Antenatal infection/inflammation and postnatal lung maturation and injury (original) (raw)
- Review
- Published: 11 January 2001
Respiratory Research volume 2, Article number: 27 (2001)Cite this article
- 11k Accesses
- 118 Citations
- Metrics details
Abstract
Chorioamnionitis is frequently associated with preterm deliveries before 30 weeks gestation. Chorioamnionitis correlates both with an increased risk of bronchopulmonary dysplasia and with a decreased risk of respiratory distress syndrome. Both interleukin-1α and endotoxin can induce inflammation in the fetal lungs and lung maturation after preterm birth when given by intra-amniotic injection. Inflammation can also result in an arrest of alveolarization, and this lung developmental abnormality is prominent in the lungs of preterm infants that die of bronchopulmonary dysplasia. The mechanisms by which infection/inflammation can have both beneficial and injurious effects on the preterm lung remain to be characterized.
Introduction
Recent information supports the concept that infection/inflammation resulting in preterm delivery can promote both early lung maturation and lung injury. As Fig. 1 illustrates, the lung continues to develop after preterm birth. The human lung undergoes a transition from the canalicular stage to the saccular stage of development at about 22 weeks, and alveolarization by secondary septal formation does not begin until about 30–32 weeks gestation [1]. Therefore, both clinical events in fetal life before preterm birth at 26 weeks gestation (65% of gestation) and preterm birth itself have the potential to adversely or beneficially influence lung function and the outcome of the infant. One of the themes of this review is that pro-inflammation is common to many of the events that occur with preterm birth, neonatal adaptation to air breathing, and subsequent lung function. The details of the modulation of lung injury and maturation by pro-inflammation have not yet been identified, and much of what follows is hypothetical and occasionally speculative. However, the epidemiologic information, some of the clinical associations, and the initial investigative work with model systems support the need for further research in this area.
Figure 1
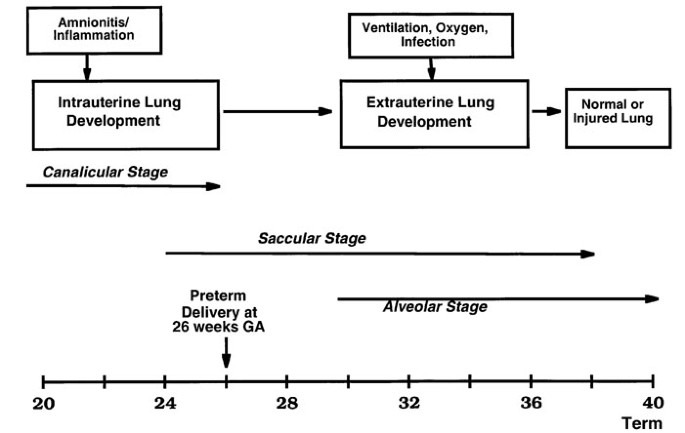
Lung development must continue for survival of the very low birth weight infant after preterm delivery at 26 weeks. Antenatal infection/inflammation associated with chorioamnionitis can modulate lung development, as can postnatal causes of inflammation such as mechanical ventilation, supplemental oxygen, or infection. The stages of lung development are modified from Burri [1].
Antenatal infection/inflammation, preterm delivery, and brain injury
Preterm fetuses infected with virulent organisms such as Escherichia coli, Group B Streptococcus or Listeria monocytogenes have sepsis/pneumonia syndromes. However, such presentations are rare before 30 weeks gestation. In contrast 50% or more of fetuses delivered before 30 weeks gestation have chronic chorioamnionitis, as indicated by histologic examination of the fetal membranes and cord, cell counts in the amniotic fluid, or culture positivity for vaginal and commensal organisms that are not generally considered pathologic [2]. More surprisingly, many of these fetuses may have been exposed to low-grade inflammation for prolonged periods, possibly beginning with colonization of the uterine tissue prior to conception. For reasons that are presently unknown, these pregnancies do not present with preterm labor before 20 weeks gestation, even if the amniotic fluid was positive for pro-inflammatory cytokines or organisms prior to the preterm labor.
