DAVID: Database for Annotation, Visualization, and Integrated Discovery (original) (raw)
- Software
- Published: 14 August 2003
- Brad T Sherman1,
- Douglas A Hosack1,
- Jun Yang1,
- Wei Gao1,
- H Clifford Lane2 &
- …
- Richard A Lempicki1
Genome Biology volume 4, Article number: R60 (2003)Cite this article
- 96k Accesses
- 7400 Citations
- 12 Altmetric
- Metrics details
Abstract
The distributed nature of biological knowledge poses a major challenge to the interpretation of genome-scale datasets, including those derived from microarray and proteomic studies. This report describes DAVID, a web-accessible program that integrates functional genomic annotations with intuitive graphical summaries. Lists of gene or protein identifiers are rapidly annotated and summarized according to shared categorical data for Gene Ontology, protein domain, and biochemical pathway membership. DAVID assists in the interpretation of genome-scale datasets by facilitating the transition from data collection to biological meaning.
Rationale
The post-genomic era has introduced high-throughput methodologies that generate experimental data at rates that exceed knowledge growth. In particular, high-density biochips including complementary deoxyribonucleic acid (cDNA) microarrays, oligonucleotide microarrays, and rapidly evolving proteomics platforms represent modern tools able to interrogate biology on a genome-wide scale and generate tens of thousands of data points simultaneously [1]. While researchers are beginning to appreciate the statistical rigors required for the analysis of genome-scale datasets, a rate-limiting step in knowledge growth occurs at the transition from statistical significance to biological discovery.
A number of public efforts are currently focusing on the annotation and curation of gene-specific functional data, including LocusLink, Protein Information Resource (PIR), GeneCards, Proteome, Kyoto Encyclopedia of Genes and Genomes (KEGG), Ensembl, and Swiss-Prot to name but a few [2–8]. These resources provide exceptional depth and coverage of the functional data available for a given gene, but are not designed to effectively explore the biological knowledge associated with hundreds or thousands of genes in parallel. In order to facilitate the functional annotation and analysis of large lists of genes we have developed a Database for Annotation, Visualization, and Integrated Discovery (DAVID), which provides a set of data-mining tools that systematically combine functionally descriptive data with intuitive graphical displays [[9](/articles/10.1186/gb-2003-4-9-r60#ref-CR9 "DAVID. [ http://www.DAVID.niaid.nih.gov
]")\]. DAVID provides exploratory visualization tools that promote discovery through functional classification, biochemical pathway maps, and conserved protein domain architectures, while simultaneously remaining linked to rich sources of biological annotation. DAVID expedites the functional annotation and analysis of any list of genes encoded by human, mouse, rat, or fly genomes. DAVID's functionality is demonstrated using the Affymetrix GeneChip data of Cicala _et al_. \[[10](/articles/10.1186/gb-2003-4-9-r60#ref-CR10 "Cicala C, Arthos J, Selig SM, Dennis G, Hosack DA, Van Ryk D, Spangler ML, Steenbeke TD, Khazanie P, Gupta N, et al: HIV envelope induces a cascade of cell signals in non-proliferating target cells that favor virus replication. Proc Natl Acad Sci. 2002, 99: 9380-9385. 10.1073/pnas.142287999.")\].System architecture and maintenance
An automated procedure written in Microsoft Visual Basic (VB) 6.0 updates DAVID weekly with the following procedures: call a series of Perl and Java applications that download public data through anonymous file transfer protocols (FTP) (Table 1); unpack and parse desired annotation data; create tab-delimited data files ready for database import; and import data into an Oracle 8i relational database management system (RDBMS) using Oracle's SQL*Loader application. Microsoft's IIE web server and Active Server Page technology are used to access the database using JavaBeans and the structured query language (SQL). LocusLink numbers for Affymetrix probe sets are derived from University of Michigan associations [[11](/articles/10.1186/gb-2003-4-9-r60#ref-CR11 "Unigene annotation for Affy chips. [ http://dot.ped.med.umich.edu:2000/ourimage/pub/shared/JMR_pub_affyannot.html
]")\] or NetAffx \[[12](/articles/10.1186/gb-2003-4-9-r60#ref-CR12 "NetAffx Analysis Center. [
http://www.affymetrix.com/analysis/index.affx
]")\]. Functional annotations and database cross-references are derived from LocusLink, which provides stable, human-curated representations of genes. For more detailed information regarding the data sources used by DAVID please see the FAQ section at \[[9](/articles/10.1186/gb-2003-4-9-r60#ref-CR9 "DAVID. [
http://www.DAVID.niaid.nih.gov
]")\].Table 1 Sources of annotation data integrated into DAVID
Analysis modules
DAVID is composed of four main modules: Annotation Tool, GoCharts, KeggCharts, and DomainCharts. The Annotation Tool is an automated method for the functional annotation of gene lists. Any combination of annotation data can be chosen from 10 options by selecting the appropriate checkboxes (Table 2). The annotations are added to the submitted gene list by selecting the upload button, which returns an HTML table containing the user's original list of identifiers appended with the chosen functional annotations. Unannotated genes are included in the output with no appended data for tracking purposes.
