Impaired Activity of the γ-Glutamyl Cycle in Nephropathic Cystinosis Fibroblasts (original) (raw)
Main
NC (MIM 219800) is an autosomal recessive disorder characterized by impaired lysosomal transport of cystine (1) and caused by mutations or deletions of the lysosomal amino acid carrier cystinosin (CTNS), encoded by the CTNS gene (17p13) (2,3).
Lysosome cystine accumulation leads to cell damage, which causes various clinical symptoms including hypothyroidism, diabetes mellitus, failure to thrive, progressive neuropathy and myopathy, and severe early-onset renal Fanconi syndrome. To date, the pathophysiology of cell damage in NC remains poorly understood.
In a previous study, we showed that patients with NC have elevated urinary pyroglutamic acid (PG) (5-oxoproline) excretion, which returns to near-normal levels after cysteamine treatment (4). PG and cytosolic cysteine represent two major metabolites of the ATP-dependent γ-glutamyl cycle, which constitutes the primary metabolic pathway for the synthesis of the soluble antioxidant agent GSH (Fig. 1) (5).
Figure 1
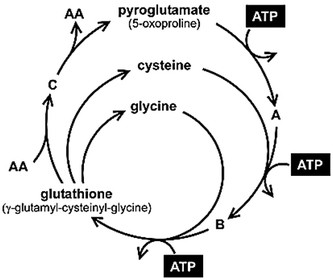
Schematic view of the γ-glutamyl cycle. A, glutamate; B, γ-glutamyl-cysteine; C, γ-glutamyl amino acid; AA, amino acid.
The aim of the present study was to further investigate the role of the γ-glutamyl cycle in NC using human fibroblast cell cultures obtained from NC patients.
METHODS
Cell cultures.
Human fibroblasts from four unrelated NC patients were kindly provided by the Cell Lines and DNA Bank of Patients Affected by Genetic Diseases (Laboratorio di Diagnosi Pre e Postnatale delle Malattie Metaboliche, Istituto G. Gaslini, Italy, partially supported by the Telethon Foundation). Control fibroblasts were isolated from skin biopsies obtained from four unrelated healthy subjects. Cells were grown at 37°C in humidified 5% CO2/95% air in Dulbecco modified Eagle medium supplemented with 10% fetal bovine serum, 100 U/mL penicillin, and 100 mg/mL streptomycin. Cell culture media and solutions were from GIBCO-Invitrogen (Paisley, UK).
Measurement of intracellular ATP.
Intracellular ATP levels were measured with the ATP Bioluminescence Assay Kit CLS II (Roche, Mannheim, Germany), according to the protocol provided by the vendor. Samples were analyzed on a Victor2 1420 Multilabel Counter (Perkin Elmer, Boston, MA). ATP concentrations were calculated from a log-log plot of the standard curve. The data were expressed as nmol/mg of proteins.
Measurement of intracellular PG.
Intracellular PG levels were determined by the SCAN method as previously described (4). Briefly, samples were subjected to oximation and extracted with ethyl acetate. The organic phase was then evaporated and the derivatized residue was injected on a gas chromatograph–mass spectrometer (GC-MS) consisting of a HP 5890 series II gas chromatograph (Hewlett-Packard, Palo Alto, CA) equipped with an Agilent Ultra 2 GC column (Agilent Technologies, Palo Alto, CA) and a HP 5972 mass spectrometer. PG was quantified by GC-MS in SIM mode by internal standard calibration (10 mmol/L tricarballylic acid). PG and tricarballylic acid were measured in electron-impact ionization mode at m/z 156–m/z 258 and m/z 302–m/z 377, respectively. Results were expressed as nmol/mg of proteins.
Detection of intracellular cysteine, γ-glutamylcysteine and GHS.