These very preterm deliveries are the major contributors to postnatal morbidities and mortality, with the most common acute morbidities being brain injury and lung injury. The infants seldom have positive blood cultures after delivery, implying that infections had been isolated to the uterus, placenta, and/or fetal membranes. However, more recent information indicates that exposure to intrauterine infection/inflammation results in more serious injury than the preterm delivery alone. Although not the topic of this review, the major concern is that indicators of intrauterine inflammation may be associated with periventricular leukomalacia and subsequently cerebral palsy. Chorioamnionitis correlates with subsequent brain injury, and the presence of the pro-inflammatory cytokines interleukin-6 (IL-6), interleukin-1β (IL-1β), or tumor necrosis factor α (TNF-α) in amniotic fluid before preterm delivery predicts subsequent brain injury [3,4]. The fetus may respond to intra-amniotic infection with a systemic inflammatory response (defined as elevated IL-6 in plasma) and preterm infants with such a response are at increased risk of subsequent brain injury [5,6]. Therefore, factors associated with antenatal infection/injury increase the risk of brain injury after preterm birth, and this association may also explain much of the cerebral palsy that occurs after term birth [7].
Antenatal infection/inflammation and lung injury
The fetal lung is also a target for factors released by antenatal infection/inflammation. Most infants born after amnionitis do not have either positive tracheal cultures or a clear pneumonic process. However, as with the brain, lung injury progressing to bronchopulmonary dysplasia (BPD) can occur after exposure to either elevated pro-inflammatory cytokines in amniotic fluid or a systemic inflammatory response [5,8]. Yoon et al correlated elevated levels of IL-6 in cord plasma with the subsequent development of BPD after preterm birth [9]. Elevated levels of IL-6, IL-1β, IL-8, and TNF-α in amniotic fluid within 5 days of preterm birth also predicted the subsequent development of BPD [8]. The most common commensal organism associated with histologic chorioamnionitis after very preterm birth is Ureaplasma urealyticum [2], which induces a robust pro-inflammatory response characterized by elevations of the previously listed cytokines in the amniotic fluid [10]. Infants colonized with Ureaplasma at birth subsequently had a 43% incidence of BPD, in contrast to a comparison group with an incidence of 19% [11]. In another report, histologic chorioamnionitis was associated with an increased incidence of BPD, while the infants that developed it had elevated cytokine levels in tracheal aspirates on the first day after birth.
There are other reports of the association between indicators of antenatal infection/inflammation and subsequent lung injuries of various types after preterm birth. Matsuda et al reported that infiltration of the cord by white blood cells predicted BPD in infants [12]. Increased leukocyte elastase in tracheal aspirates at birth predicted the subsequent development of pulmonary emphysema, characteristic of the Wilson-Mikity variant of BPD [13]. Raised levels of amniotic fluid TNF-α correlated with an increased incidence of respiratory distress syndrome (RDS) [14]. In an autopsy series, about 50% of very low birth weight infants that died shortly after birth had histopathologic evidence of pulmonary infection/inflammation [15]. Not all reports found a striking relationship between antenatal infection/inflammation and subsequent lung injury. Variable outcomes probably result from the amount and duration of antenatal exposure to inflammation/infection and the fetal responses.
The link between inflammation/infection and BPD
The initial description of BPD by Northway, Rosan, and Porter [16] identified barotrauma and oxygen exposure as the causes of airway injury, fibrosis, and emphysema in ventilated preterm infants. Experimental models demonstrated that either barotrauma or oxygen exposure could cause lung injuries similar to BPD. Recently, inflammation characterized by activated recruitment of white blood cells to the lungs and pro-inflammatory cytokine production has been identified as common to both oxidant injury and barotrauma in the lungs of the adult and the developing infant [17,18,19]. Most infants that develop BPD weigh less than 1 kg at birth and are born before 28 weeks gestation. The unique aspect that distinguishes BPD from other lung injuries is that the injury is superimposed on a lung that is in the saccular stage of development. Fibrosis, airway injury, and emphysema are all minimal in infants who have died of BPD, with the dominant abnormality being an arrest in alveolar and microvascular development [20,21].
These anatomic findings can be reproduced in experimental models by oxygen exposure and ventilation. For example, newborn mice exposed to 85% oxygen develop inflammation and an arrest in secondary septation [19]. Preterm baboons ventilated for 7 days with 100% oxygen from 140-day gestation have fewer and larger alveoli, when evaluated as young adults [22]. Therefore high and prolonged oxygen exposure can arrest alveolar development. More clinically relevant are the observations that an arrest in alveolar and vascular development occurs in surfactant-treated preterm lambs and 125-day gestation baboons that are chronically ventilated with just sufficient pressure to maintain normal gas exchange, using supplemental oxygen at less than 50% [23,24]. Airway samples from these animals contain elevated levels of a variety of pro-inflammatory mediators and inflammatory cells. Therefore, ventilatory support similar to that used for preterm infants can arrest lung development.
