MAPK:
= mitogen activated protein kinase
PI3K:
= phosphotidylinositol 3-kinase
ROCK:
= Rho kinase.
References
- Kleer CG, van Golen KL, Merajver SD: Molecular biology of breast cancer metastasis. Inflammatory breast cancer: clinical syndrome and molecular determinants. Breast Cancer Res. 2000, 2: 423-429. 10.1186/bcr89.
Article CAS PubMed PubMed Central Google Scholar - Breast Cancer Facts and Figures 2001–2002. American Cancer Society, Atlanta, GA. 2002, [http://www.cancer.org/downloads/STT/BrCaFF2001.pdf]
- van Golen KL, Davies S, Wu ZF, Wang Y, Bucana CD, Root H, Chandrasekharappa S, Strawderman M, Ethier SP, Merajver SD: A novel putative low-affinity insulin-like growth factor-binding protein, LIBC (lost in inflammatory breast cancer), and RhoC GTPase correlate with the inflammatory breast cancer phenotype. Clin Cancer Res. 1999, 5: 2511-2519.
CAS PubMed Google Scholar - van Golen KL, Wu ZF, Qiao XT, Bao LW, Mearajver SD: RhoC GTPase, a novel transforming oncogene for human mammary epithelial cells that partially recapitulates the inflammatory breast cancer phenotype. Cancer Res. 2000, 60: 5832-5838.
CAS PubMed Google Scholar - Sahai E, Marshall CJ: Rho-GTPases and cancer. Nat Rev Cancer. 2002, 2: 133-142. 10.1038/nrc725.
Article PubMed Google Scholar - Fritz G, Just I, Kaina B: Rho GTPases are over-expressed in human tumors. Int J Cancer. 1999, 81: 682-687. 10.1002/(SICI)1097-0215(19990531)81:5<682::AID-IJC2>3.0.CO;2-B.
Article CAS PubMed Google Scholar - Ridley AJ: Rho GTPases and cell migration. J Cell Sci. 2001, 114: 2713-2722.
CAS PubMed Google Scholar - Kjoller L, Hall A: Signaling to Rho GTPases. Exp Cell Res. 1999, 253: 166-179. 10.1006/excr.1999.4674.
Article CAS PubMed Google Scholar - Ren XD, Kiosses WB, Schwartz MA: Regulation of the small GTP-binding protein Rho by cell adhesion and the cytoskeleton. EMBO J. 1999, 18: 578-585. 10.1093/emboj/18.3.578.
Article CAS PubMed PubMed Central Google Scholar - Lin R, Cerione RA, Manor D: Specific contributions of the small GTPases Rho, Rac, and Cdc42 to Dbl transformation. J Biol Chem. 1999, 274: 23633-23641. 10.1074/jbc.274.33.23633.
Article CAS PubMed Google Scholar - Symons M, Settleman J: Rho family GTPases: more than simple switches. Trends Cell Biol. 2000, 10: 415-419. 10.1016/S0962-8924(00)01832-8.
Article CAS PubMed Google Scholar - Geyer M, Wittinghofer A: GEFs, GAPs, GDIs and effectors: taking a closer (3D) look at the regulation of Ras-related GTP-binding proteins. Curr Opin Struct Biol. 1997, 7: 786-792. 10.1016/S0959-440X(97)80147-9.
Article CAS PubMed Google Scholar - Roof RW, Haskell MD, Dukes BD, Sherman N, Kinter M, Parsons SJ: Phosphotyrosine (p-Tyr)-dependent and -independent mechanisms of p190 RhoGAP-p120 RasGAP interaction: Tyr 1105 of p190, a substrate for c-Src, is the sole p-Tyr mediator of complex formation. Mol Cell Biol. 1998, 18: 7052-7063.
Article CAS PubMed PubMed Central Google Scholar - Moores SL, Selfors LM, Fredericks J, Breit T, Fujikawa K, Alt FW, Brugge JS, Swat W: Vav family proteins couple to diverse cell surface receptors. Mol Cell Biol. 2000, 20: 6364-6373. 10.1128/MCB.20.17.6364-6373.2000.
Article CAS PubMed PubMed Central Google Scholar - Bustelo XR: Vav proteins, adaptors and cell signaling. Oncogene. 2001, 20: 6372-6381. 10.1038/sj.onc.1204780.