These measurements were performed according to a previously reported protocol (6) with minor modifications. Briefly, cells were divided into two aliquots, one of which was treated with _N_-ethylmaleimide (NEM) to a final concentration of 5 mmol/L for the measurement of the oxidized forms of thiols. The untreated aliquot was used to measure total [reduced + oxidized] thiols. Cells were sonicated on ice and cell lysates were treated with sulfosalicylic acid for protein precipitation. Protein concentrations were measured with a BCA assay (Pierce, Rockford, IL). The acid-soluble fractions were derivatized and analyzed by high-performance liquid chromatography/fluorescence detection (FD) on an Agilent Technologies Model 1100 instrument (Agilent Technologies), equipped with a Hypersil-ODS chromatographic column (Thermo-Hypersil, Bellefonte, PA). Results were expressed as nmol/mg of proteins.
Respiratory chain enzyme activity.
The enzymatic activity of the respiratory chain components was determined by spectrophotometric measurements performed on 80–100 μg of proteins obtained from digitonin-treated fibroblasts as previously reported (7). Briefly, citrate synthase (CS) activity was measured on cell extracts incubated for 3 min at 30°C in 80 mmol/L Tris-HCl pH 8.0, 100 mmol/L 5,5′-dithiobis(2-nitrobenzoic acid), 0.1% Triton X 100, 400 mmol/L acetyl-CoA, in a final volume of 1 mL. Oxaloacetate (500 mmol/L) was then added and citrate synthesis was followed at 412 nm (extinction coefficient 13.6 mmol/L−1 cm−1) on a Beckman DU640 spectrophotometer (Beckman Coulter, Fullerton, CA). For cytochrome c oxidase (COX) activity, cell extracts were incubated in 10 mmol/L potassium phosphate pH 7.4, 1 mg/mL cytochrome c reduced with sodium dithionite, and 0.025% lauryl maltoside, in a final volume of 1 mL. The oxidation of cytochrome c was followed at 550 nm (extinction coefficient 18.5 mmol/L−1 cm−1). Results were expressed as nmol/min/mg of protein and normalized to CS activity.
Measurement of apoptotic and necrotic cell death.
Apoptotic and necrotic cell death was determined by flow cytometry performed with the Annexin V-FITC Apoptosis Detection Kit (Oncogene Research Products, Boston, MA) according to manufacturer's specifications. Briefly, cells were harvested and resuspended in phosphate-buffered saline to approximately 106 cells/mL. After 15-min incubation in the dark in the presence of 0.5 mg/mL Annexin V-FITC, cells were centrifuged at 1000 × g for 5 min, stained with 0.6 mg/mL propidium iodide, and immediately analyzed on a FACSCalibur System equipped with Cell Quest Pro software (Becton Dickinson, San Jose, CA). Apoptosis and/or necrosis were expressed as a percentage of the total cell count.
Chemicals.
Unless otherwise specified, all chemicals were of analytic grade and purchased from Sigma Chemical Co. (St. Louis, MO).
Calculations and statistics.
All experiments were performed in triplicate. Values were expressed as mean ± SE in figures and mean ± SD in the text. Statistical analysis was performed with the two-way t test. A p value <0.05 was considered significant.
RESULTS
Intracellular ATP, PG, and GSH levels in human fibroblasts.
The activity of the γ-glutamyl cycle was analyzed in fibroblasts isolated from four unrelated NC patients and four control subjects. Intracellular one-half cystine concentrations were 23 ± 5 nmol/mg of proteins in NC fibroblasts and 1.8 ± 0.4 nmol/mg of proteins in controls (p < 0.0001). These elevated cystine levels did not allow the measurement of cell cysteine content, especially in conditions characterized by low cysteine concentrations (cystine/cysteine ratio >10).
Under basal conditions, PG concentrations were 0.65 ± 0.08 nmol/mg of proteins in NC cells and 0.35 ± 0.06 nmol/mg of proteins in control cells (p < 0.01) (Fig. 2B). Intracellular ATP and GSH levels were approximately 10% lower in NC fibroblasts, but these differences were not statistically significant (Fig. 2A, C). GSSG concentrations were 1.46 ± 0.24 nmol/mg of proteins in NC cells and 0.97 ± 0.18 nmol/mg of proteins in controls (p = not significant [NS]).