The mediators that interfere with lung development are not known, nor are the normal modulators of the alveolarization process. However, there is information from transgenic mouse models that inflammatory mediators can interfere with postnatal alveolarization. Overexpression of TNF-α, transforming growth factor α (TGF-α), IL-11, or IL-6 in the lungs of developing mice results in variable pathologies that have in common an interference with alveolarization [25]. Infants destined to develop BPD have elevated levels of IL-1β, TNF-α, IL-8, and of multiple other cytokines, chemokines, and other promotors and products of inflammation in their airways soon after birth, and these markers of inflammation persist [26]. Preterm infants who develop sepsis or sepsis/pneumonia also are at increased risk of developing BPD [27]. Those born after exposure to amnionitis who also have a systemic inflammatory response, as defined by elevated IL-6 levels in cord blood, have an increased risk of developing BPD [9], as do infants born after exposure to elevated levels of IL-6, IL-8, IL-1β, and TNF-α in the amniotic fluid [8]. The anti-inflammatory cytokine IL-10 was undetectable in airway samples of preterms, which may strengthen the predisposition to persistence of chronic inflammation [28].
The inflammatory response is complex and no single component has been identified as being of primary importance in promoting lung injury or inhibiting alveolarization in the preterm. The preterm lung will differ in injury and immune responses from the adult lung because it contains almost no macrophages and has low levels of innate host defense elements, such as the surfactant proteins SP-A and SP-D, defensins, and lysozyme. Also, the white blood cells of the preterm show less response to inflammatory stimuli than their counterparts in the adult. There is a lack of information about how the preterm lung initiates and sustains inflammatory responses: information that is central to understanding injury progression to BPD, resulting in a disruption of alveolar and vascular development. The common theme is that inflammation interferes with alveolarization, but cause and effect have not been demonstrated. Also, alveolarization can be delayed by anti-inflammatory effectors such as glucocorticoids and by non-specific stresses such as caloric deficits. Alveolarization can also be stimulated or promoted by agents such as plasma thyroxine (T4) and retinoic acid [29]. The regulation of alveolarization remains to be understood.
Infection/inflammation and lung maturation
Fetal or neonatal exposure that can result in lung and brain injury is consistent with the general understanding that severe and chronic inflammation is damaging. However, the preterm fetus can apparently also benefit from an antenatal inflammatory stimulus. The clinical associations are not consistent in all reports, but are striking in several series. In a recent report of the outcomes of all infants born in England at less than 26 weeks gestation in 1995, chorioamnionitis was independently associated with decreased death (odds ratio 0.51, P < 0.005) [30]. Watterberg et al [31] reported that infants born to women with histologic chorioamnionitis had a decreased incidence of RDS but an increased incidence of BPD, relative to infants born to women without chorioamnionitis. The infants from the pregnancies with chorioamnionitis had elevated IL-1β levels in tracheal aspirates. Ureaplasma, the organism most frequently associated with chorioamnionitis, elevates cytokines in amniotic fluid, and colonization of preterm infants has been associated in some series with BPD. Hannaford et al [11] found that infants colonized at birth with Ureaplasma had significantly less RDS in the early neonatal period, but progressed to having significantly more BPD than uncolonized preterm infants. These selected examples of an association between inflammation and decreased RDS after preterm delivery but increased BPD are counterintuitive because in general RDS is thought to be the major predictor of BPD. However, it is now common in clinical practice to encounter very preterm infants with little or no RDS who progress to BPD [32,33]. The antenatal histories of these infants have not been well described, but it would seem reasonable to hypothesize that antenatal infection/inflammation may induce a pro-inflammatory response that is not appropriately suppressed after delivery, resulting in BPD.