Article CAS PubMed Google Scholar - Sahai E, Marshall CJ: ROCK and mDia have opposing effects on adherens junctions downstream of Rho. Nat Cell Biol. 2002, 4: 408-415. 10.1038/ncb796.
Article CAS PubMed Google Scholar - Itoh K, Yoshioka K, Akedo H, Uehata M, Ishizaki T, Narumiya S: An essential part for Rho-assoicated kinase in the transcellular invasion of tumor cells. Nat Med. 1999, 5: 221-225. 10.1038/5587.
Article CAS PubMed Google Scholar - Bishop AL, Hall A: Rho GTPases and their effector proteins. Biochem J. 2000, 348 (pt 2): 241-255. 10.1042/0264-6021:3480241.
Article CAS PubMed PubMed Central Google Scholar - Tikoo A, Czekay S, Viars C, White S, Heath JK, Arden K, Maruta H: p190-A, a human tumor suppressor gene, maps to the chromosomal region 19q13.3 that is reportedly deleted in some gliomas. Gene. 2000, 257: 23-31. 10.1016/S0378-1119(00)00387-5.
Article CAS PubMed Google Scholar - Sachdev P, Jiang YX, Li W, Miki T, Maruta H, Nur-E-Kamal MS, Wang LH: Differential requirement for Rho family GTPases in an oncogenic insulin-like growth factor-I receptor-induced cell transformation. J Biol Chem. 2001, 276: 26461-26471. 10.1074/jbc.M010995200.
Article CAS PubMed Google Scholar - Lee RJ, Albanese C, Fu M, D'Amico M, Lin B, Watanabe G, Haines GK, Siegel PM, Hung MC, Yarden Y, Horowitz JM, Muller WJ, Pestell RG: Cyclin D1 is required for transformation by activated Neu and is induced through an E2F-dependent signaling pathway. Mol Cell Biol. 2000, 20: 672-683. 10.1128/MCB.20.2.672-683.2000.
Article CAS PubMed PubMed Central Google Scholar - Bernards R, Weinberg RA: A progression puzzle. Nature. 2002, 418: 823-10.1038/418823a.
Article CAS PubMed Google Scholar - Ren XD, Kiosses WB, Sieg DJ, Otey CA, Schlaepfer DD, Schwartz MA: Focal adhesion kinase suppresses Rho activity to promote focal adhesion turnover. J Cell Sci. 2000, 113: 3673-3678.
CAS PubMed Google Scholar - Guvakova MA, Surmacz E: The activated insulin-like growth factor I repeptor induces depolarization in beast epithelial cells charaterized by actin filament disassembly and tyrosine dephosphorylation of FAK, Cas, and paxillin. Exp Cell Res. 1999, 251: 244-255. 10.1006/excr.1999.4566.
Article CAS PubMed Google Scholar - Chen BH, Tzen JTC, Bresnick AR, Chen HC: Role of Rho-associated kinase and myosin light chain kinase in morphologic and migratory defects of focal adhesion kinase-null cells. J Biol Chem. 2002, 277: 33857-33863. 10.1074/jbc.M204429200.
Article CAS PubMed Google Scholar - Guvakova MA, Boettiger D, Adams JC: Induction of fascin spikes in breast cancer cells by activation of the insulin-like growth factor-I receptor. Int J Biochem Cell Biol. 2002, 34: 685-698. 10.1016/S1357-2725(01)00160-1.
Article CAS PubMed Google Scholar - Guvakova MA, Adams JC, Boettiger D: Functional role of α-actinin, PI 3-kinase and MEK 1/2 in insulin-like growth factor I receptor kinase regulated motility of human breast carcinoma cells. J Cell Sci. 2002, 115: 4149-4165. 10.1242/jcs.00104.
Article CAS PubMed Google Scholar - Sander EE, ten Klooster JP, van Delft S, van der Kammen RA, Collard JG: Rac downregulates Rho activity: reciprocal balance between both GTPases determines cellular morphology and migratory behavior. J Cell Biol. 1999, 147: 1009-1027. 10.1083/jcb.147.5.1009.
Article CAS PubMed PubMed Central Google Scholar - Adams JC, Schwartz MA: Stimulation of fascin spikes by thrombospondin-1 is mediated by the GTPases Rac and Cdc42. J Cell Biol. 2000, 150: 807-822. 10.1083/jcb.150.4.807.