Figure 2
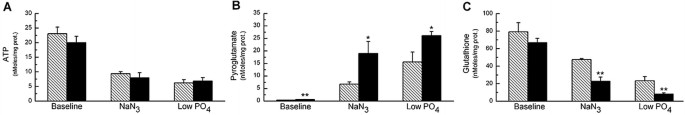
Intracellular ATP (A), PG (B), and total GSH (C) levels in control human fibroblasts and NC fibroblasts. Cells were incubated for 72 h in low-phosphate medium or with low concentration of NaN3 to obtain a similar decrease in ATP levels of approximately 65% (see text for details). CTNS+/+ (hatched columns); CTNS −/− (solid columns). *p < 0.05; **p < 0.01. n = 12.
To test the ATP sensitivity of the γ-glutamyl cycle, cells were incubated in culture conditions known to reduce intracellular ATP concentrations without causing other prominent cytotoxic effects. Preliminary tests were performed to identify optimal NaN3 or phosphate concentrations in culture medium and duration of exposure (data not shown). When cells were incubated for 72 h in the presence of 70 μmol/L NaN3 or in medium with phosphate concentrations reduced to 65 μmol/L, ATP levels were reduced by approximately 55% (p < 0.001), without significant differences between NC and control cells (Fig. 2A). Under these experimental conditions, cell growth was hindered, but no appreciable cell death was observed.
ATP depletion reduced the activity of the γ-glutamyl cycle in both NC and control fibroblasts, as revealed by a marked increase in intracellular PG levels and a decrease in GSH concentrations. These changes were significantly more pronounced in NC cells (Fig. 2B, C). A similar inverse correlation between GSH and PG levels was observed in the two groups of cells (R = −0.98, p < 0.0001), indicating that PG accumulation reflects the degree of inhibition of the γ-glutamyl cycle similarly in both cell types, but that ATP depletion inhibits GSH synthesis considerably more in NC cells.
GSH synthesis after exposure to oxidative stress.
Because GSH is one of the main cellular antioxidants, cell response to a prolonged oxidative stress was directly tested by exposing fibroblasts for 72 h to 100 mmol/L H2O2. As shown in Figure 3, intracellular ATP was reduced by approximately 10% in both cell types, when compared with control conditions (p = NS). Control fibroblasts responded to the oxidative stress by upregulating GSH synthesis (+110 ± 18%, p < 0.005), whereas in NC cells, GSH synthesis was increased only by 17 ± 5% (p = NS). Oxidized GSH (GSSG) remained below 4% of total GSH. Intracellular levels of γ-glutamyl cysteine, the direct precursor of GSH, ranged from 9 to 11 nmol/mg of proteins in all experiments, with no significant differences between control and NC fibroblasts or between treated and untreated cells. H2O2 treatment produced a 2.2-fold increase in PG levels in both cell types. Absolute PG concentrations were 85% higher in NC cells than in controls.
Figure 3
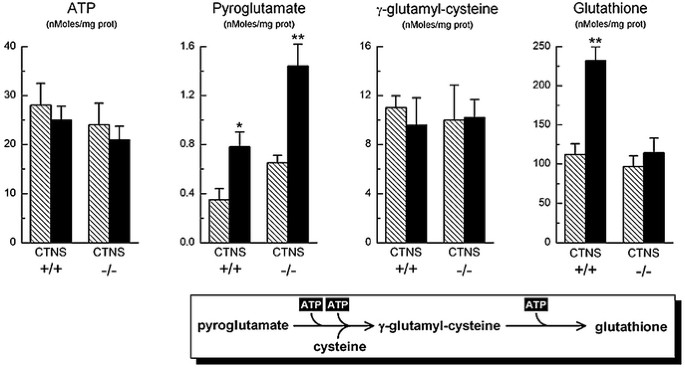
Activation of the γ-glutamyl cycle after exposure to oxidative stress. Control and NC fibroblasts were cultured in the presence of 100 mmol/L H2O2 for 72 h. Cells were then harvested and divided into aliquots for the determination of the reported metabolites. As shown, fibroblasts from patients with NC were unable to increase significantly their synthesis of GSH. To facilitate data reading, the flow chart at the bottom summarizes a partial sequence of the γ-glutamyl cycle (see also Fig. 1). Baseline (hatched columns); +H2O2 (solid columns). *p < 0.05; **p < 0.01; n = 12.
