Neutralization of pro‐inflammatory monocytes by targeting TLR2 dimerization ameliorates colitis (original) (raw)
Introduction
Intestinal immune cells play a fundamental role in the maintenance of gut homeostasis and gut defense against pathogen invasion (Kabat et al, 2014). Gut health relies on the establishment of quiescent coexistence of immune cells and the commensal bacteria. Dysregulation of the response toward commensals results in the development of inflammatory bowel disease (IBD) (Casellas et al, 1998; Strober et al, 2002; Cho, 2008; Sun et al, 2011; Zhang & Li, 2014). Human IBD includes Crohn's disease (CD) and ulcerative colitis (UC), which differ in the location of the maladies.
Ulcerative colitis is characterized by pronounced inflammation of the intestinal mucosa, generally restricted to the colon. Until recently, UC pathogenesis was thought to be largely driven by an aggressive adaptive immune response against luminal bacterial antigens. However, more recent studies and genomewide association studies (GWAS) suggest uncontrolled reactivity of innate immune cells as prime cause of chronic inflammation in UC (Jostins et al, 2012; Corridoni et al, 2014). Specifically, mouse UC models have highlighted a prominent of influx of Ly6Chi blood monocytes into the tissue. In the healthy lamina propria, these cells maintain the intestinal macrophage compartment (Varol et al, 2009; Bain et al, 2014) and differentiate into quiescent intestinal macrophages (Rivollier et al, 2012; Zigmond et al, 2012). Under pathological conditions however, the same cells fail to be appeased, but respond to bacterial stimuli by acquiring pro‐inflammatory activities and, as a result, actively promote disease (Rivollier et al, 2012; Zigmond et al, 2012).
Toll‐like receptors (TLR) are membrane pattern recognition receptors (PRRs) that recognize a wide range of microbial products (Kang & Lee, 2011). TLR activation by certain ligands induces the critical formation of TLR dimers with unique specificities (Botos et al, 2011). Upon binding to di‐ and tri‐acylated lipoproteins, TLR2 dimerizes, for instance, with TLR6 and TLR1, respectively (Ozinsky et al, 2000; Takeuchi et al, 2002). TLR dimerization is coordinated through ligand binding to the extracellular domain of one receptor, leading to their lateral movement toward the partner molecule resulting in conformational changes throughout the proteins (Akira & Takeda, 2004). Once in the complex, TLR2 interacts with co‐receptors allowing recruitment of adaptor proteins, such as MyD88 or TRIF (Deguine & Barton, 2014; Narayanan & Park, 2015) and stimulating signaling pathways (Kawai & Akira, 2011), which ultimately induce a range of inflammatory cytokines and chemokines (Kaisho & Akira, 2006; Fukata & Arditi, 2013).
During acute and chronic inflammation, TLR engagement by commensal‐derived products triggers the production of pro‐inflammatory agents. The latter notion is supported by abounding evidence of TLR over‐expression and activity in tissues obtained from IBD patients (Hausmann et al, 2002; Canto et al, 2006). Specifically, TLR2 was shown to be significantly more active in peripheral mononuclear cells from UC patients than those obtained from healthy individuals (Toiyama et al, 2006; Frolova et al, 2008). Moreover, the dimerization of TLR2 with TLR6 was shown to play an important role in colitis, as compared to TLR2–TLR1 dimerization (Morgan et al, 2014). The critical role of TLR2–TLR6 dimers in disease development is further supported by the finding that TLR6‐deficient mice are resistant to colitis (Morgan et al, 2014). Collectively, these results highlight the importance of TLR2 and particularly its assembly with TLR6 in UC development.
Ly6Chi monocytes that enter inflamed gut tissue upregulate expression of TLR2, TLR6, and NOD2 (Zigmond et al, 2012) that are involved in the induction of pro‐inflammatory cytokines, such as IL‐6, IL‐23, and IL‐1β (Zigmond et al, 2012; Neurath, 2014).
Here, we investigated the role of TLR signaling in monocytes in the development of acute murine colitis. We show that TLR2 heterodimerization plays a fundamental role in the activation of the pro‐inflammatory potential of Ly6Chi monocytes. Specifically, inhibition of TLR2 dimer formation using the TLR2 transmembrane domain (TMD)‐derived peptide, TLR2‐p (Fink et al, 2013), ameliorated DSS‐induced acute colitis by interfering with Ly6Chi monocyte activation without affecting monocyte recruitment. Altogether, our data reveal that TLR2 heterodimerization plays a fundamental role in the activation of pathogenic pro‐inflammatory Ly6Chi monocytes in UC and that its inhibition might have therapeutic potential in the treatment of acute gut inflammation.
Results
The TLR2 TMD‐derived peptide, TLR2‐p, inhibits TLR2 signaling by interacting with its reciprocal receptors within the membrane
Hetero‐ and homodimerization of TLR are critical steps in the activation of these PRRs (Akira & Takeda, 2004; Kaisho & Akira, 2006; Botos et al, 2011; Kang & Lee, 2011; Irvine et al, 2013). To specifically target TLR2 and modulate its activities, we synthesized a peptide derived from the C‐terminus of the TLR2 TMD region (TLR2‐p) and established that it impaired induced TLR2 signaling in a sepsis model (Fink et al, 2013). To investigate whether interference with TLR2 activation by the TLR2‐p peptide is due to peptide–protein interactions within the membrane, we analyzed its co‐localization with TLR2 and TLR1 using confocal microscopy. Bone marrow (BM) culture‐derived macrophages were probed with an antibody against TLR2, followed by staining with a secondary APC‐labeled antibody (Fig 1A) or with an antibody against TLR1, followed by staining with a secondary FITC‐labeled antibody (Fig 1B). For membrane staining, cells were labeled with the lipid dye DiD (Fig 1C). The stained cells were then treated with either a rhodamine‐labeled TLR2‐p peptide or scrambled TLR2 peptide (scrTLR2‐p), as control. Merge images revealed that TLR2‐p localized to the cell membrane and in the same regions as TLR2 and TLR1, unlike the scrTLR2‐p peptide that was found throughout the cell (Fig 1A and B). The co‐localization of the TLR2‐p with TLR2, TLR1, or the membrane was significantly higher, than with the control peptide.
Figure 1
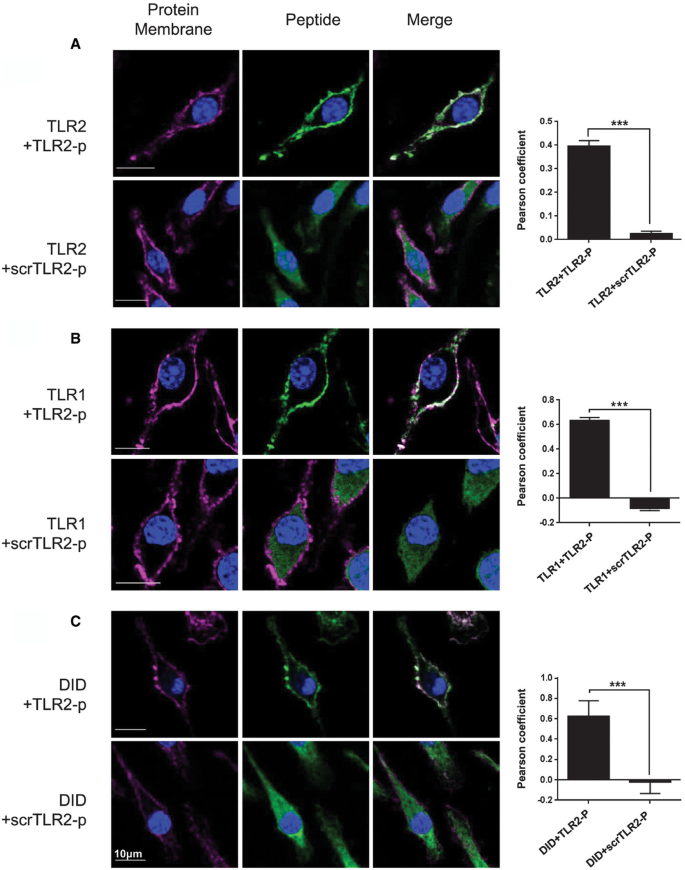
TLR2 inhibitor peptide co‐localized with its corresponding TLR
A–C Representative images of cellular localization of TLR2‐p and scrTLR2‐p peptides in BM‐derived macrophages were observed using confocal microscopy. (A) Cells were probed with anti‐TLR2 antibody followed by staining with APC‐labeled secondary antibody or (B) with anti‐TLR1 antibody followed by staining with FITC‐labeled secondary antibody (left panels, red). For membrane staining, cells were labeled with lipid dye, DiD (C). Then, rhodamine‐labeled fluorescent peptide was added to the cells (middle panels, green). Merged images are shown in the right panel. Scale bars, 10 μm. The mean Pearson correlation coefficient observed for TLR2‐p or scrTLR2‐p peptides with TLR2, TLR1, and DiD is presented as mean of two independent experiments ± SEM (***P < 0.001, n = 19–61).