Table 2 Options provided by the Annotation Tool
The GoCharts module graphically displays the distribution of differentially expressed genes among functional categories using the controlled vocabulary of the Gene Ontology Consortium (GO), which provides a structured language that can be applied to the functions of genes and proteins in all organisms even as knowledge continues to accumulate and change [13]. The language is structured in a directed acyclic graph (DAG), wherein term specificity increases and genome coverage decreases as one moves down the hierarchy. In contrast with a true hierarchy, child terms in a DAG may have more than one parent term and may have a different class of relationship with its different parents. The structure of GO starts with three main categories, Biological Process, Molecular Function, and Cellular Component. Biological Process includes broad biological goals, such as mitosis or purine metabolism, that are accomplished by ordered assemblies of molecular functions. Molecular Function describes the tasks performed by individual gene products; examples are transcription factor and DNA helicase. The Cellular Component classification type involves subcellular structures, locations, and macromolecular complexes; examples include nucleus, telomere, and origin recognition complex. After choosing a classification type, levels that determine list coverage and specificity are chosen by selecting the appropriate radio button. Level 1 provides the highest list coverage with the least amount of term specificity. With each increasing level coverage decreases while specificity increases so that level 5 provides the least amount of coverage with the highest term specificity.
Classification data is displayed as a bar chart, where the length of the bar represents the number of gene identifiers in each category. The user can set visualization parameters for sorting output data and displaying categories that contain at least a minimum number of genes. Selecting an individual bar opens a new HTML table displaying the gene identifier, LocusLink number, gene name, the current classification, and other classifications for each gene in that category. A 'Show All' button opens a new HTML table displaying all classification data and a 'Show Chart Data' button opens an HTML table containing the underlying chart data, thus allowing users to recreate customized chart graphics in a spreadsheet program. A new chart can be displayed for any subset of genes by selecting the classification type and level using the checkboxes and radio buttons available within the user's current page that allow for drill-down capabilities. A count of the number of genes annotated is included in the output, and unannotated genes are binned into the 'unclassified' category, thus providing users with an automated tracking system for genes not annotated.
KeggCharts graphically display the distribution of differentially expressed genes among KEGG biochemical pathways. Each pathway is linked to the KEGG pathway map, wherein differentially expressed genes from the original list are highlighted in red. In this view genes are further linked to additional annotations available through KEGG's DBGET retrieval system [6]. As with GoCharts, the user can set visualization parameters for sorting output data and displaying categories that contain at least a minimum number of genes and the KeggCharts visualization inherits all of the dynamic features of GoCharts.
DomainCharts display the distribution of differentially expressed genes among PFAM protein domains [14]. Each domain designation is linked to the Conserved Domain Database (CDD) of the National Center for Biotechnology Information (NCBI), where details regarding domain function, structure and sequence are readily available. As with GoCharts and KeggCharts, the user can set visualization parameters for sorting output data and displaying categories that contain at least a minimum number of genes and the DomainCharts visualization inherits all of the dynamic features of GoCharts and KeggCharts. For further information regarding the functionality of DAVID visit the FAQ section at [[9](/articles/10.1186/gb-2003-4-9-r60#ref-CR9 "DAVID. [ http://www.DAVID.niaid.nih.gov
]")\].Using DAVID to mine functional annotation
To demonstrate the functionality of DAVID we analyzed a list of genes differentially expressed in human peripheral blood mononuclear cells (PBMCs) after incubation with HIV-1 envelope proteins. Details of the experimental, RNA preparation, and GeneChip hybridization procedures, along with details of the chip-to-chip normalizations and statistical analysis of differential gene expression are provided in Cicala et al. [10]. Briefly, primary human PBMCs and monocyte-derived macrophages were incubated for 16 hours with HIV-1 envelope protein (gp120). High-density oligonucleotide microarrays (Affymetrix HU-95A GeneChip) were used to monitor gp120-induced transcriptional events. This analysis resulted in the identification of 402 differentially expressed genes.
Whereas 16 genes modulated by HIV-1 gp120 have previously been associated with HIV replication and/or envelope signaling, the remaining genes are of unknown function or have never been associated with HIV-1 or gp120. Converting this list of genes into biological meaning requires the gathering of pertinent information from several data repositories. For many researchers this process consists of iterative browsing through several databases for each gene, manually gathering gene-specific information regarding sequence, function, pathway, and disease association. In contrast, the systematic approach of DAVID simultaneously adds biologically rich information derived from several public data sources to lists of genes in parallel. Selecting DAVID's Annotation Tool and uploading the list of 402 differentially expressed genes initiates the functional annotation and analysis of the entire dataset. Once submitted, the gene list is stored for the entire analysis session, allowing users to switch between modules without having to resubmit data.