Respiratory chain enzyme activity and apoptotic cell death. Because GSH plays an important role in the prevention of mitochondrial oxidative damage, the activities of the mitochondrial enzymes CS and COX were measured in cells cultured for 72 h under the previously described experimental conditions (i.e. 70 μmol/L NaN3, 100 mmol/L H2O2, 65 μmol/L phosphate). Results revealed no significant differences between NC and control fibroblasts, neither in the activities of these enzymes nor in the COX/CS ratios (data not shown). Incubation in low-phosphate medium triggered apoptosis in both NC and control fibroblasts but without differences between the two groups of cells (NC cells: baseline, 8.7 ± 5.9%; low phosphate, 21.4 ± 2.1%; p < 0.02; control cells: baseline, 8.2 ± 7.3%; low phosphate, 19.2 ± 1.0%; p < 0.02). Similarly, no differences in the rates of apoptosis were found between NC and control cells incubated in medium containing 70 μmol/L NaN3 or 100 mmol/L H2O2 (data not shown).
DISCUSSION
Although important progress have been made in the understanding of the molecular determinants of NC (8,9), the sequence of events leading to cell dysfunction still remains poorly understood.
The present study originates from the observation of increased PG excretion in urine of cystinotic patients at the onset of their renal symptoms (4). A number of in vitro data suggest that proximal tubular ATP depletion, impaired oxidative metabolism, and mitochondrial damage play a central role in the development of NC-associated Fanconi syndrome (10,11). Therefore, pyroglutamic aciduria in NC may purely indicate inhibition of the ATP-dependent γ-glutamyl cycle in renal tubular cells.
The activity of the γ-glutamyl cycle is also known to depend on the availability of cytosolic cysteine, which constitutes a limiting factor for the de novo synthesis of GSH (5). The original data from Oshima et al. (12) show that cultured human fibroblasts can use the cysteine-derived form of cystine reduction for GSH or protein synthesis. Cysteine is also transported into lysosomes by a cysteine-specific lysosomal carrier (13). Once in the lysosome, cysteine reacts with protein disulfide bridges leading to the formation of cystine (14), which is transported out of the lysosome by the CTNS carrier and again reduced into cysteine by GSH (13). This complex recycling process through the lysosomal pathway is also indirectly suggested by the observation that cystine-depleted NC fibroblasts incubated for 30 min in the presence of 35S-cystine show a 35% reduction in the 35S-cysteine content (12). More recently, Chol et al. (15) have also shown that decreased GSH content in NC fibroblasts can be restored by exogenous precursors of cysteine, which further indicates that cystine derived from lysosomal efflux is a valuable physiologic source of cytosolic cysteine.
We therefore measured PG and GSH levels in primary fibroblast cell cultures obtained from cystinotic patients and control subjects. Although under standard culture conditions, PG concentrations in cystinotic fibroblasts were low, they were significantly higher than in control cells. Such limited increase in PG concentrations is most likely irrelevant in terms of cell metabolism but may indicate impaired activity of the γ-glutamyl cycle.
To further investigate the activity of the γ-glutamyl cycle, we manipulated intracellular ATP concentrations. For this purpose, we had to meet three conditions. First, ATP levels had to be decreased to the same extent in both groups of cells. Second, drug-induced toxic effects had to be avoided as much as possible. Finally, cells had to be treated for a prolonged period of time to detect significant changes in GSH levels. We have been able to meet these conditions by limiting inorganic phosphate in the culture medium or by inhibiting oxidative phosphorylations with very low doses of NaN3 for 72 h. Under these conditions, our data showed that PG accumulates significantly more in NC fibroblasts than in control cells, reflecting a more severe state of GSH depletion. Although these results were obtained in nonphysiologic conditions, they highlight the close metabolic relationships that link GSH, cysteine, and ATP, as illustrated in Figure 4.