The specific interaction of the TLR2‐p with its native reciprocal TLR was further investigated by co‐immunoprecipitation assay. BM macrophage lysates were incubated with either rhodamine‐labeled TLR2‐p or control peptide and then subjected to immunoprecipitation using specific TLR antibodies bound to protein G beads. Following SDS–PAGE, proteins bound to rhodamine‐labeled peptides were detected by a fluorescent scanner (Fig 2A). TLR2‐p efficiently precipitated with TLR2, TLR1, and TLR6 displaying preference for the latter, while the scrTLR2‐p peptide showed only low interaction. To validate the specificity of TLR2‐p peptide co‐precipitation with its reciprocal receptor, we probed TLR2‐p interactions with TLR4 that does not bind TLR2. TLR2‐p showed low affinity to TLR4, similar to the levels observed with the control peptide. TLR2‐p peptide was further tested for its inhibitory activity in vitro. Primary murine CD115+ monocytes cells treated with lipoteichoic acid (LTA) (TLR2‐6 ligand) and PAM3CSK (TLR2‐1 ligand) expressed reduced levels of the pro‐inflammatory cytokines TNF‐α and IL‐6 following treatment with TLR2‐p, while the control peptide showed no inhibition (Fig 2B). The same cells, when treated with LPS (TLR4 ligand), showed no differences in their secreted pro‐inflammatory cytokines following treatment with TLR2‐p. Collectively, this establishes the specific interaction of the TLR2‐p peptide with its corresponding receptors, TLR1, TLR2, and TLR6, and provides critical mechanistic insights into the action of TLR2‐p and its potential to block its dimerization and activation (Figs 1 and 2).
Figure 2
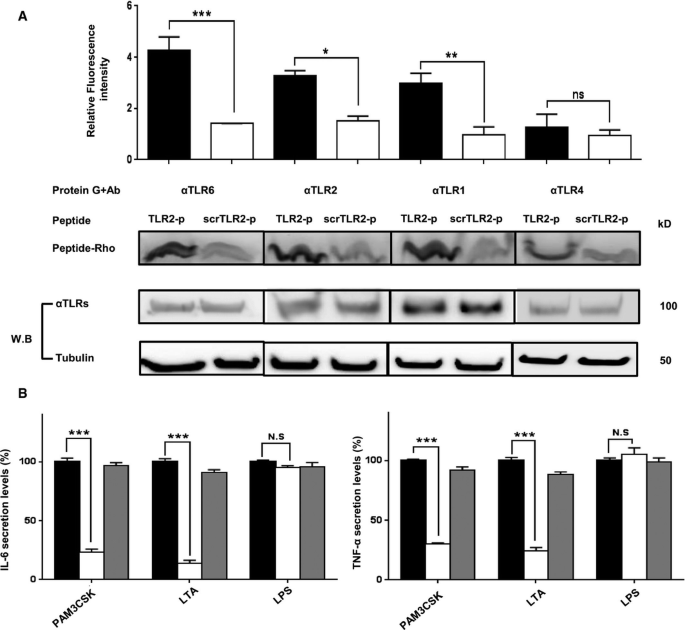
TLR2 inhibitor peptide physically interacts with its corresponding TLR
A. Biochemical analyses of peptide interaction with TLR. BM‐derived macrophages were incubated with 1 μM fluorescently labeled rhodamine (Rho)‐TLR2‐p or its mutant Rho‐ScrTLR2‐p peptide. After 1 h of incubation at 37°C, the cells were lysed. Soluble fraction was used for immunoprecipitation with the indicated TLR antibodies. Protein samples were resolved by SDS–PAGE, and the presence of the labeled peptide was detected with a fluorescent scanner. Subsequently, the gel was transferred to a membrane and subjected to Western blotting for TLR6, TLR1, TLR2, and TLR4 in the appropriate samples. Equal loading was measured by detecting of anti‐tubulin in the cell lysate. The results are presented as the mean fluorescent intensity from two independent experiments. Non‐specific binding of the peptides to G protein beads was subtracted. The results are mean ± SEM of at least two independent experiments (*P < 0.05, **P < 0.01, ***P < 0.001).
B. Protein analysis of secreted pro‐inflammatory cytokines, IL‐6 and TNF‐α from primary CD115+ cells. Cells were incubated with 20 μM of TLR2‐p or scrTLR2‐p peptide for 0.5 h and then washed and incubated with LTA (5 μg/ml), PAM3CSK (0.5 μg/ml), or LPS (0.2 μg/ml) at 37°C for 5 h (for TNF‐α detection) and 22 h (for IL‐6 detection). The results are mean ± SEM of two independent experiments (***P < 0.001, n = 8).
TLR2‐p peptide inhibits the dimerization of TLR2 and TLR6, resulting in attenuation of downstream signaling
To test whether the TLR2‐p peptide inhibits TLR2–TLR6 dimerization, we performed a fluorescence resonance energy transfer (FRET) assay using the ImageStreamX imaging flow cytometer. BM macrophages were decorated with anti‐TLR6 antibodies conjugated to PE, used as a donor (yellow, middle column) and antibodies against TLR2 followed by staining with a secondary APC‐labeled antibody, acting as acceptor (see scheme in Fig 3A). LTA exposure induced an APC‐FRET signal, indicating TLR6–TLR2 dimerization. Cells pre‐incubated with TLR2‐p for half an hour prior to LTA activation showed reduced levels of FRET signal similar to non‐stimulated cells (Fig 3B). Note that in accordance with earlier reports, we observed weak association between TLR2 and TLR6 before LTA stimulation (Triantafilou et al, 2006). In contrast, control peptide had no effect on dimerization and showed the same FRET signal as LTA‐stimulated cells. To verify that the signal observed was due to direct TLR2–TLR6 interactions, we measured FRET between TLR2 and another surface receptor expressed by macrophages, the surface glycoprotein EMR1 (recognized by the F4/80 antibody). No increase of FRET signal was observed in this setting upon LTA stimulus (Fig 3C). Addition of TLR2‐p to BM macrophages also efficiently blocked LTA‐induced ERK phosphorylation (Fig 3D). Activation of cells pre‐treated with scrTLR2‐p was indistinguishable from LTA‐stimulated cells. Collectively, these results establish that TLR2‐p blocks TLR2 signaling in in vitro cultures by inhibiting critical TLR2–TLR6 dimerization following LTA stimulation.