Annotation Tool
The Annotation Tool provides several annotation options and builds a tabular view of the users gene list and the available annotations (Table 2). Choosing the annotation fields Gene Symbol, LocusLink, OMIM, Unigene, Reference Sequence, and Gene Name followed by selecting the 'Upload' button produces an HTML table in the web browser containing all genes and their available annotations, where gene identifiers, descriptive and classification data are pulled from the database and appended to the gene list (Figure 1). Gene identifiers such as Gene Symbol and LocusLink are hyperlinked to additional gene-specific data available at their original sources, thus providing in-depth gene-specific details and annotation pedigrees. Classification data and functional summaries can be used to quickly scan for information relevant to the researcher's experimental system. The server time required for execution of this module correlates linearly with the size of the gene list and takes less than 45 seconds for lists of up to 1,000 genes (Figure 2, numbers in parentheses represent r2 values). These results demonstrate the power and efficiency of an integrated approach to the functional annotation of large datasets.
Figure 1

Ouput of Annotation Tool. Shown are appended annotations for the first several Affymetrix probe sets in an HTML table containing all 402 entries. Categorical information about the experimental conditions were submitted along with the Affymetrix probe-set identifiers and included in the output in the value column. Identifiers such as Symbol, LocusLink, OMIM, RefSeq, and Unigene accessions are hyper-linked to their origin sources for more detailed information. Text included in summary fields is derived from descriptive, functional information provided in NCBI's LocusLink reports.
Figure 2
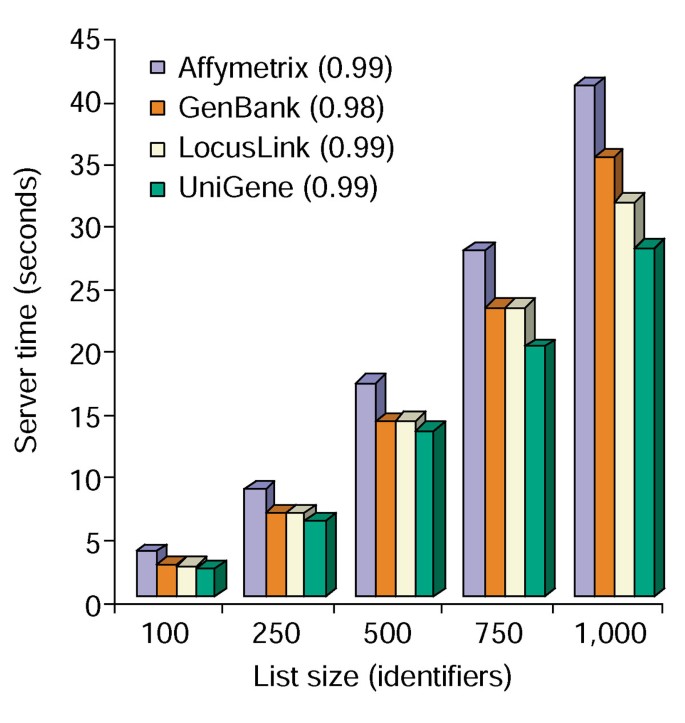
Time analysis of Annotation Tool. Server time required (y axis) to simultaneously append all 10 annotation options to gene lists ranging in size from 100 to 1,000 (x axis). The average of three trials for gene lists containing Affymetrix, GenBank, LocusLink, and UniGene identifiers are shown and the numbers in parentheses represent r2 value of the correlation between gene-list size and the server time required for annotation.
GoCharts
Choosing the GoCharts module opens a new window with a variety options. Users choose between three general types of classification (biological process, molecular function, and cellular component) and five levels of annotation that represent term coverage and specificity (see Analysis Modules section). Any combination of classification and coverage level can be specified. Also included are options to annotate gene lists with all GO terms available or only the most specific terms, which are referred to as terminal nodes. The option to choose different levels of term specificity provides needed flexibility and thus allows researchers to determine dynamically which level of coverage and specificity best suits their data and stage of analysis. For instance, early-stage analyses may consist of annotating gene lists with very general terms in order to gain a broad understanding of the data. In this case, selecting biological process and level 1 classifies genes using general terms such as 'death' and 'cell communication'. Using increased term specificity facilitates the extraction of more detailed functional information. In this case selecting biological process and level 5 classifies genes using terms such as 'apoptotic mitochondrial changes' and 'chemosensory perception'.