The mechanisms that explain how inflammation/infection can induce lung maturation remain to be worked out, but there is some information demonstrating that pro-inflammatory cytokines can modulate lung development. Bry et al reported that intra-amniotic IL-1α induced the surfactant protein mRNAs for SP-A and SP-B, increased surfactant lipids, and improved the pressure-volume curves of preterm rabbits in a dose-dependent fashion [34]. In vitro IL-1α induces the mRNAs for SP-A, SP-B, and SP-C in rabbit lung explants from 19-day and 22-day fetuses, but suppresses surfactant mRNAs in lung explants from 27-day or 30-day fetuses, demonstrating that the cytokine effect is gestation-specific [35]. In an initial experiment, 125 μ g of human recombinant IL-1α, given by intra-amniotic injection, increased surfactant lipids and improved the pressure-volume curves of preterm sheep, without increasing fetal cortisol or altering postnatal kidney function [36]. Therefore, a single pro-inflammatory cytokine can promote changes consistent with lung maturation. However, IL-1α and endotoxin share common signaling pathways, and when intra-amniotic recombinant sheep IL-1α or IL-1β is given to sheep, it causes amnionitis (Jobe and Ikegami, unpublished data). Other mediators induced as part of this response could be modulating lung development.
In experiments designed to model clinical chorioamnionitis in sheep and to evaluate the lung responses, we used E. coli 055/β 5 endotoxin as the pro-inflammatory stimulus [37]. The intra-amniotic endotoxin induces IL-1β, IL-6, IL-8, and to a lesser extent TNF-α in the amnion/chorion within 5 hours. This cytokine response extinguishes within 2 days, but the numbers of inflammatory cells in the amnion/chorion remain elevated for many days [38]. Cytokine mRNA elevations are detected in the fetal lung 5 hours after exposure to intra-amniotic endotoxin and the greatest increases in cytokine mRNA are at 1–2 days, with baseline levels measured by 4 days. The systemic response to the intra-amniotic endotoxin is very modest, with no increases in fetal plasma cortisol and a small decrease in circulating white blood cells at 1–2 days, followed by an increase by 4–7 days.
Several aspects of the inflammatory response to intra-amniotic endotoxin are remarkable: there is no cytokine response in the placenta, liver, or gut, and there is no response at all if the endotoxin was injected into allantoic fluid. The fetal lung responds with increased mRNAs for SP-A, SP-B, SP-C, and SP-D by 1 day (a 6-fold increase for SP-A), with these increased mRNA levels persisting for 15 days after a single intra-amniotic injection of 20 mg of endotoxin [39]. The surfactant proteins in alveolar washes are beginning to increase 4 days after the endotoxin stimulus and have increased 100-fold from control values by 7 days. The fetal sheep at 125-day gestation has very little SP-B in the airspaces and only about 25% of this is the fully processed mature peptide. Intra-amniotic endotoxin induces processing of SP-B and a 100-fold increase in mature SP-B in the airspaces within 7 days. The saturated phosphatidylcholine in alveolar washes increases in parallel with the surfactant proteins, and compliance and pressure-volume curves are improved by 4 days after exposure to intra-amniotic endotoxin [40]. The lung responses are equivalent over a dose range of 1–100 mg of endotoxin, and the lung responses indicative of maturation correlate with inflammatory responses in the amnion/chorion and in lung tissue. These studies suggest that components of the inflammatory response are inducing lung maturation, although we are a long way from demonstrating cause and effect.
Antenatal glucocorticoids are inducers of early lung maturation that are used clinically. Watterberg et al [41] proposed that lung maturation was induced by the elevation of fetal cortisol levels by stress from amnionitis. At least in the fetal sheep model, intra-amniotic endotoxin does not increase fetal plasma cortisol [40], which means that other mediators are responsible for the effects of endotoxin on the fetal lung.
These experiments are consistent with the clinical observations that amnionitis can be associated with a decrease in RDS. The relationship between fetal exposure to inflammation and its subsequent association with BPD needs to be clarified. In adult lungs, prior exposure to endotoxin will result in an amplified inflammatory response to mechanical ventilation [42]. The initiation of mechanical ventilation of the preterm lung will recruit activated granulocytes to it, even with surfactant treatment and gentle ventilation. It can be hypothesized that ventilation or oxygen exposure of an inflamed preterm lung may aggravate and perpetuate the injury process [43]. The irony is that this effect may occur to a lung that has profited in the short term by being more mature, as a result of exposure to inflammation in utero.