Figure 3
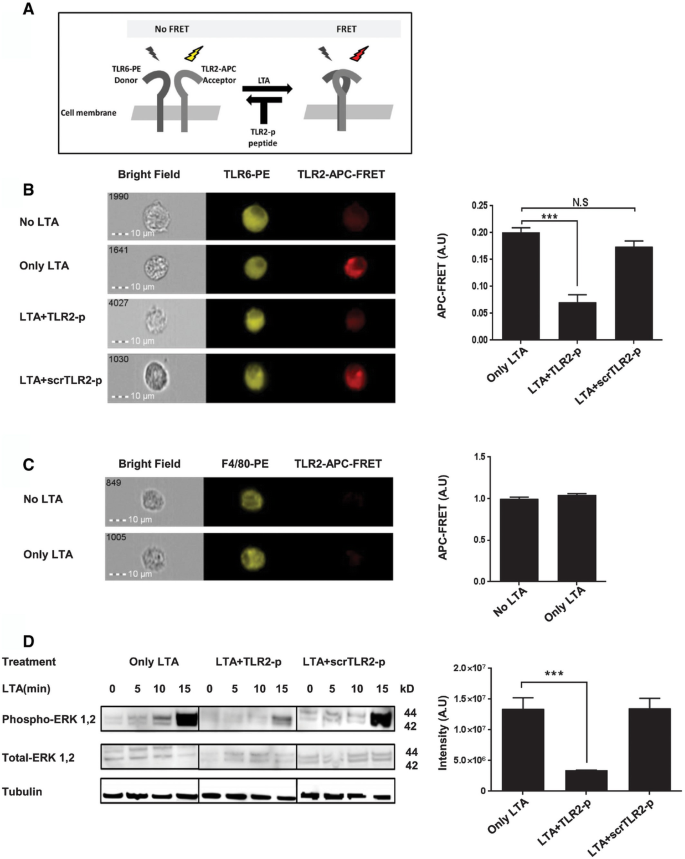
TLR2‐p peptide directly inhibits the dimerization of TLR2 and TLR6 after LTA treatment resulting in reduced levels of ERK1/2 signaling
A A scheme showing the FRET reaction.
B, C Representative images of cellular interaction between TLR2 and TLR6 (A) and between F4/80 and TLR2 (B) in BM‐derived macrophages with the indicated treatments were observed by fluorescence resonance energy transfer (FRET) using ImageStreamX. Scale bars, 10 μm. Cells were incubated with 20 μM of TLR2‐p or scrTLR2‐p peptide for 0.5 h and then washed and incubated with 500 ng/ml LTA for another 0.5 h at 37°C. Cells were probed with anti‐TLR6‐PE‐ or anti‐F4/80‐PE‐conjugated antibody (donor) and anti‐TLR2 antibody following by staining with APC‐labeled secondary antibody (acceptor). PE intensity (middle panel) and FRET intensity (right panel) were measured via ImageStreamX imaging flow cytometer. The FRET intensity is shown as mean ± SEM of two independent experiments (***P < 0.001, n = 2,579–15,567).
D Representative image of ERK1/2 phosphorylation levels in CD115+ BM monocytes upon LTA exposure. Cells were pre‐treated for 0.5 h with 20 μM of TLR2‐p peptide, scrTLR2‐p peptide or untreated and then washed and incubated with 500 ng/ml LTA for the indicated times. ERK1/2 phosphorylation levels and total ERK1/2 levels were detected by Western blotting. Equal loading was detected by measuring tubulin. Results are representative data of two independent experiments. The band intensity was quantified for p‐ERK levels after 15 min of LTA exposure. Results are normalized to tubulin levels and are the mean of two experiments ± SD (***P < 0.001).
Inhibition of TLR2 dimerization ameliorates acute colitis
Signals that emanate from TLR2 after its obligatory pairing with TLR6 or TLR1 are thought to contribute to gut inflammation. Accordingly, strategies interfering with TLR2 activation improve disease scores in UC animal models, although the exact mechanisms of action and cell types targeted by these regimen remain mostly undefined (Hausmann et al, 2002; Pierik et al, 2006; Toiyama et al, 2006; Heimesaat et al, 2007; Frolova et al, 2008). To investigate the therapeutic potential of TLR2 dimerization inhibition, we tested the effect of TLR2‐p in an acute colitis model, in which mice are exposed to an oral dextran sodium sulfate (DSS) regimen (Okayasu et al, 1990). Mice were exposed to DSS‐containing drinking water for 7 days, with or without intraperitoneal (IP) injections of TLR2‐p every second day (see scheme, Fig 4A). Mice treated with DSS only, or with DSS and the scrambled control peptide (scrTLR2‐p), exhibited weight loss and severe colitis, as evaluated by colonoscopy (Becker et al, 2006) (Fig 4B–D). In contrast, TLR2‐p‐treated mice exhibited significant mitigation in the severity of colitis upon DSS challenge. Moreover, TLR2‐p‐treated mice also displayed milder mucosal pathology when compared to scrTLR2‐p/DSS or DSS only‐treated mice, as evidenced by decreased luminal blood and reduced edema and lesion formation (Fig 4E and F). Furthermore, analysis of supernatants of colon explant cultures showed that TLR2‐p‐treated animals displayed reduced levels of IL‐6, IL‐1β, and IFN‐λ (Fig 5A). The latter finding was supported by results obtained by qRT–PCR analysis of the respective colonic tissue (Fig 5B). Collectively, these results establish that inhibition of TLR2 dimerization by TLR2‐p reduces acute inflammation in DSS‐challenged animals.
Figure 4
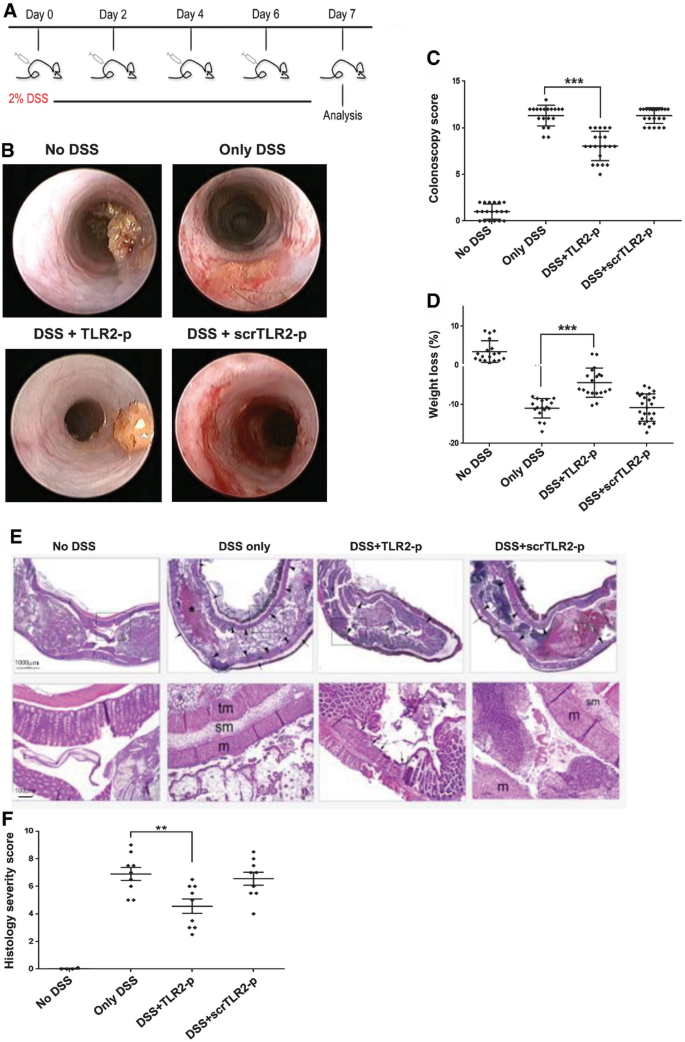
TLR2‐p inhibitor peptide ameliorated DSS‐induced colitis
A. A scheme showing peptide and DSS administration regime.
B. Representative colonoscopy images of the indicated treatments (day 7). C57BL mice were treated with 2% of dextran sulfate sodium (DSS) in drinking water for 7 days. An amount of 5 mg/kg of TLR2‐p and scrTLR2‐p peptides were injected IP every following day from day one.
C. A graphical summary of endoscopic colitis grades assessed on day 7 after treated with DSS only, DSS with TLR2‐p peptide, DSS with scrTLR2‐p peptide, and untreated mice (n = 19, 21, 22 and 19, respectively). Results represent the mean ± SD of three independent experiments (***P < 0.001).