However, increased term specificity comes a cost, in that as it increases list coverage decreases (Figure 3). In our studies we find that level 2 typically maintains good coverage while also providing meaningful term specificity. Figure 4a illustrates how the GoCharts visualization quickly reveals that 35 differentially expressed genes are involved in 'stress responses'. Each GO term can be viewed in the tree or DAG views by hyperlinks to QuickGO [[15](/articles/10.1186/gb-2003-4-9-r60#ref-CR15 "QuickGO. [ http://www.ebi.ac.uk/ego/
]")\].Figure 3
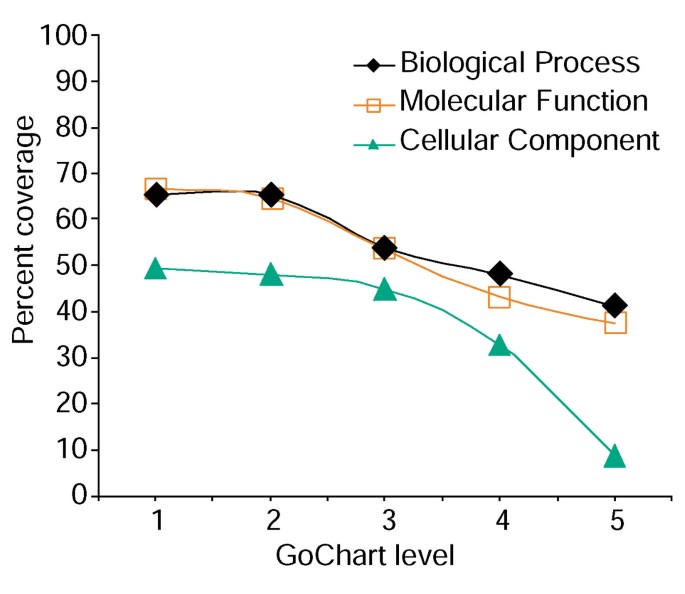
Analysis of gene-list coverage using GoCharts. A list of 402 Affymetrix probe set identifiers were annotated with the Proteome assigned functional classifications provided by LocusLink. Percent coverage represents the number of genes out of 402 that were annotated at a term-specificity level within the Biological Process, Molecular Function, and Cellular Component classification types. Percent coverage decreases as term specificity increases.
Figure 4
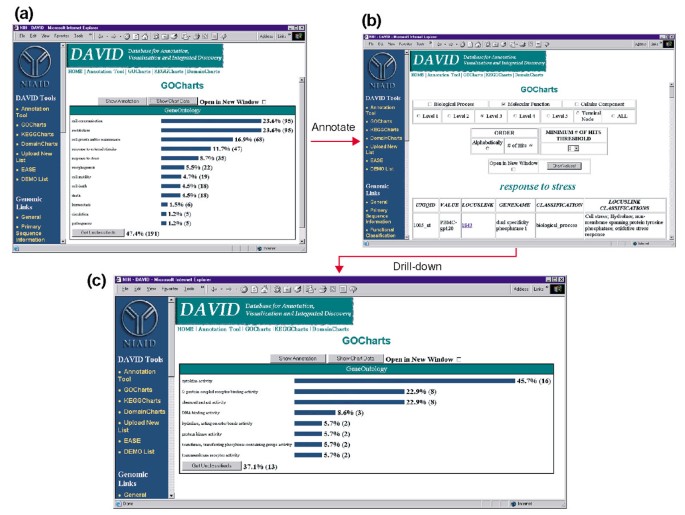
Ouput of GoCharts. (a) A bar chart showing the distribution of differentially expressed genes among Gene Ontology (GO) Biological Processes. Parameters were set to GO level 2, a hit threshold of five, and output was sorted by hit count. Blue bars are linked to additional annotation data shown in (b). Selecting the blue bar in (a) corresponding to 'response to stress' opens an HTML table showing the LocusLink, gene name, current classification, and other classification data for the genes in that category. (c) This subset of genes involved in 'stress response' was further characterized by selecting GO Molecular Function, GO level 3, a hit threshold of 2, and sorted by hit count. Selecting the 'Chart Values' button creates a new histogram revealing that 16 of the 35 stress-response genes encode proteins possessing cytokine activity.
Because HIV-1 has a major impact on the function of cells of the immune system and their ability to carry out stress responses, we selected the histogram bar representing the number of genes involved in stress response, which opens an HTML table containing the Affymetrix identifier, LocusLink number, gene name, the current classification, and other classifications for all 35 genes (Figure 4b). Now that we have reduced our gene list to those genes involved in stress responses, we further characterized this subset by repeating the GoCharts procedure available at the top of the stress-response HTML table. Choosing molecular function, level 3 produces a new histogram that quickly reveals that nearly half (16/35) of the stress-response genes possess cytokine activity (Figure 4c). Indeed, cytokines have been shown to play an important part in the HIV-1 life cycle and the results obtained here suggest that treatment of PBMCs with HIV-1 envelope proteins significantly modulates the transcription of numerous cytokine genes. The efficiency with which GoCharts systematically summarized this large dataset with graphic visualizations, while remaining linked to primary data and external resources drastically improved the discovery process.
KeggCharts
Figure 5a depicts the output of KeggCharts with a histogram displaying the distribution of differentially expressed genes among biochemical pathways. The chart shows that a KEGG pathway of apoptosis includes five genes induced by HIV-1 gp120. Selecting the pathway name opens the corresponding KEGG biochemical pathway map and highlights in red outline the differentially expressed genes functioning in that pathway (Figure 5b). In this view genes are further linked to additional annotations available through KEGG's DBGET retrieval system [6]. Note that only four genes in the KEGG apoptosis pathway are highlighted in red, while the KeggCharts tool mapped five Affymetrix probe sets to the apoptosis pathway. This difference is due to the fact that two of the Affymetrix probesets are targeting the same 'TNF-alpha' gene.