BPD in tiny preterm infants is primarily an arrest of alveolar development. Antenatal glucocorticoids can arrest alveolarization in animals that initiate alveolarization before birth [29]. In sheep, alveolar development is arrested comparably by maternal betamethasone or intra-amniotic endotoxin given 7 days before preterm delivery [44]. The components of the inflammatory response that disrupt alveolar development in the fetal lung remain to be identified. With a bit of luck, the factors that favor maturation may be different from those that promote injury. However, the basic biologic question is: Can the lung have induced maturation without its subsequent development suffering? Put in practical terms: Does the lung that matures early have the same number of alveoli and capillaries as the normal adult lung and does it respond similarly to stresses? Answers to these questions are not available.
References
- Burri PH: Structural aspects of prenatal and postnatal development and growth of the lung. In: Lung Growth and Development. Edited by McDonald JA. New York: Marcel Dekker, Inc,. 1997, 1-35.
Google Scholar - Goldenberg RL, Hauth JC, Andrews WW: Intrauterine infection and preterm delivery. N Engl J Med. 2000, 342: 1500-1507. 10.1056/NEJM200005183422007.
Article CAS Google Scholar - Alexander JM, Gilstrap LC, Cox SM, McIntire DM, Leveno KJ: Clinical chorioamnionitis and the prognosis for very low birth weight infants. Obstet Gynecol. 1998, 91: 725-729. 10.1016/S0029-7844(98)00056-8.
Article CAS Google Scholar - Yoon BH, Jun JK, Romero R, Park KH, Gomez R, Choi JH, Kim IO: Amniotic fluid inflammatory cytokines (interleukin-6, interleukin-1beta, and tumor necrosis factor-alpha), neonatal brain white matter lesions, and cerebral palsy. Am J Obstet Gynecol. 1997, 177: 19-26.
Article CAS Google Scholar - Gomez R, Romero R, Ghezzi F, Yoon BH, Mazor M, Berry SM: The fetal inflammatory response syndrome. Am J Obstet Gynecol. 1998, 179: 194-202.
Article CAS Google Scholar - Leviton A, Paneth N, Reuss ML, Susser M, Allred EN, Dammann O, Kuban K, Van Marter LJ, Pagano M, Hegyi T, Hiatt M, Sanocka U, Shahrivar F, Abiri M, Disalvo D, Doubilet P, Kairam R, Kazam E, Kirpekar M, Rosenfeld D, Schonfeld S, Share J, Collins M, Genest D, Shen-Schwarz S: Maternal infection, fetal inflammatory response, and brain damage in very low birth weight infants. Developmental Epidemiology Network Investigators. Pediatr Res. 1999, 46: 566-575.
Article CAS Google Scholar - Grether JK, Nelson KB, Dambrosia JM, Phillips TM: Interferons and cerebral palsy. J Pediatr. 1999, 134: 324-332.
Article CAS Google Scholar - Yoon BH, Romero R, Jun JK, Park KH, Park JD, Ghezzi F, Kim BI: Amniotic fluid cytokines (interleukin-6, tumor necrosis factor-alpha, interleukin-1 beta, and interleukin-8) and the risk for the development of bronchopulmonary dysplasia. Am J Obstet Gynecol. 1997, 177: 825-830.
Article CAS Google Scholar - Yoon BH, Romero R, Kim KS, Park JS, Ki SH, Kim BI, Jun JK: A systemic fetal inflammatory response and the development of bronchopulmonary dysplasia. Am J Obstet Gynecol. 1999, 181: 773-779.
Article CAS Google Scholar - Yoon BH, Romero R, Park JS, Chang JW, Kim YA, Kim JC, Kim KS: Microbial invasion of the amniotic cavity with Ureaplasma urealyticum is associated with a robust host in fetal, amniotic, and maternal compartments. Am J Obstet Gynecol. 1998, 179: 1254-1260.
Article CAS Google Scholar - Hannaford K, Todd DA, Jeffery H, John E, Blyth K, Gilbert GL: Role of ureaplasma urealyticum in lung disease of prematurity. Arch Dis Child Fetal Neonatal Ed. 1999, 81: F162-167.
Article CAS Google Scholar - Matsuda T, Nakajima T, Hattori S, Hanatani K, Fukazawa Y, Kobayashi K, Fujimoto S: Necrotizing funisitis: Clinical significance and association with chronic lung disease in premature infants. Am J Obstet Gynecol. 1997, 177: 1402-1407.
Article CAS Google Scholar - Fujimura M, Kitajima H, Nakayama M: Increased leukocyte elastase of the tracheal aspirate at birth and neonatal pulmonary emphysema. Pediatrics. 1993, 92: 564-569.