D. A graphical summary of changes in body weight (day 6) of the indicated groups. Results are the mean ± SD of two independent experiments (n = 6–8) (***P < 0.001).
E. Representative H&E histological images for distal colon section (day 7). Black asterisk indicates large collection of blood in the lumen, arrowheads indicate widespread mucosal collapse and ulceration, and arrows indicate edema in the sub mucosa. Scale bars, 100 μm (lower row) and 1,000 μm (upper row).
F. A graphical summary of histological severity score of the indicated treatments. Results are the mean ± SD of two independent experiments (n = 9–10) (**P < 0.01).
Figure 5
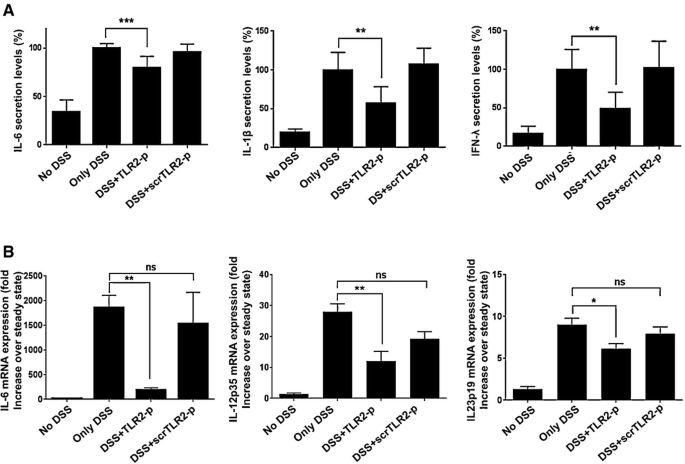
TLR2‐p inhibitor peptide downregulates pro‐inflammatory cytokines in the colonic mucosa
A, B Protein and RNA samples of colon tissue were extracted from mice with the indicated treatments. (A) Cytokine levels of IL‐6, IL‐1β, and IFN‐λ were determined by ELISA (**P < 0.01, ***P < 0.001, n = 4–7) and (B) mRNA expression levels of IL‐6, IL‐12p35, and IL‐23p19 were assessed by quantitative RT–PCR and normalized to TBP (*P < 0.05, **P < 0.01, n = 4–6). Results represent two or more independent experiments.
TLR2‐p peptide inhibits TLR2 signaling in Ly6Chi monocytes without affecting their recruitment to the inflamed gut
DSS colitis is associated with a massive tissue infiltration of Ly6Chi monocytes that acquire a pronounced pro‐inflammatory signature (Zigmond et al, 2012). Ablation or sequestration of Ly6Chi monocytes ameliorates acute colitis establishing these cells or their derivatives as critical drivers of gut inflammation (Zigmond et al, 2012; Getts et al, 2014). Monocyte infiltrates display prominent expression of TLR2 and TLR6 (Zigmond et al, 2012). We therefore reasoned that the TLR2‐p regimen might improve the DSS colitis scores by targeting Ly6Chi monocytes and inhibiting their pro‐inflammatory response. First, we evaluated colonic monocyte infiltration. With DSS, the frequency of Ly6Chi cells within the total CD11b+ pool increased in the colon, corroborating earlier studies (Waddell et al, 2011; Zigmond et al, 2012). Monocyte infiltrates of DSS/TLR2‐p‐ and DSS/scrTLR2‐p‐treated mice were comparable to non‐peptide‐treated controls, establishing that TLR2‐p does not affect monocyte recruitment (Fig 6A and B). Next, we investigated whether TLR2‐p blocks the pro‐inflammatory activity of Ly6Chi monocytes. To compensate for in vivo peptide degradation and to increase binding probability of the peptides to Ly6Chi monocytes, we performed an additional injection on day 5 and sorted Ly6Chi monocytes on day 6 of DSS challenge (see scheme, Fig 7A). Also this modified protocol improved colitis scores of DSS‐treated animals (Fig 7B). To probe for a direct effect of the TLR2‐p on Ly6Chi monocytes, we isolated the latter from the different animal groups (Fig EV1) and subjected the cells to qRT–PCR analysis for IL‐6, IL‐12, and IL‐23 production. As shown in Fig 7C, TLR2‐p treatment significantly impaired monocyte production of pro‐inflammatory cytokines. Taken together, this provides a mechanistic explanation for the fact that TLR2‐p treatment ameliorates acute colitis development. Specifically, we show that the agent interferes with the TLR2‐triggered activation of Ly6Chi monocytes that infiltrate the gut tissue, thereby curbing the pro‐inflammatory reaction.
Figure 6
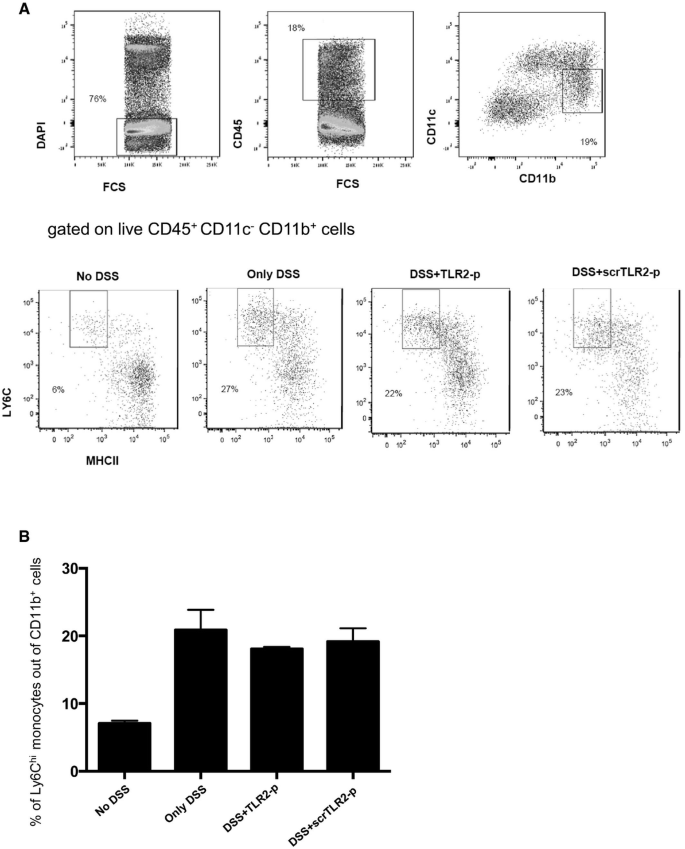
TLR2 inhibitor peptide does not have an effect on the recruitment of pro‐inflammatory monocytes
A. Flow cytometry analysis of colonic lamina propria CD11c− CD11b+Ly6Chi MHCII‐monocytes from steady state and DSS day 7 treated with TLR2 peptide or scrTLR2 peptide, showing comparable monocyte infiltrates into the colitis colon. Plots were pre‐gated on live CD45+ cells.
B. Graphical summary of monocytes presented as % out of CD11b + cells. Results are the mean ± SEM of three independent experiments (n = 3 per group).