Figure 4
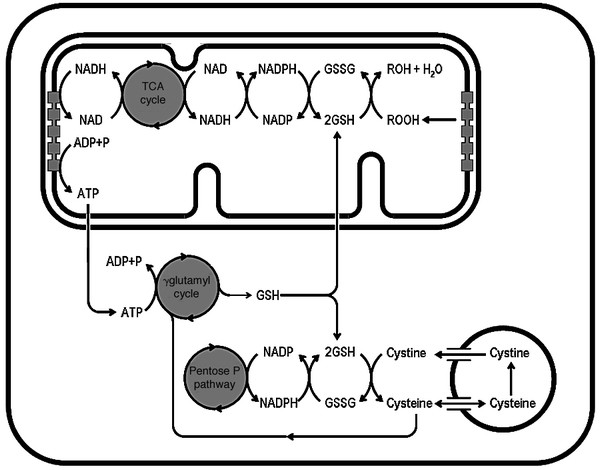
Schematic representation of the metabolic interconnections between cysteine, GSH metabolism, and ATP synthesis. Mitochondrial GSH is essential for neutralizing free radicals released during oxidative phosphorylations, which generate ATP. ATP in turn is required for GSH synthesis by the γ-glutamyl cycle and for cystine efflux from lysosomes (not shown). Cysteine is, on the one hand, the major limiting substrate for GSH synthesis. On the other hand, cysteine is generated by GSH-mediated reduction of cystine, which is in part supplied to the cytosol by lysosomes through the CTNS pathway. This pathway is also responsible for recycling cysteine that has been sequestered in the lysosomal compartment after transport through a cysteine-specific carrier.
Because GSH is a major cellular antioxidant, cell response to hydrogen peroxide was studied according to previously reported experimental protocols (16,17). Our data show that cystinotic fibroblasts are unable to increase GSH synthesis in response to oxidative stress. Stimulation of the γ-glutamyl cycle in NC cells produced a moderate increase in PG levels, but γ-glutamyl cysteine concentrations remained unchanged, indicating that the cycle was impaired at a step downstream of PG synthesis and upstream of the binding of glycine with γ-glutamyl cysteine. This step corresponds to the entry of cysteine into the cycle. This finding further supports the hypothesis that GSH synthesis is limited by decreased cysteine availability in cystinotic fibroblasts exposed to oxidative stress. Unfortunately, the large cystine content of cystinotic cells did not allow accurate evaluation of cysteine concentrations by subtraction of the NEM-reactive fraction from the total cysteine/cystine pool.
In addition, Chol et al. (15) have recently reported similar activities of GSH reductase and GSH peroxidase in cystinotic and control fibroblasts. These findings further support the hypothesis that decreased GSH synthesis primarily results from lack of precursors. Similarly, Oshima et al. (12) have also provided indirect evidence indicating normal activity of the γ-glutamyl cysteine synthetase in NC fibroblasts.
In our experimental conditions, no significant differences were detected in the activities of the mitochondrial enzymes COX and CS or in the rate of apoptosis between NC and control fibroblasts. These results are in contrast to previously published data showing severe mitochondrial damage during GSH depletion (18) or increased rate of apoptosis in cystinotic fibroblasts exposed to various apoptotic stimuli (19). These discrepancies are most likely due to differences in the experimental conditions, which in the present study were primarily aimed at studying the γ-glutamyl cycle under minimal cell damage conditions.
Based on the present data, it can be speculated that several clinical and biologic symptoms of NC, including altered oxidative metabolism of phagocytic cells (20), increased rate of apoptosis (19), central and peripheral nervous system complications (21,22), glucose intolerance (23,24), and the overall accelerated aging process that we have observed in a number of patients, could be, at least in part, attributed to increased susceptibility to oxidative stress. On the other hand, since these cells are the primary sites for cystine reabsorption, it is difficult to envision that the activity of the γ-glutamyl cycle is impaired in proximal tubular epithelia solely as a consequence of substrate depletion. More work is still needed to elucidate the complex sequence of events leading from lysosomal cystine accumulation to proximal tubular cell dysfunction and Fanconi syndrome in NC.