CAS Google Scholar - Hitti J, Krohn MA, Patton DL, Tarczy-Hornoch P, Hillier SL, Cassen EM, Eschenbach DA: Amniotic fluid tumor necrosis factor-alpha and the risk of respiratory distress syndrome among preterm infants. Am J Obstet Gynecol. 1997, 177: 50-56.
Article CAS Google Scholar - Barton L, Hodgman JE, Pavlova Z: Causes of death in the extremely low birth weight infant. Pediatrics. 1999, 103: 446-451.
Article CAS Google Scholar - Northway WH, Rosan RC, Porter DY: Pulmonary disease following respirator therapy of hyaline-membrane disease: bronchopulmonary dysplasia. N Engl J Med. 1967, 276: 357-368.
Article Google Scholar - Chiumello D, Pristine G, Slutsky AS: Mechanical ventilation affects local and systemic cytokines in an animal model of acute respiratory distress syndrome. Am J Respir Crit Care Med. 1999, 160: 109-116.
Article CAS Google Scholar - Carlton DP, Albertine KH, Cho SC, Lont M, Bland RD: Role of neutrophils in lung vascular injury and edema after premature birth in lambs. J Appl Physiol. 1997, 83: 1307-1317.
CAS Google Scholar - Warner BB, Stuart LA, Papes RA, Wispe JR: Functional and pathological effects of prolonged hyperoxia in neonatal mice. Am J Physiol. 1998, 275: L110-117.
CAS Google Scholar - Hussain NA, Siddiqui NH, Stocker JR: Pathology of arrested acinar development in postsurfactant bronchopulmonary dysplasia. Hum Pathol. 1998, 29: 710-717.
Article Google Scholar - Coalson JJ: Pathology of chronic lung disease of early infancy. In Chronic Lung Disease in Early Infance. Edited by Bland RD, Coalson JJ. New York: Marcel Dekker, Inc,. 2000, 85-124.
Google Scholar - Coalson JJ, Winter V, deLemos RA: Decreased alveolarization in baboon survivors with bronchopulmonary dysplasia. Am J Respir Crit Care Med. 1995, 152: 640-646.
Article CAS Google Scholar - Coalson JJ, Winter VT, Siler-Khodr T, Yoder BA: Neonatal chronic lung disease in extremely immature baboons. Am J Respir Crit Care Med. 1999, 160: 1333-1346.
Article CAS Google Scholar - Albertine KH, Jones GP, Starcher BC, Bohnsack JF, Davis PL, Cho S, Carlton DP, Bland RD: Chronic lung injury in preterm lambs. Am J Respir Crit Care Med. 1999, 159: 945-958.
Article CAS Google Scholar - Jobe AH: The New BPD: An arrest of lung development. Pediatr Res. 1999, 46: 641-643.
Article CAS Google Scholar - Speer CP, Groneck P, Speer CP: Oxygen radicals, cytokines, adhesion molecules and lung injury in neonates. Seminars in Neonatology. 1998, 3: 219-228.
Article Google Scholar - Rojas MA, Gonzalez A, Bancalari E, Claure N, Poole C, Silva-Neto G: Changing trends in the epidemiology and pathogenesis of neonatal chronic lung disease. J Pediatr. 1995, 126: 605-610.
Article CAS Google Scholar - Jones CA, Cayabyab RG, Kwong KYC, Stotts C, Wong B, Hamdan H, Minoo P, deLemos RA: Undetectable interleukin (IL)-10 and persistent IL-8 expression early in Hyaline Membrane Disease: A possible development basis for the predisposition to chronic lung inflammation in preterm newborns. Pediatr Res. 1996, 39: 966-975.
Article CAS Google Scholar - Massaro DJ, Massaro GD: The regulation of the formation of pulmonary alveoli. In: Chronic Lung Disease in Early Infancy. Edited by Bland RD, Coalson JJ. New York: Marcel Dekker, Inc,. 2000, 479-492.
Google Scholar - Costeloe K, Hennessy E, Gibson AT, Marlow N, Wilkinson AR: The EPICure study: outcomes to discharge from hospital for infants born at the threshold of viability. Pediatrics. 2000, 106: 659-671.
Article CAS Google Scholar - Watterberg KL, Demers LM, Scott SM, Murphy S: Chorioamnionitis and early lung inflammation in infants in whom bronchopulmonary dysplasia develops. Pediatrics. 1996, 97: 210-215.