Figure 7
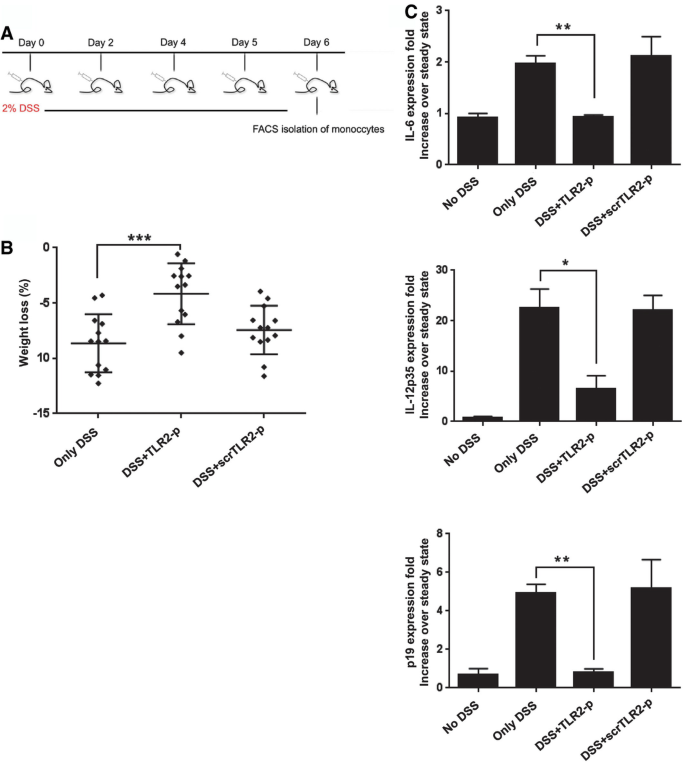
TLR2‐p inhibitor peptide downregulates pro‐inflammatory cytokines expressed by pro‐inflammatory monocytes
A. A scheme showing peptide and DSS administration regime.
B. Weight loss of DSS‐challenged mice on day 6 (***P < 0.001). Results are the mean ± SD of three independent experiments (n = 4–5).
C. Real‐time PCR analysis showing mRNA expression levels of IL‐6, IL‐12p35, and IL‐23p19 of FACS‐isolated monocytes collected from mice with the indicated treatments. Results are the mean ± SEM of two to three independent experiments (*P < 0.05, **P < 0.01, n = 6–7 mice pooled per group).
Discussion
Monocytes have emerged as critical drivers of acute gut inflammation. Here, we show that neutralization of the pro‐inflammatory activities of Ly6Chi monocytes by targeting TLR signaling ameliorated DSS‐induced acute colitis. Specifically, we used peptide‐based interference with specific TLR dimerization to manipulate the differentiation of recruited monocytes. Collectively, we establish the critical role of TLR2 dimer formation in the local generation of pro‐inflammatory cells in acute gut inflammation.
Ly6Chi monocytes entering the healthy colon acquire a non‐inflammatory gene expression profile (Zigmond et al, 2012). In contrast, when entering inflamed tissue, the differentiation of these cells into the quiescent, non‐inflammatory CX3CR1hi macrophages is blocked and diverted to a distinct fate (Rivollier et al, 2012; Zigmond et al, 2012). Ly6Chi monocytes then respond to the bacterial products they encounter with the production of pro‐inflammatory cytokines, such as IL‐23 and IL‐6 (Zigmond et al, 2012). These effector monocytes actively promoted gut inflammation, as their ablation using an anti‐CCR2 regimen (Zigmond et al, 2012), or microparticle‐mediated sequestration (Getts et al, 2014) ameliorates DSS colitis. Given their plasticity and role as macrophage precursors, Ly6Chi monocytes, and their equivalent in the human, the classical CD14+ CD16+/− monocytes, have emerged as attractive targets for cellular therapy. However, approaches interfering with monocyte recruitment, such as ablation or sequestration, or targeting of critical chemokine receptors (Leuschner et al, 2011), also inherently prevent the re‐establishment of non‐inflammatory CX3CR1hi macrophages and thus by themselves compromise gut homeostasis. Manipulations that specifically interfere with the local development of pro‐inflammatory effector monocytes are hence to be favored. Here, we highlight the potential of such an approach by showing that specific neutralization of TLR2 signaling by inhibiting critical TLR dimerization blocks monocyte differentiation into pro‐inflammatory cells.
Interference with TLR2 signaling has been proposed to have therapeutic potential in the treatment of gut inflammation and UC (Hausmann et al, 2002; Pierik et al, 2006; Toiyama et al, 2006; Heimesaat et al, 2007; Frolova et al, 2008). To transmit a signal upon ligand engagement, TLR2 needs to form heterodimers with TLR6 or TLR1 (Irvine et al, 2013; Ozinsky et al, 2000; Fukata & Arditi, 2013; Botos et al, 2011; Morgan et al, 2014). TLR assembly is partially mediated by interactions of their TMDs (Reuven et al, 2014). Accordingly, exogenously added peptides derived from receptor TMD regions can inhibit assembly and activation (Gerber et al, 2004; Yin et al, 2007; He et al, 2011; Reuven et al, 2014). Here, we utilized this strategy to modulate TLR2 signaling in the context of gut inflammation by directly abrogating TLR2 dimerization within the membrane, using a TLR2 TMD‐derived peptide. The inhibitory activity of TLR2‐p relied on its ability to bind to its corresponding reciprocal receptors, TLR6 and TLR1, and thus maintaining TLR2 in inactive monomeric form (Fink et al, 2013).
To investigate mechanistic aspects of TLR2‐p and shed light on its potential in vivo mode of action, we studied its effect on BM macrophages. Specifically, we show (i) by immunofluorescence that the TLR2‐p peptide localizes to the cell membrane in the same regions as TLR2, and (ii) by immunoprecipitation that the TLR2‐p peptide physically interacts with TLR2, TLR1, and TLR6, with preference to latter. Moreover, using a FRET assay, we directly established that TLR2‐p interferes with LTA‐induced TLR2–TLR6 assembly inhibiting signaling and ERK phosphorylation.
The therapeutic effect of TLR2‐p was tested in vivo in the DSS colitis model that mimics clinical and histological features of UC (Laroui et al, 2012). Intraperitoneal TLR2‐p injection significantly ameliorated colitis as assessed by histology, colonoscopy, and body weight measurements. Whole tissue analysis through colon explant cultures and RT–PCR showed that TLR2‐p inhibited induction of the pro‐inflammatory cytokines IL‐6, IL‐12, and IL‐1β that were shown to play a fundamental role in colitis (Neurath, 2014). TLR2‐p also reduced IL‐23 and IFN‐λ expression. Interestingly though, irrespective of the peptide treatment, Ly6Chi monocytes infiltrated the colon. qRT–PCR analysis of these monocyte infiltrates however showed that the TLR2‐p peptide blocked pro‐inflammatory cytokine production by these cells. We thereby established that TLR2 activation and dimerization induced by local stimuli are critical for the differentiation of monocytes toward pro‐inflammatory cells. Of note, TLR2 heterodimerization blockade selectively impaired the pro‐inflammatory activity of the monocytes without ablating them. Although not specifically addressed in this study, the latter might be of benefit as it preserves regulatory and protective activities of these cells. Thus, monocyte‐derived PGE2 was for instance shown to inhibit the pathogenic potential of neutrophil at mucosal sites (Grainger et al, 2013). We show that TLR2‐p blocks the TLR2 ligand induced production of pro‐inflammatory cytokine by monocytes in in vitro cultures. Moreover, we previously showed that TLR2 expression contributes to the acquisition of a pro‐inflammatory signature by these cells in the DSS colitis model (Zigmond et al, 2012). In light of these data and the results of the ablation experiments, we consider it likely that TLR2‐p acts directly on the recruited monocytes in the present study. However, we cannot rule out that TLR2‐p also interfered with TLR2 signaling in other immune cells, or even non‐hematopoietic cells that might contribute to the prevention of colitis.
Taken together, the present study identified an essential role for TLR2 dimerization in the development of acute colitis, specifically in Ly6Chi subpopulation. Therapeutically, our study highlights TLR2‐p peptide as a potential therapeutic modality to treat UC, but may have broader implications in other clinical situations mediated by TLR assembly.
Materials and Methods
Peptide synthesis, purification, and fluorescent labeling
TLR2‐p peptide (kkLILLVGALAHHFHGLWkk) and scrTLR2‐p peptide (kkGFGLHIRVWLHLLTLCkk) were synthesized by a 9‐fluorenylmethoxycarbonyl (Fmoc) solid‐phase method on Rink amide MBHA resin (Calbiochem‐Novabiochem, San Diego, California) by using an ABI 433A automatic peptide synthesizer (Applied Biosystems, Foster City, CA) as described previously (Merrifield et al, 1982). Following, the peptides were cleaved from the resin by incubation for 2 h with 95% TFA, 2.5% H2O, and 2.5% triethylsilane. Purification of the crude peptides was performed by RP‐HPLC (> 98%) on a Vydac C4 column (Grace Discovery Sciences, Deerfield, IL). The cleaning of the peptide was then identified by electrospray mass spectroscopy. For assays with fluorescent peptides, addition of Rho (rhodamine) 5(6)‐carboxytetramethylrhodamine _N_‐succinimidyl ester (TAMRA, BioChemika) fluorescent probe to the N‐terminus of the peptides was performed by standard Fmoc chemistry. Peptides used for in vivo assay were treated twice with 20% acetic acid for the replacement of the trifluoroacetate anion added during HPLC purification.