Figure 5
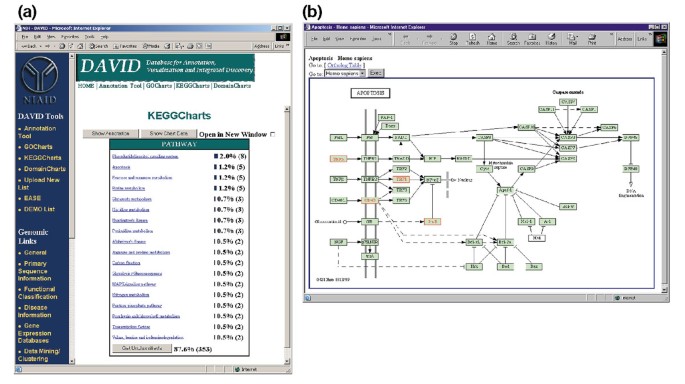
Ouput of KeggCharts. (a) Visualization chart showing the distribution of 402 genes among KEGG biochemical pathways. The hit threshold was set to three and the output was sorted by hit count. The large number of unclassified identifiers is due to the fact that KEGG is biochemical-pathway centric and thus provides low coverage of gene lists. Similarly to the output of GoCharts, blue bars represent the number of genes in each pathway. Selecting a blue bar opens an HTML table showing the LocusLink, gene name, current classification, and other classification data for the genes in that pathway (data not shown). (b) The KEGG biochemical pathway that appears following the selection of the pathway name 'apoptosis' in (a) depicts four differentially expressed genes within the apoptosis pathway by highlighting them in light green and red. The fact that the KEGG pathway highlights only four genes whereas the KeggChart maps five Affymetrix probe sets to the apoptosis pathway is due to the fact that two probe sets target the same 'TNF-alpha' gene.
DomainCharts
DomainCharts are operationally akin to both KeggCharts and GoCharts, except that the results visually depicting the distribution of genes among PFAM protein domains (Figure 6a). The DomainCharts histogram identifies 16 genes with kinase domains (pkinase), probably reflecting the effects of HIV-1 gp120 on the signal transduction machinery. The chart also identifies six genes with interleukin-8 domains (IL-8), a domain that represents a highly conserved motif among stress-response cytokines. Selecting the domain name 'IL8' opens the Conserved Domain Database (CDD) page corresponding to that PFAM domain (Figure 6b). This page provides detailed sequence, structure, and functional information about the IL-8 domain and the proteins that contain it.
Figure 6
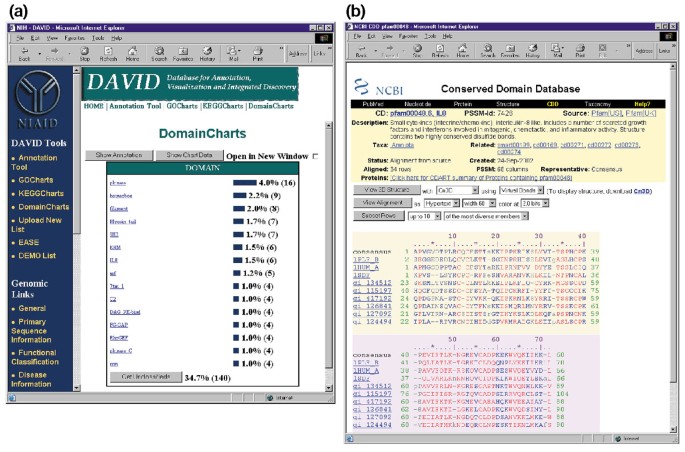
Ouput of DomainCharts. (a) Visualization chart showing the distribution of 402 genes among protein domains. The parameters were set to a minimum hit threshold of four and output was sorted by hit count. Similar to the output of GoCharts and KeggCharts, blue bars represent the number of genes containing that particular domain. Selecting a blue bar opens an HTML table showing the LocusLink, gene name, current classification, and other classification data for the genes in that pathway (data not shown). (b) Selecting the domain name 'IL8' in (a), which contains six differentially expressed genes, brings the user to a new page containing the output from the Conserved Domain Database (CDD) of NCBI, which provides detailed information about the IL-8 domain, including structural information, multiple sequence alignments, and descriptive information about the domain and the proteins that possess it.