CAS Google Scholar - Charafeddine L, D'Angio CT, Phelps DL: Atypical chronic lung disease patterns in neonates. Pediatrics. 1999, 103: 759-765.
Article CAS Google Scholar - Bancalari E, Gonzalez A: Clinical course and lung function abnormalities during development of neonatal chronic lung disease. In: Chronic Lung Disease in Early Infancy. Edited by Bland RD, Coalson JJ. New York: Marcel Dekker, Inc,. 2000, 41-64.
Google Scholar - Bry K, Lappalainen U, Hallman M: Intra-amniotic interleukin-1 accelerates surfactant protein synthesis in fetal rabbits and improves lung stability after premature birth. J Clin Invest. 1997, 99: 2992-2999.
Article CAS Google Scholar - Glumoff V, Vayrynen O, Kangas T, Hallman M: Degree of lung maturity determines the direction of the interleukin-1-induced effect on the expression of surfactant proteins. Am J Respir Cell Mol Biol. 2000, 22: 280-288.
Article CAS Google Scholar - Emerson GA, Bry K, Hallman M, Jobe AH, Wada N, Ervin MG, Ikegami M: Intra-amniotic interleukin-1 alpha treatment alters postnatal adaptation in premature lambs. Biol Neonate. 1997, 72: 370-379.
Article CAS Google Scholar - Jobe AH, Newnham JP, Willet KE, Sly P, Ervin MG, Bachurski C, Possmayer F, Hallman M, Ikegami M: Effects of antenatal endotoxin and glucocorticoids on the lungs of preterm lambs. Am J Obstet Gynecol. 2000, 182: 401-408.
Article CAS Google Scholar - Kallapur SG, Willet KE, Jobe AH, Ikegami M, Bachurski C: Intra-amniotic endotoxin: chorioamnionitis precedes lung maturation in preterm lambs. Am J Physiol (Lung). 2000.
Google Scholar - Bachurski CJ, Ross GF, Ikegami M, Kramer BW, Jobe AH: Intra-amniotic endotoxin increases pulmonary surfactant components and induces SP-B processing in fetal sheep. Am J Physiol (Lung). 2000.
Google Scholar - Jobe AH, Newnham JP, Willet KE, Moss TJ, Ervin MG, Padbury JF, Sly PD, Ikegami M: Endotoxin induced lung maturation in preterm lambs. Am J Respirt Crit Care Med. 2000.
Google Scholar - Watterberg KL, Scott SM, Naeye RL: Chorioamnionitis, cortisol, and acute lung disease in very low birth weight infants. Pediatrics. 1997, 99: E6-
Article CAS Google Scholar - Tremblay LN, Slutsky AS, Dreyfuss D, Saumon G: Ventilator-induced lung injury: Mechanisms and clinical correlates. In: Physiological Basis of Ventilatory Support Edited by Marini JJ, Slutsky AS. New York: Marcel Dekker, Inc,. 1998, 395-451.
Google Scholar - Jobe AH, Ikegami M: Mechanisms initiating lung injury in the preterm. Early Hum Dev. 1998, 53: 81-94. 10.1016/S0378-3782(98)00045-0.
Article CAS Google Scholar - Willet K, Jobe A, Ikegami M, Brennan S, Newnham J, Sly P: Antenatal endotoxin and glucocorticoid effects on lung morphometry in preterm lambs. Pediatr Res. 2000.
Google Scholar
Acknowledgements
This work was supported in part by Grants HD-12714 and HL-65397 from the National Institutes of Health and Human Services.
Author information
Authors and Affiliations
- Children's Hospital Medical Center, 3333 Burnet Avenue, Cincinnati, Ohio, 45229-3039, USA
Alan H Jobe MD, PhD & Machiko Ikegami
Authors
- Alan H Jobe MD, PhD
You can also search for this author inPubMed Google Scholar - Machiko Ikegami
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toAlan H Jobe MD, PhD.
Rights and permissions
About this article
Cite this article
Jobe, A.H., Ikegami, M. Antenatal infection/inflammation and postnatal lung maturation and injury.Respir Res 2, 27 (2001). https://doi.org/10.1186/rr35
- Received: 06 November 2000
- Accepted: 13 December 2000
- Published: 11 January 2001
- DOI: https://doi.org/10.1186/rr35