Bone marrow macrophage and monocyte isolation
Femora and tibiae BM cells were collected and cultured in RPMI medium containing FBS (10%), L‐glutamine (1%), sodium pyruvate (1%), Pen‐strep (1%), and 10 ng/ml recombinant CSF‐1 (Peprotech). For isolation of CD115+ monocytes, ficoll density gradients were used to remove erythrocytes followed by MACS cell separation with CD115‐biotin (AFS98, Biolegend) and streptavidin‐conjugated magnetic beads (Miltenyi Biotech). At day 3, half the medium was replaced, and on day 7, cells were used for the different in vitro assays.
Co‐localization of peptides with TLR molecules and lipid dye
BM‐derived macrophages (5 × 104) were fixed with 3% paraformaldehyde for 20 min and washed with PBS. The cells were then blocked for unspecific binding with 10% FCS in PBS at room temperature (RT) for 30 min. For labeling of TLR2, mouse anti‐TLR2 was added (1:25) overnight at 4°C (Biolegend), followed by staining with a secondary anti‐mouse‐APC (1:100) for 2 h at RT (Biolegend). For labeling of TLR1, rat anti‐TLR1 was added (1:50) overnight at 4°C (eBioscience), followed by staining with a secondary anti‐rat‐FITC (1:100) for 2 h at RT (Santa Cruz Biotechnology). For labeling of the membrane, fluorescence lipid dye 1,1′‐dioctadecyl‐3,3,3′,3′‐tetramethylindodicarbocyanine‐5,5′‐disulfonic acid, DiD (0.5 μg/ml, Invitrogen) was added for 10 min at RT. The Rho‐labeled fluorescent peptide (1 μM) was added during the last 1 min of incubation. Afterwards, cells were washed with PBS and deposited onto a glass slide using Fluoromount‐G (Southern Biotech) overnight at RT. The labeled cell samples were observed under a fluorescence confocal microscope, Olympus IX81 FV10‐ASW, objective 60× (oil), NA:1.35 (due to color corrected). DiD and TLR2‐APC were observed under fluorescence intensity of 645 nm, TLR1‐FITC under fluorescence intensity of 488 nm, and rodamine‐labeled peptides under fluorescence intensity of 560 nm. The levels of co‐localization were measured by Pearson correlation coefficient using the Imaris analyzing software (Bitplane).
Immunoprecipitation of fluorescently labeled peptides with TLR
BM‐derived macrophages (2 × 106) were incubated with 1 μM rhodamine‐labeled peptide for 1 h at 37°C. Then, the cells were washed and lysed with ice‐cold radio immunoprecipitation (RIPA) assay buffer (50 mM Tris–HCl pH 7.5, 150 mM NaCl, 1% Nonidet P‐40, 0.5% sodium deoxycholate, 0.1% SDS). After 15 min of centrifugation at 14,000 rpm, 4°C, the supernatants were incubated with TLR antibodies including anti‐TLR2, anti‐TLR6, anti‐TLR1, and anti‐TLR4 (Santa Cruz Biotechnology) over night at 4°C. Following, the samples were added to protein G beads (Santa Cruz Biotechnology) for a second overnight incubation at 4°C. The beads were then washed with cold PBS, diluted 1:2 with SDS tricine buffer, and boiled for 10 min at 95°C. Protein supernatants were subjected to 12% SDS–PAGE and the presence of co‐immunoprecipitated peptide was detected with a Typhoon 9400 variable mode imager. Excitation was set at 532 nm and emission at 585 nm. Western blot analysis was performed with a secondary antibody conjugated to HRP. To verify equal loading amount, tubulin was blotted using an anti‐tubulin antibody (Santa Cruz Biotechnology).
Fluorescence resonance energy transfer (FRET) between TLR2 and TLR6
BM‐derived macrophages (4 × 106) were stimulated with 20uM of TLR2‐p peptide or scrTLR2‐p peptide for 0.5 h at 37°C, washed three times with PBS, and stimulated with 500 ng/ml LTA for 0.5 h at 37°C. Following, cells were fixed with 3.7% paraformaldehyde for 20 min at RT and washed with PBS. The cells were then permeabilized and blocked for unspecific binding with 5% donkey serum, 1 mg/ml BSA, and 0.1% Triton in PBS at RT for 15 min. For labeling of TLR6, rabbit anti‐TLR6‐PE‐conjugated polyclonal antibody was added (1:50) over night at 4°C (Bioss, Inc.) TLR2 staining was performed as described above. For F4/80 labeling, rat anti‐F4/80‐PE‐conjugated monoclonal antibody was added (1:100) for 0.5 h at RT (Serotec). Cells were imaged using multispectral imaging flow cytometry (ImageStreamX Mark II, Amnis Corp), objective 60×, NA:0.9 at RT. Cells in PBS were collected from each sample, and data were analyzed using image analysis software (dedicated software; Amnis Corp). Images were compensated for fluorescent dye overlap by using single‐stain controls. Cells were gated for single cells using the area and aspect ratio features, and for focused cells using the Gradient RMS feature, as previously described (George et al, 2006). The instrument is composed of two cameras, which allows separation of the signals—camera 1 for 488‐nm and 561‐nm lasers (channels 1–6), and camera 2 for the 405‐nm and 642‐nm lasers (channels 7–12). For FRET measurements, the samples were illuminated with the 488‐nm laser. The PE staining was measured on channel 3 (560–595 nm) and the FRET resulting fluorescence was collected on channel 5 (640–745 nm). As a control for staining intensity, the APC staining was also read using the 642‐nm laser on channel 11 (640–745). The FRET was calculated for each cell by the FRET intensity. Results are normalized to untreated cells.
Phospho‐ERK detection assay
BM‐derived macrophages (1 × 106) were pre‐treated with growing medium containing 0.1% serum overnight. Then, the cells were treated with the peptides for 1 h (20 μM), washed with PBS, and stimulated with LTA at 37°C (500 ng/ml) for the indicated times. Following this, cell lysates were prepared in RIPA buffer containing 50 mM NaF, 2 mM Na3VO4, protease and phosphatase inhibitors (Sigma‐Aldrich). Protein concentration was measured using the BCA Protein Assay Kit (Pierce Chemical Co.), and 50 μg of protein was loaded and separated on 12% SDS–PAGE. Afterwards, the samples were transferred onto PVDF membrane. The immunoblot was incubated overnight with blocking solution (2% BSA in Tris‐buffered saline and Tween‐20, TBST) at 4°C and then incubated with primary antibodies, phospho‐ERK1/2, total‐ERK1/2, or tubulin, for 1 h at RT (Sigma). After washing with PBST, HRP‐conjugated secondary antibody was applied for 2 h at RT. The blots were developed on an enhanced chemiluminescence (ECL) detection system.
Mice
All animal experiments were approved by the Institutional Animal Care and Use Committee. C57BL/6 mice were purchased from Harlan (Rehovot, Israel). Animals were kept in specific pathogen‐free (SPF) environment.
DSS‐induced experimental colitis
After 7 days of exposure to 2% DSS in the drinking water, C57 black mice developed colitis. An amount of 5 mg/kg of TLR2 and scrTLR2 peptides dissolved in saline was injected IP every following day from day one. Control C57 black mice were given regular drinking water and injections of saline.