Comparison of DAVID with related programs
Several other programs have overlapping and related functionality when compared with DAVID, but none combines all of DAVID's features within a single platform. These programs include ENSMART [[16](/articles/10.1186/gb-2003-4-9-r60#ref-CR16 "ENSMART. [ http://www.ensembl.org/EnsMart
]")\], FatiGO \[[17](/articles/10.1186/gb-2003-4-9-r60#ref-CR17 "FatiGO. [
http://fatigo.bioinfo.cnio.es
]")\], GeneLynx \[[18](/articles/10.1186/gb-2003-4-9-r60#ref-CR18 "GeneLynx. [
http://www.genelynx.org
]")\], GoMiner \[[19](/articles/10.1186/gb-2003-4-9-r60#ref-CR19 "GoMiner. [
http://discover.nci.nih.gov/gominer/index.jsp
]")\], MAPPFinder \[[20](/articles/10.1186/gb-2003-4-9-r60#ref-CR20 "GenMAPP including MAPPFinder. [
http://www.genmapp.org
]")\], MatchMiner \[[21](/articles/10.1186/gb-2003-4-9-r60#ref-CR21 "MatchMiner. [
http://discover.nci.nih.gov/matchminer/html/index.jsp
]")\], Resourcerer \[[22](/articles/10.1186/gb-2003-4-9-r60#ref-CR22 "Resourcerer. [
http://pga.tigr.org/tigr-scripts/magic/r1.pl
]")\] and Source \[[23](/articles/10.1186/gb-2003-4-9-r60#ref-CR23 "Source. [
http://source.stanford.edu/cgi-bin/SourceSearch
]")\], which collectively fall into two general categories: exploratory tools, defined as combining functional annotation with some form of graphical representation of summarized data; and annotation tools, defined as providing query-based access to functional annotation and producing a tabular output. FatiGO, GoMiner, and MAPPFinder are exploratory tools, whereas ENSMART, GeneLynx, MatchMiner, Resourcerer, and Source are strictly annotation tools that produce tabular output. A major advantage of DAVID is that it combines features of both categories, with GoCharts, KeggCharts, and DomainCharts representing exploratory tools, while DAVID's Annotation Tool produces a tabular output of functional annotation. We compared DAVID and these related programs on the basis of their available implementations and documentation as of May 2003, and the distribution of DAVID's functional features among these programs is shown in Table [3](/articles/10.1186/gb-2003-4-9-r60#Tab3).Table 3 Distribution of DAVID's features among related programs
Exploratory tools
FatiGO is a web-accessible application that functions in much the same way as DAVID's GoCharts, including the ability to specify term-specificity level. Unlike DAVID, FatiGO does not allow the setting of a minimum hit threshold for simplified viewing of only the most highly represented functional categories. Likewise, FatiGO limits the graphical output to only one top-level GO category at a time, whereas DAVID allows the combined viewing of biological process, molecular function, and cellular component annotations simultaneously. FatiGO's static barchart output looks very similar to DAVID's GoChart; an important distinction is that DAVID's GoCharts are dynamic, allowing users to drill-down and traverse the GO hierarchy for any subset of genes, view the underlying chart data and associated annotations, and link out to external data repositories including LocusLink and QuickGO. As shown in Table 3 the majority of accession types accepted and functional annotations offered by DAVID are not available from FatiGO.
GoMiner is a standalone Java application that requires downloading of the program itself along with at least two auxiliary files, one for DAG visualization and another for protein structural visualization. The remote database queried by GoMiner is reported to be updated every six months. It has been our experience that, to accurately reflect the current knowledge associated with a given gene, functional annotation data must be updated far more frequently. If users wish to use GoMiner with a local copy of its annotation database, they must also download and install a local copy of the MySQL database and the required drivers, a process that may be difficult for inexperienced users of MySQL. In contrast, DAVID is web-accessible and updated weekly. The functionality of GoMiner is most similar to DAVID's GoCharts module. An enhanced feature of GoMiner is that it provides intuitive tree and DAG views of genes embedded within the GO hierarchy. DAVID has the ability to display such views through hyperlinks of GO terms to QuickGO's tree and DAG views. A unique function provided by DAVID is the ability to drill-down and traverse the GO hierarchy for any subset of genes sharing a common classification, as demonstrated by the identification of stress response genes with cytokine activity. Neither the tree nor DAG view of GoMiner provides this functionality.
The body of biological knowledge associated with any list of genes extends far beyond the structured vocabulary of GO. DAVID provides, in addition to GoCharts, two additional analysis modules that utilize PFAM protein domain designations and KEGG biochemical pathways to graphically summarize the distribution of genes among functional domains and pathways. Moreover, DAVID highlights pathway members within the biochemical pathways provided by KEGG. Whereas GoMiner provides hyperlinks to pathway databases such as BioCarta and KEGG for individual genes, lists of genes can only be batch processed in the context of GO. In addition to providing hyperlinks to external data repositories for each gene, DAVID provides links to primary sequence information available at NCBI and human-curated functional summaries parsed from LocusLink. These features are not available in GoMiner. DAVID can be used to collect, analyze and explore functional annotation associated with human, mouse, rat, and Drosophila gene lists, whereas GoMiner is restricted to analyzing human data. Another restrictive feature of GoMiner is that it only takes HUGO gene symbols as input. This is problematic in that many genes and expressed sequence tags (ESTs) do not have HUGO symbols. Moreover, this restriction requires the translation of every gene list into HUGO symbols.