Abbreviations
COX:
cytochrome c oxidase
CS:
citrate synthase
CTNS:
cystinosin
GSH:
glutathione
GSSG:
oxidized glutathione
NC:
nephropathic cystinosis
PG:
pyroglutamic acid
References
- Gahl WA, Bashan N, Tietze F, Bernardini I, Schulman JD 1982 Cystine transport is defective in isolated leukocyte lysosomes from patients with cystinosis. Science 217: 1263–1265
Article CAS Google Scholar - Town M, Jean G, Cherqui S, Attard M, Forestier L, Whitmore SA, Callen DF, Gribouval O, Broyer M, Bates GP, van't Hoff W, Antignac C 1998 A novel gene encoding an integral membrane protein is mutated in nephropathic cystinosis. Nat Genet 18: 319–324
Article CAS Google Scholar - Mason S, Pepe G, Dall'Amico R, Tartaglia S, Casciani S, Greco M, Bencivenga P, Murer L, Rizzoni G, Tenconi R, Clementi M 2003 Mutational spectrum of the CTNS gene in Italy. Eur J Hum Genet 11: 503–508
Article CAS Google Scholar - Rizzo C, Ribes A, Pastore A, Dionisi-Vici C, Greco M, Rizzoni G, Federici G 1999 Pyroglutamic aciduria and nephropathic cystinosis. J Inherit Metab Dis 22: 224–226
Article CAS Google Scholar - Lu SC 1999 Regulation of hepatic glutathione synthesis: current concepts and controversies. FASEB J 13: 1169–1183
Article CAS Google Scholar - Pastore A, Massoud R, Motti C, Lo Russo A, Fucci G, Cortese C, Federici G 1998 Fully automated assay for total homocysteine, cysteine, cysteinylglycine, glutathione, cysteamine, and 2-mercaptopropionylglycine in plasma and urine. Clin Chem 44: 825–832
CAS PubMed Google Scholar - Robinson BH, Glerum DM, Chow W, Petrova-Benedict R, Lightowlers R, Capaldi R 1990 The use of skin fibroblast cultures in the detection of respiratory chain defects in patients with lacticacidemia. Pediatr Res 28: 549–555
Article CAS Google Scholar - Kalatzis V, Antignac C 2003 New aspects of the pathogenesis of cystinosis. Pediatr Nephrol 18: 207–215
PubMed Google Scholar - Baum M 1998 The Fanconi syndrome in cystinosis: insights into the physiopathology. Pediatr Nephrol 12: 492–497
Article CAS Google Scholar - Coor C, Salmon RF, Quigley R, Marver D, Baum M 1991 Role of adenosine triphosphate (ATP) and Na-K-ATPase in the inhibition of proximal tubular transport with intracellular cystine loading. J Clin Invest 87: 955–961
Article CAS Google Scholar - Sakarcan A, Aricheta R, Baum M 1992 Intracellular cystine loading causes proximal tubule respiratory dysfunction: effect of glycine. Pediatr Res 32: 710–713
Article CAS Google Scholar - Oshima RG, Rhead WJ, Thoene JG, Schneider JA 1976 Cystine metabolism in human fibroblasts. Comparison of normal, cystinotic, and gamma-glutamylcysteine synthetase-deficient cells. J Biol Chem 251: 4287–4293
CAS PubMed Google Scholar - Pisoni RL, Acker TL, Lisowski KM, Lemons RM, Thoene JG 1990 A cysteine-specific lysosomal transport system provides a major route for the delivery of thiol to human fibroblast lysosomes: possible role in supporting lysosomal proteolysis. J Cell Biol 110: 327–335
Article CAS Google Scholar - Lloyd JB 1986 Disulphide reduction in lysosomes. The role of cysteine. Biochem J 237: 271–272
Article CAS Google Scholar - Chol M, Nevo N, Cherqui S, Antignac C, Rustin P 2004 Glutathione precursors replenish decreased glutathione pool in cystinotic cell lines. Biochem Biophys Res Commun 324: 231–235