Murine colonoscopy
High‐resolution murine video endoscopic system was used to score mice for colitis severity. The system consist a miniature probe (1.9 mm outer diameter), a xenon light source, a triple chip HD camera, and an air pump (“Coloview”, Karl Storz) to achieve regulated inflation of the mouse colon. Windows Movie Maker software (Microsoft) was used to process the digitally recorded video files. Grading of colitis scores was conducted as previously described (Becker et al, 2006).
Histology
Four percent of paraformaldehyde was used to fix the tissues overnight at 4°C, and tissues were embedded in paraffin, sectioned, and stained with H and E. Slides were evaluated using an Olympus BX51 microscope, and image acquisition was conducted with the Olympus DP70 camera and DP‐Manager software. Grading of histology scores was done by a pathologist and conducted as follows: (i) amount of inflammation: 0—none, 1—mild, 2—moderate, 3—severe, and 4—significant accumulation of inflammatory cells in the gut lumen, (ii) depth of inflammation: 0—none, 1—mucosa or submucosa only, 2—mucosa and submucosa, 3—limited transmural (2.5 in < 20% of inflamed segment), and 4—transmural, (iii) nature of changes: 0—ulcer < 30% of affected mucosa and 1—ulcers > 30% of affected mucosa, (iv) re‐epithelialization: if widespread > 50% of affected mucosa: −1, and (v) the extent of the combined lesion: 1— 10%, 2— 10–30%, 3— 30–50%, 4— 50–90%, and 5— > 90%.
ELISA
For the in vitro experiment, 1 × 106 primary CD115+ cells were treated with TLR2‐p or scrTLR2‐p peptide for 2 h at 37°C. Then, the peptide was washed and the cells were stimulated with either LTA (TLR2‐TLR6 ligand) or PAM3CSK (TLR2‐TLR1 ligand) for 5 h (for TNF‐α) and 22 h (for IL‐6). For in vivo experiments, colons of mice were flushed with PBS and opened longitudinally. Subsequently, 3‐mm2 punch biopsies were obtained from the distal colon and incubated for 24 h in RPMI including 10% fetal calf serum and antibiotics (100 ml medium/one punch) at 37°C. Supernatants were collected and kept at −20°C until assessment. Quantitative evaluations of IL‐6, IL‐1β, and IFN‐γ levels in supernatants were done using a DuoSet ELISA kit (R&D Systems).
cDNA Synthesis and Real‐Time RT–PCR was performed following the method established previously (Aychek et al, 2015).
Real‐time RT–PCR primers
The sequences for primers used in this study are as follows:
Lamina propria monocytes isolation and flow cytometric analysis cell sorting
Isolation of lamina propria cells (LPCs) was performed following the method established previously (Aychek et al, 2015). The following fluorochrome‐labeled monoclonal antibodies and staining reagents were used according to the manufacturer's protocols: Monocytes isolated lamina propria were stained with CD11c (N418), IAb (AF6‐120.1), CD45 (30‐F11), CD11b (M1/70), Ly6C (HK1.4) purchased from BioLegend and F4/80‐ (CI:A3‐1) purchased from Serotec. Cells were analyzed on LSR cytometer (BD) using FlowJo software (Treestar). Cells were sorted with FACSAria machine (BD).
Statistical analysis
Samples sizes were chosen with adequate statistical power on the basis of past experience and literature. Differences between group means were tested with a _t_‐test when the experiment contained two groups, or one‐way ANOVA (followed by a Tukey post hoc test) when the experiment contained more than two groups. In cases where the experiment was run in two batches, “batch” was entered as a random categorical factor into a 2‐way ANOVA. Residuals were tested for normal distribution using Shapiro–Wilk tests, and homogeneity of variances was tested using Levenes' tests or F‐test where only two variances were compared. Analysis were done using STATISTICA (data analysis software system), version 12 StatSoft, Inc. All exclusion/inclusion criteria were pre‐established according to the IACUC. The colonoscopy and histology experiments were performed with blinding of the investigators. Data are presented as mean ± SEM. Values of P < 0.05 were considered statistically significant. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0005.
References
- Akira S, Takeda K (2004) Toll‐like receptor signalling. Nat Rev Immunol 4: 499–511
Google Scholar - Aychek T, Mildner A, Yona S, Kim KW, Lampl N, Reich‐Zeliger S, Boon L, Yogev N, Waisman A, Cua DJ, Jung S (2015) IL‐23‐mediated mononuclear phagocyte crosstalk protects mice from Citrobacter rodentium‐induced colon immunopathology. Nat Commun 6: 6525
Google Scholar - Bain CC, Bravo‐Blas A, Scott CL, Gomez Perdiguero E, Geissmann F, Henri S, Malissen B, Osborne LC, Artis D, Mowat AM (2014) Constant replenishment from circulating monocytes maintains the macrophage pool in the intestine of adult mice. Nat Immunol, 15: 929–937
Google Scholar - Becker C, Fantini MC, Neurath MF (2006) High resolution colonoscopy in live mice. Nat Protoc 1: 2900–2904
Google Scholar - Botos I, Segal DM, Davies DR (2011) The structural biology of Toll‐like receptors. Structure 19: 447–459
Google Scholar - Canto E, Ricart E, Monfort D, Gonzalez‐Juan D, Balanzo J, Rodriguez‐Sanchez JL, Vidal S (2006) TNF alpha production to TLR2 ligands in active IBD patients. Clin Immunol 119: 156–165
Google Scholar - Casellas F, Borruel N, Papo M, Guarner F, Antolin M, Videla S, Malagelada JR (1998) Antiinflammatory effects of enterically coated amoxicillin‐clavulanic acid in active ulcerative colitis. Inflamm Bowel Dis 4: 1–5
Google Scholar - Cho JH (2008) The genetics and immunopathogenesis of inflammatory bowel disease. Nat Rev Immunol 8: 458–466
Google Scholar - Corridoni D, Arseneau KO, Cominelli F (2014) Inflammatory bowel disease. Immunol Lett 161: 231–235
Google Scholar - Deguine J, Barton GM (2014) MyD88: a central player in innate immune signaling. F1000Prime Rep 6: 97
Google Scholar - Fink A, Reuven EM, Arnusch CJ, Shmuel‐Galia L, Antonovsky N, Shai Y (2013) Assembly of the TLR2/6 transmembrane domains is essential for activation and is a target for prevention of sepsis. J Immunol 190: 6410–6422
Google Scholar - Frolova L, Drastich P, Rossmann P, Klimesova K, Tlaskalova‐Hogenova H (2008) Expression of Toll‐like receptor 2 (TLR2), TLR4, and CD14 in biopsy samples of patients with inflammatory bowel diseases: upregulated expression of TLR2 in terminal ileum of patients with ulcerative colitis. J Histochem Cytochem 56: 267–274
Google Scholar - Fukata M, Arditi M (2013) The role of pattern recognition receptors in intestinal inflammation. Mucosal Immunol 6: 451–463
Google Scholar - George TC, Fanning SL, Fitzgerald‐Bocarsly P, Medeiros RB, Highfill S, Shimizu Y, Hall BE, Frost K, Basiji D, Ortyn WE, Morrissey PJ, Lynch DH (2006) Quantitative measurement of nuclear translocation events using similarity analysis of multispectral cellular images obtained in flow. J Immunol Methods 311: 117–129
Google Scholar - Gerber D, Sal‐Man N, Shai Y (2004) Two motifs within a transmembrane domain, one for homodimerization and the other for heterodimerization. J Biol Chem 279: 21177–21182