Like GoMiner, MAPPFinder is a stand-alone, exploratory tool for the analysis of lists of genes within the context of GO. The downloadable program comes with a copy of the supporting relational database of gene to GO-term associations. However, as with GoMiner there are important considerations regarding the installation, support, and updating of the software and underlying database, as indicated by the documentation and bug reports listed on their website. Importantly, in addition to the batch processing of gene lists within the context of GO, MAPPFinder provides functionality similar to that of DAVID's KeggCharts, providing the ability to view lists of genes within the context of biochemical pathways. However, in order to use this functionality through MAPPFinder, users must download additional programs and files, including the GenMAPP program and its associated MAPP files, whereas the KeggCharts module of DAVID is easily accessible at the click of a button.
Annotation tools
ENSMART is a web-accessible application that integrates an enormous amount of functional annotation for numerous species. ENSMART takes as input lists of several accession types, including Affymetrix probe sets, making it quite flexible. Database cross-references provided by ENSMART cover a broad spectrum of functional annotations pertaining to gene- and protein-specific attributes as well as disease and cross-species attributes. However, users are limited to a maximum of three cross-references for a given gene list. Unlike DAVID, ENSMART does not provide graphic summaries of GO categories, protein domains, or biochemical pathway membership, nor does ENSMART provide the ability to drill-down within groups of genes sharing common functional features.
GeneLynx and Source are highly similar web-accessible annotation tools that provide a wealth of gene-specific information for individual genes and both are flexible in that they take as input several different accession types. However, the rich information and available hyperlinks provided in single-gene mode is lost when either GeneLynx or Source are used to batch process lists of genes. The output of batch processing with Source is a text-style table that is feasible for download and automated processing, but provides little utility for interactive exploration. Although GeneLynx can perform batch searching for a list of genes, functional annotations must be viewed one gene at a time.
MatchMiner is a companion program of GoMiner that performs the translations of gene accession types into the HUGO symbols required by GoMiner. MatchMiner is simply a web-accessible resource for translating accession types. It takes several accession types but does not take LocusLink numbers, and although it was reported to accept identifiers from Affymetrix chip sets, MatchMiner returned no data for several gene lists composed of HuFL6800 probe sets. Notably, MatchMiner does not provide any functional annotation and is restricted to human data. Thus, within the context of the other exploratory and annotation tools discussed here, MatchMiner's utility is limited, or supportive, at best.
Resourcerer is a web-accessible application for comparing and annotating human, mouse, and rat GeneChip and microarray platforms. A major feature of Resourcerer is its broad coverage of microarray platforms and its ability to identify overlapping gene targets between chips, even across technology platforms and species barriers. Resourcerer's output is in tabular form and provides hyperlinks to accession cross-references such as GenBank and UniGene. Resourcerer does not provide graphic summaries or annotations from GO, PFAM, KEGG, or any other resource, thus limiting its utility as a tool for functional annotation.
Conclusions
In conclusion, the development of any complete, in-silico discovery system requires full, query-based access to an integrated, up-to-date view of all relevant information, regardless of its physical location and content structure. Still in its infancy, DAVID represents the foundation of our continued development efforts that aim to integrate information-rich data sources and provide quantitative summaries and analysis methods. In addition to the functionality reported here, the methods of Hosack et al. [24] have been incorporated into a DAVID analysis module called EASEonline, which allows users to identify statistically over-represented functional categories within a given list of genes. Committed to maintaining a system able to coevolve with technological advances and the new forms of data that are sure to follow, DAVID's current design elements provide automated solutions that enable researchers to rapidly discover biological themes in large datasets consisting of lists of genes.
References
- Quackenbush J: Computation analysis of microarray data. Nat Rev Genet. 2001, 2: 418-427. 10.1038/35076576.
Article PubMed CAS Google Scholar - Wheeler DL, Chappey C, Lash AE, Leipe DD, Madden TL, Schuler GD, Tatusova TA, Rapp BA: Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2000, 28: 10-14. 10.1093/nar/28.1.10.
Article PubMed CAS PubMed Central Google Scholar - Wu CH, Huang H, Arminski L, Castro-Alvear J, Chen Y, Hu Z-Z, Ledley RS, Lewis KC, Mewes H-W, Orcutt BC, et al: The Protein Information Resource: an integrated public resource of functional annotation of proteins. Nucleic Acids Res. 2002, 30: 35-37. 10.1093/nar/30.1.35.
Article PubMed CAS PubMed Central Google Scholar - Rebhan M, Chalifa-Caspi V, Prilusky J, Lancet D: GeneCards: a novel functional genomics compendium with automated data mining and query reformulation support. Bioinformatics. 1998, 14: 656-664. 10.1093/bioinformatics/14.8.656.
Article PubMed CAS Google Scholar - Costanzo MC, Crawford ME, Hirschman JE, Kranz JE, Olsen P, Robertson LS, Skrzypek MS, Braun BR, Hopkins KL, Kondu P, et al: YPD™, PombePD™, and WormPD™: model organism volumes of the BioKnowledge™ library, an integrated resource for protein information. Nucleic Acids Res. 2001, 29: 75-79. 10.1093/nar/29.1.75.
Article PubMed CAS PubMed Central Google Scholar - Kanehisa M, Goto S: KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28: 27-30. 10.1093/nar/28.1.27.