Article CAS Google Scholar - Wong A, Yang J, Cavadini P, Gellera C, Lonnerdal B, Taroni F, Cortopassi G 1999 The Friedreich's ataxia mutation confers cellular sensitivity to oxidant stress which is rescued by chelators of iron and calcium and inhibitors of apoptosis. Hum Mol Genet 8: 425–430
Article CAS Google Scholar - Campo GM, D'Ascola A, Avenoso A, Campo S, Ferlazzo AM, Micali C, Zanghi L, Calatroni A 2004 Glycosaminoglycans reduce oxidative damage induced by copper (Cu+2), iron (Fe+2) and hydrogen peroxide (H2O2) in human fibroblast cultures. Glycoconj J 20: 133–141
Article CAS Google Scholar - Martensson J, Meister A 1989 Mitochondrial damage in muscle occurs after marked depletion of glutathione and is prevented by giving glutathione monoester. Proc Natl Acad Sci U S A 86: 471–475
Article CAS Google Scholar - Park M, Helip-Wooley A, Thoene J 2002 Lysosomal cystine storage augments apoptosis in cultured human fibroblasts and renal tubular epithelial cells. J Am Soc Nephrol 13: 2878–2887
Article CAS Google Scholar - Pintos Morell G, Niaudet P, Jean G, Descamps-Latscha B 1985 Altered oxidative metabolism, motility, and adherence in phagocytic cells from cystinotic children. Pediatr Res 19: 1318–1321
Article CAS Google Scholar - Fink JK, Brouwers P, Barton N, Malekzadeh MH, Sato S, Hill S, Cohen WE, Fivush B, Gahl WA 1989 Neurologic complications in long-standing nephropathic cystinosis. Arch Neurol 46: 543–548
Article CAS Google Scholar - Vogel DG, Malekzadeh MH, Cornford ME, Schneider JA, Shields WD, Vinters HV 1990 Central nervous system involvement in nephropathic cystinosis. J Neuropathol Exp Neurol 49: 591–599
Article CAS Google Scholar - Robertson RP, Harmon J, Tran PO, Poitout V 2004 Beta-cell glucose toxicity, lipotoxicity, and chronic oxidative stress in type 2 diabetes. Diabetes 53: S119–S124
Article CAS Google Scholar - Broyer M, Tete MJ, Gubler MC 1987 Late symptoms in infantile cystinosis. Pediatr Nephrol 1: 519–524
Article CAS Google Scholar
Author information
Authors and Affiliations
- Department of Nephrology and Urology, Bambino Gesù Children's Hospital and Research Institute, Rome, 00165, Italy
Liliana Mannucci, Gianfranco Rizzoni & Francesco Emma - Laboratory of Biochemistry, Bambino Gesù Children's Hospital and Research Institute, Rome, 00165, Italy
Anna Pastore - Department of Neuroscience, Bambino Gesù Children's Hospital and Research Institute, Rome, 00165, Italy
Cristiano Rizzo - Molecular Medicine Unit, Bambino Gesù Children's Hospital and Research Institute, Rome, 00165, Italy
Fiorella Piemonte
Authors
- Liliana Mannucci
You can also search for this author inPubMed Google Scholar - Anna Pastore
You can also search for this author inPubMed Google Scholar - Cristiano Rizzo
You can also search for this author inPubMed Google Scholar - Fiorella Piemonte
You can also search for this author inPubMed Google Scholar - Gianfranco Rizzoni
You can also search for this author inPubMed Google Scholar - Francesco Emma
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toFrancesco Emma.
Additional information
This work was supported by the Italian Telethon Foundation, grant E0899.
Rights and permissions
About this article
Cite this article
Mannucci, L., Pastore, A., Rizzo, C. et al. Impaired Activity of the γ-Glutamyl Cycle in Nephropathic Cystinosis Fibroblasts.Pediatr Res 59, 332–335 (2006). https://doi.org/10.1203/01.pdr.0000196370.57200.da
- Received: 21 March 2005
- Accepted: 29 September 2005
- Issue Date: 01 February 2006
- DOI: https://doi.org/10.1203/01.pdr.0000196370.57200.da