Article CAS PubMed PubMed Central Google Scholar - Pennisi PA, Barr V, Nunez NP, Stannard B, Le Roith D: Reduced expression of insulin-like growth factor I receptors in MCF-7 breast cancer cells leads to a more metastatic phenotype. Cancer Res. 2002, 62: 6529-6537.
CAS PubMed Google Scholar - Meyer GE, Shelden E, Kim B, Feldman EL: Insulin-like growth factor I stimulates motility in human neuroblastoma cells. Oncogene. 2001, 20: 7542-7550. 10.1038/sj.onc.1204927.
Article CAS PubMed Google Scholar - Tanno S, Tanno S, Mitsuuchi Y, Altomare DA, Xiao GH, Testa JR: AKT activation up-regulates insulin-like growth factor I receptor expression and promotes invasiveness of human pancreatic cancer cells. Cancer Res. 2001, 61: 589-593.
CAS PubMed Google Scholar - Lopez T, Hanahan D: Elevated levels of IGF-1 receptor convey invasive and metastatic capability in a mouse model of pancreatic islet tumorigenesis. Cancer Cell. 2002, 1: 339-353. 10.1016/S1535-6108(02)00055-7.
Article CAS PubMed Google Scholar - Jackson JG, Zhang X, Yoneda T, Yee D: Regulation of breast cancer cell motility by insulin receptor substrate-2 (IRS-2) in metastatic variants of human breast cancer cell lines. Oncogene. 2001, 20: 7318-7325. 10.1038/sj.onc.1204920.
Article CAS PubMed Google Scholar - Zhang X, Yee D: Insulin-like growth factor binding protein-1 (IGFBP-1) inhibits breast cancer cell motility. Cancer Res. 2002, 62: 4369-4375.
CAS PubMed Google Scholar - Kleer CG, van Golen KL, Zhang Y, Wu ZF, Rubin MA, Merajver SD: WISP3 is a novel tumor suppressor gene of inflammatory breast cancer. Oncogene. 2002, 21: 3172-3180. 10.1038/sj.onc.1205462.
Article CAS PubMed Google Scholar - Lemoine NR, Hughes CM, Barton CM, Poulsom R, Jeffery RE, Kloppel G, Hall PA, Gullick WJ: The epidermal growth factor receptor in human pancreatic cancer. J Pathol. 1992, 166: 7-12.
Article CAS PubMed Google Scholar - Kleer CG, van Golen KL, Zhang Y, Wu ZF, Rubin MA, Merajver SD: Characterization of RhoC expression in benign and malignant breast disease: a potential new marker for small breast carcinomas with metastatic ability. Am J Pathol. 2002, 160: 579-584.
Article CAS PubMed PubMed Central Google Scholar - Bouzahzah B, Albanese C, Ahmed F, Pixley F, Lisanti MP, Segall JD, Condeelis J, Joyce D, Minden A, Der CJ, Chan A, Symons M, Pestell RG: Rho family GTPases regulate mammary epithelium cell growth and metastasis through distinguishable pathways. Mol Med. 2001, 7: 816-830.
CAS PubMed PubMed Central Google Scholar - Kusama T, Mukai M, Iwasaki T, Tatsuta M, Matsumoto Y, Akedo H, Nakamura H: Inhibition of epidermal growth factor-induced RhoA translocation and invasion of human pancreatic cancer cells by 3-hydroxy-3-methylglutaryl-coenzyme a reductase inhibitors. Cancer Res. 2001, 61: 4885-4891.
CAS PubMed Google Scholar - Wang Z, Tung PS, Moran MF: Association of p120 ras GAP with endocytic components and colocalization with epidermal growth factor (EGF) receptor in response to EGF stimulation. Cell Growth Differ. 1996, 7: 123-133.
CAS PubMed Google Scholar
Author information
Authors and Affiliations
- Department of Internal Medicine, Division of Hematology/Oncology, The University of Michigan Comprehensive Cancer Center, Ann Arbor, MI, 48109-0548, USA
Kenneth L van Golen
Authors
- Kenneth L van Golen
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toKenneth L van Golen.
Additional information
Competing interests
None declared.
Rights and permissions
About this article
Cite this article
van Golen, K.L. Inflammatory breast cancer: Relationship between growth factor signaling and motility in aggressive cancers.Breast Cancer Res 5, 174 (2003). https://doi.org/10.1186/bcr598
- Published: 01 June 2003
- DOI: https://doi.org/10.1186/bcr598