Google Scholar - Getts DR, Terry RL, Getts MT, Deffrasnes C, Muller M, Van Vreden C, Ashhurst TM, Chami B, McCarthy D, Wu H, Ma J, Martin A, Shae LD, Witting P, Kansas GS, Kuhn J, Hafezi W, Campbell IL, Reilly D, Say J et al (2014) Therapeutic inflammatory monocyte modulation using immune‐modifying microparticles. Sci Transl Med 6: 219ra7
Google Scholar - Grainger JR, Wohlfert EA, Fuss IJ, Bouladoux N, Askenase MH, Legrand F, Koo LY, Brenchley JM, Fraser ID, Belkaid Y (2013) Inflammatory monocytes regulate pathologic responses to commensals during acute gastrointestinal infection. Nat Med 19: 713–721
Google Scholar - Hausmann M, Kiessling S, Mestermann S, Webb G, Spottl T, Andus T, Scholmerich J, Herfarth H, Ray K, Falk W, Rogler G (2002) Toll‐like receptors 2 and 4 are up‐regulated during intestinal inflammation. Gastroenterology 122: 1987–2000
Google Scholar - He L, Shobnam N, Hristova K (2011) Specific inhibition of a pathogenic receptor tyrosine kinase by its transmembrane domain. Biochim Biophys Acta 1808: 253–259
Google Scholar - Heimesaat MM, Fischer A, Siegmund B, Kupz A, Niebergall J, Fuchs D, Jahn HK, Freudenberg M, Loddenkemper C, Batra A, Lehr HA, Liesenfeld O, Blaut M, Gobel UB, Schumann RR, Bereswill S (2007) Shift towards pro‐inflammatory intestinal bacteria aggravates acute murine colitis via Toll‐like receptors 2 and 4. PLoS One 2: e662
Google Scholar - Irvine KL, Hopkins LJ, Gangloff M, Bryant CE (2013) The molecular basis for recognition of bacterial ligands at equine TLR2, TLR1 and TLR6. Vet Res 44: 50
Google Scholar - Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, Lee JC, Schumm LP, Sharma Y, Anderson CA, Essers J, Mitrovic M, Ning K, Cleynen I, Theatre E, Spain SL, Raychaudhuri S, Goyette P, Wei Z, Abraham C et al (2012) Host‐microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 491: 119–124
Google Scholar - Kabat AM, Srinivasan N, Maloy KJ (2014) Modulation of immune development and function by intestinal microbiota. Trends Immunol 35: 507–517
Google Scholar - Kaisho T, Akira S (2006) Toll‐like receptor function and signaling. J Allergy Clin Immunol 117: 979–987; quiz 988
Google Scholar - Kang JY, Lee JO (2011) Structural biology of the Toll‐like receptor family. Annu Rev Biochem 80: 917–941
Google Scholar - Kawai T, Akira S (2011) Toll‐like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 34: 637–650
Google Scholar - Laroui H, Ingersoll SA, Liu HC, Baker MT, Ayyadurai S, Charania MA, Laroui F, Yan Y, Sitaraman SV, Merlin D (2012) Dextran sodium sulfate (DSS) induces colitis in mice by forming nano‐lipocomplexes with medium‐chain‐length fatty acids in the colon. PLoS One 7: e32084
Google Scholar - Leuschner F, Dutta P, Gorbatov R, Novobrantseva TI, Donahoe JS, Courties G, Lee KM, Kim JI, Markmann JF, Marinelli B, Panizzi P, Lee WW, Iwamoto Y, Milstein S, Epstein‐Barash H, Cantley W, Wong J, Cortez‐Retamozo V, Newton A, Love K et al (2011) Therapeutic siRNA silencing in inflammatory monocytes in mice. Nat Biotechnol 29: 1005–1010
Google Scholar - Merrifield RB, Vizioli LD, Boman HG (1982) Synthesis of the antibacterial peptide cecropin A (1‐33). Biochemistry 21: 5020–5031
Google Scholar - Morgan ME, Koelink PJ, Zheng B, den Brok MH, van de Kant HJ, Verspaget HW, Folkerts G, Adema GJ, Kraneveld AD (2014) Toll‐like receptor 6 stimulation promotes T‐helper 1 and 17 responses in gastrointestinal‐associated lymphoid tissue and modulates murine experimental colitis. Mucosal Immunol 7: 1266–1277
Google Scholar - Narayanan KB, Park HH (2015) Toll/interleukin‐1 receptor (TIR) domain‐mediated cellular signaling pathways. Apoptosis 20: 196–209
Google Scholar - Neurath MF (2014) Cytokines in inflammatory bowel disease. Nat Rev Immunol 14: 329–342
Google Scholar - Okayasu I, Hatakeyama S, Yamada M, Ohkusa T, Inagaki Y, Nakaya R (1990) A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology 98: 694–702
Google Scholar - Ozinsky A, Underhill DM, Fontenot JD, Hajjar AM, Smith KD, Wilson CB, Schroeder L, Aderem A (2000) The repertoire for pattern recognition of pathogens by the innate immune system is defined by cooperation between toll‐like receptors. Proc Natl Acad Sci USA 97: 13766–13771
Google Scholar - Pierik M, Joossens S, van Steen K, van Schuerbeek N, Vlietinck R, Rutgeerts P, Vermeire S (2006) Toll‐like receptor‐1, ‐2, and ‐6 polymorphisms influence disease extension in inflammatory bowel diseases. Inflamm Bowel Dis 12: 1–8
Google Scholar - Reuven EM, Fink A, Shai Y (2014) Regulation of innate immune responses by transmembrane interactions: lessons from the TLR family. Biochim Biophys Acta 1838: 1586–1593
Google Scholar - Rivollier A, He J, Kole A, Valatas V, Kelsall BL (2012) Inflammation switches the differentiation program of Ly6Chi monocytes from antiinflammatory macrophages to inflammatory dendritic cells in the colon. J Exp Med 209: 139–155
Google Scholar - Strober W, Fuss IJ, Blumberg RS (2002) The immunology of mucosal models of inflammation. Annu Rev Immunol 20: 495–549
Google Scholar - Sun L, Nava GM, Stappenbeck TS (2011) Host genetic susceptibility, dysbiosis, and viral triggers in inflammatory bowel disease. Curr Opin Gastroenterol 27: 321–327
Google Scholar - Takeuchi O, Sato S, Horiuchi T, Hoshino K, Takeda K, Dong Z, Modlin RL, Akira S (2002) Cutting edge: role of Toll‐like receptor 1 in mediating immune response to microbial lipoproteins. J Immunol 169: 10–14
Google Scholar - Toiyama Y, Araki T, Yoshiyama S, Hiro J, Miki C, Kusunoki M (2006) The expression patterns of Toll‐like receptors in the ileal pouch mucosa of postoperative ulcerative colitis patients. Surg Today 36: 287–290
Google Scholar - Triantafilou M, Gamper FG, Haston RM, Mouratis MA, Morath S, Hartung T, Triantafilou K (2006) Membrane sorting of toll‐like receptor (TLR)‐2/6 and TLR2/1 heterodimers at the cell surface determines heterotypic associations with CD36 and intracellular targeting. J Biol Chem 281: 31002–31011
Google Scholar - Varol C, Vallon‐Eberhard A, Elinav E, Aychek T, Shapira Y, Luche H, Fehling HJ, Hardt WD, Shakhar G, Jung S (2009) Intestinal lamina propria dendritic cell subsets have different origin and functions. Immunity 31: 502–512
Google Scholar - Waddell A, Ahrens R, Steinbrecher K, Donovan B, Rothenberg ME, Munitz A, Hogan SP (2011) Colonic eosinophilic inflammation in experimental colitis is mediated by Ly6C(high) CCR2(+) inflammatory monocyte/macrophage‐derived CCL11. J Immunol 186: 5993–6003
Google Scholar - Yin H, Slusky JS, Berger BW, Walters RS, Vilaire G, Litvinov RI, Lear JD, Caputo GA, Bennett JS, Degrado WF (2007) Computational design of peptides that target transmembrane helices. Science 315: 1817–1822
Google Scholar - Zhang YZ, Li YY (2014) Inflammatory bowel disease: pathogenesis. World J Gastroenterol 20: 91–99
Google Scholar - Zigmond E, Varol C, Farache J, Elmaliah E, Satpathy AT, Friedlander G, Mack M, Shpigel N, Boneca IG, Murphy KM, Shakhar G, Halpern Z, Jung S (2012) Ly6C hi monocytes in the inflamed colon give rise to proinflammatory effector cells and migratory antigen‐presenting cells. Immunity 37: 1076–1090
Google Scholar