Article PubMed CAS PubMed Central Google Scholar - Hubbard T, Barker D, Birney E, Cameron G, Chen Y, Clark L, Cox T, Cuff J, Curwen V, Down T, et al: The Ensembl genome database project. Nucleic Acids Res. 2002, 30: 38-41. 10.1093/nar/30.1.38.
Article PubMed CAS PubMed Central Google Scholar - Gasteiger E, Jung E, Bairoch A: SWISS-PROT: connecting biomolecular knowledge via a protein database. Curr Issues Mol Biol. 2001, 3: 47-55.
PubMed CAS Google Scholar - DAVID. [http://www.DAVID.niaid.nih.gov]
- Cicala C, Arthos J, Selig SM, Dennis G, Hosack DA, Van Ryk D, Spangler ML, Steenbeke TD, Khazanie P, Gupta N, et al: HIV envelope induces a cascade of cell signals in non-proliferating target cells that favor virus replication. Proc Natl Acad Sci. 2002, 99: 9380-9385. 10.1073/pnas.142287999.
Article PubMed CAS PubMed Central Google Scholar - Unigene annotation for Affy chips. [http://dot.ped.med.umich.edu:2000/ourimage/pub/shared/JMR_pub_affyannot.html]
- NetAffx Analysis Center. [http://www.affymetrix.com/analysis/index.affx]
- The Gene Ontology Consortium: Gene ontology: tool for the unification of biology. Nature Genet. 2000, 25: 25-29. 10.1038/75556.
Article PubMed Central Google Scholar - Sonnhammer ELL, Eddy SR, Durbin R: Pfam: A comprehensive database of protein domain families based on seed alignments. Proteins. 1997, 28: 405-420. 10.1002/(SICI)1097-0134(199707)28:3<405::AID-PROT10>3.0.CO;2-L.
Article PubMed CAS Google Scholar - QuickGO. [http://www.ebi.ac.uk/ego/]
- ENSMART. [http://www.ensembl.org/EnsMart]
- FatiGO. [http://fatigo.bioinfo.cnio.es]
- GeneLynx. [http://www.genelynx.org]
- GoMiner. [http://discover.nci.nih.gov/gominer/index.jsp]
- GenMAPP including MAPPFinder. [http://www.genmapp.org]
- MatchMiner. [http://discover.nci.nih.gov/matchminer/html/index.jsp]
- Resourcerer. [http://pga.tigr.org/tigr-scripts/magic/r1.pl]
- Source. [http://source.stanford.edu/cgi-bin/SourceSearch]
- Hosack DA, Dennis G, Sherman BT, Lane HC, Lempicki RA: Identifying biological themes within lists of genes with EASE. Genome Biol. 2003, 4: P4-10.1186/gb-2003-4-6-p4.
Article Google Scholar - Searching GenBank. [http://www.ncbi.nlm.nih.gov/Genbank/GenbankSearch.html]
- UniGene. [http://www.ncbi.nlm.nih.gov/UniGene]
- RefSeq. [http://www.ncbi.nlm.nih.gov/RefSeq/]
- LocusLink. [http://www.ncbi.nlm.nih.gov/LocusLink]
- KEGG. [http://www.genome.ad.jp/kegg]
- OMIM. [http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=OMIM]
- Gene Ontology. [http://www.geneontology.org]
Acknowledgements
The authors are grateful to the referees for their constructive comments and thank Bill Wilton and Mike Tartakovsky for information technology and network support. The project has been funded with federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, under Contract No. NO1-C0-56000. The contents of this tool do not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products or organizations imply endorsement by the United States government.
Author information
Authors and Affiliations
- Science Applications International Corporation - Frederick, Clinical Services Program, Laboratory of Immunopathogenesis and Bioinformatics, National Cancer Institute at Frederick, MD, 21702, USA
Glynn Dennis Jr, Brad T Sherman, Douglas A Hosack, Jun Yang, Wei Gao & Richard A Lempicki - Laboratory of Immunoregulation, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD, 20892, USA
H Clifford Lane
Authors
- Glynn Dennis Jr
You can also search for this author inPubMed Google Scholar - Brad T Sherman
You can also search for this author inPubMed Google Scholar - Douglas A Hosack
You can also search for this author inPubMed Google Scholar - Jun Yang
You can also search for this author inPubMed Google Scholar - Wei Gao
You can also search for this author inPubMed Google Scholar - H Clifford Lane
You can also search for this author inPubMed Google Scholar - Richard A Lempicki
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toRichard A Lempicki.
Rights and permissions
About this article
Cite this article
Dennis, G., Sherman, B.T., Hosack, D.A. et al. DAVID: Database for Annotation, Visualization, and Integrated Discovery.Genome Biol 4, R60 (2003). https://doi.org/10.1186/gb-2003-4-9-r60
- Received: 04 April 2003
- Revised: 06 June 2003
- Accepted: 04 July 2003
- Published: 14 August 2003
- DOI: https://doi.org/10.1186/gb-2003-4-9-r60