High Mobility Group Box 1 Contributes to the Pathogenesis of Experimental Pulmonary Hypertension via Activation of Toll-like Receptor 4 (original) (raw)
- Research Article
- Open access
- Published: 20 December 2012
- Richard Shapiro1,
- Han Zheng1,
- Ferhaan Ahmad2,3,4,
- David Ishizawar2,
- Suzy A Comhair5,
- Serpil C Erzurum5,
- Timothy R Billiar1 &
- …
- Philip M Bauer1,4,6
Molecular Medicine volume 18, pages 1509–1518 (2012)Cite this article
- 971 Accesses
- 89 Citations
- 3 Altmetric
- Metrics details
Abstract
Survival rates for patients with pulmonary hypertension (PH) remain low, and our understanding of the mechanisms involved are incomplete. Here we show in a mouse model of chronic hypoxia (CH)-induced PH that the nuclear protein and damage-associate molecular pattern molecule (DAMP) high mobility group box 1 (HMGB1) contributes to PH via a Toll-like receptor 4 (TLR4)-dependent mechanism. We demonstrate extranuclear HMGB1 in pulmonary vascular lesions and increased serum HMGB1 in patients with idiopathic pulmonary arterial hypertension. The increase in circulating HMGB1 correlated with mean pulmonary artery pressure. In mice, we similarly detected the translocation and release of HMGB1 after exposure to CH. HMGB1-neutralizing antibody attenuated the development of CH-induced PH, as assessed by measurement of right ventricular systolic pressure, right ventricular hypertrophy, pulmonary vascular remodeling and endothelial activation and inflammation. Genetic deletion of the pattern recognition receptor TLR4, but not the receptor for advanced glycation end products, likewise attenuated CH-induced PH. Finally, daily treatment of mice with recombinant human HMGB1 exacerbated CH-induced PH in wild-type (WT) but not _Tlr4_−/− mice. These data demonstrate that HMGB1-mediated activation of TLR4 promotes experimental PH and identify HMGB1 and/or TLR4 as potential therapeutic targets for the treatment of PH.
Introduction
Pulmonary hypertension (PH) is a devastating disease characterized by a sustained increase in pulmonary arterial pressure. This increase in pressure is primarily due to vasoconstriction and remodeling of small pulmonary arteries, resulting in increased pulmonary vascular resistance, right ventricular failure and ultimately death. PH can be idiopathic, familial, associated with connective tissue disease or a result of drug or toxin exposure (1). Patients with these various forms of PH exhibit similar clinical, functional and hemodynamic characteristics and poor survival (2–5).
Recently, the development of therapies for the management of PH has focused on promoting vasodilation (6). While use of drugs promoting vasodilation leads to improvements in functional capacity and modest decreases in pulmonary artery pressure, recent metaanalysis of controlled trials using these agents demonstrated only limited benefit in terms of mortality (7,8).
One significant barrier to the treatment of PH is the relative lack of understanding of the role of immune mechanisms in the development and progression of the disease. Over the past decade, a new class of proinflammatory molecules, termed damage-associated molecular pattern molecules (DAMPs), has emerged as important inflammatory mediators (9). DAMPs are endogenous molecules, ranging from proteins to small molecules, that are actively secreted or passively released by damaged cells. Once in the extracellular milieu, DAMPs promote inflammation and tissue repair.
High mobility group box 1 (HMGB1) is a prototypical and highly conserved DAMP. Under normal conditions, HMGB1 is a nuclear protein, where it functions to stabilize nucleosomes (10). However, on release into the extracellular environment, HMGB1 acts as a pro-inflammatory cytokine by activating pattern recognition receptors (PRRs), including Toll-like receptor 4 (TLR4) and the receptor for advanced glycation end products (RAGE) (11–13). Evidence suggests that in response to persistent tissue injury, HMGB1, via activation of PRRs, acts as a key initiating molecule of innate immunity, orchestrating inflammation, stem cell recruitment/activation and tissue remodeling (13). Herein, we examined the role of HMGB1, TLR4 and RAGE in the development of chronic hypoxia (CH)-induced PH in mice.
Materials and Methods
Human Subjects
Human tissue was obtained in compliance with University of Pittsburgh and Cleveland Clinic Institutional Review Board guidelines.
Animal Use
All animal procedures were carried out in accordance with the guidelines set by the University of Pittsburgh Institutional Animal Care and Use Committee (IACUC) and with IACUC approval.
Mice
C57BL/6J mice were purchased from The Jackson Laboratory. Tlr4 −/− mice and Rage −/− mice were generated on a C57BL/6J background as previously described (14,15). Age-matched 8- to 12-wk-old male mice were used for the studies.
Cell Culture
Human pulmonary artery endothelial cells (HPAECs), human pulmonary artery smooth muscle cells (HPASMCs) and cell media were from Lonza. For experiments, HPAECs were serumstarved overnight, and HPASMCs were serum-starved for 24 h in minimal media. Cells were used between passage 4 and 9.
Materials
Mouse anti-human TLR4 neutralizing antibody and nonimmune mouse IgG were from eBioscience (San Diego, CA, USA). Polyclonal antibody against HMGB1 (provided by Kevin Tracey, North Shore-Long Island Jewish Health System, Feinstein Institute for Medical Research) was prepared as described previously (16). Neutralizing activity of anti-HMGB1 was confirmed in HMGB1-stimulated macrophage cultures by assay of tumor necrosis factor (TNF) release. In the presence of anti-HMGB1 antibody, neutralizing antibody was defined as inhibiting >80% of HMGB1-induced TNF release. Nonimmune rabbit IgG was purchased from Sigma-Aldrich. For Western blots, antibody against HMGB1 (#3935) was from Cell Signaling Technology (Danvers, MA, USA).
HMGB1 Isolation and Purification
The His-tagged full-length cDNA for human HMGB1 subcloned into the secretion signal of the FLAG expression vector YEpFLAG (Sigma) was transformed into the protease-deficient yeast strain BJ3505. The yeast was propagated, and HIS-tagged HMGB1 was purified as previously described (17). After purification, the protein was dialyzed versus 25 mmol/L Tris, 150 mmol/L KCl (pH 8.0), aliquoted and snap frozen at −80°C.
Hypoxic Exposure and Physiologic Measurements
Mice were exposed to CH (10% O2) for the indicated times, with normoxic mice serving as control. Right ventricular systolic pressure (RVSP) was measured essentially as described (18). Briefly, mice were anesthetized with sodium pentobarbital (60 mg/kg intraperitoneally [IP]) and ventilated via tracheotomy with room air (175 breaths per minute, 175 µL tidal volume). Body temperature was monitored and regulated with a rectal probe and heating pad. RVSP was determined by placing a 1 F solid-state pressure transducing catheter (Millar Instruments, Houston, TX, USA) directly into the right ventricle (RV). Data were acquired by using a PowerLab data acquisition system and LabChart Pro software (AD Instruments). Blood was collected via cardiac puncture. Bronchoalveolar lavage (BAL) was obtained by washing the lung via the trachea three times with 0.5 mL phosphate-buffered saline (PBS). The vasculature was flushed with PBS, the heart was excised and right heart hypertrophy was determined by the ratio of the weight of the RV to the left ventricle (LV) plus septum (Fulton index). The right lung was tied off, dissected and flash frozen, and the left lung was perfused with paraformaldehyde (4%) for embedding into paraffin.
Immunohistochemistry
Paraffin-embedded lung sections (5 µm) were baked 60 min at 55°C, deparaffinized in xylenes and rehydrated through decreasing alcohol concentrations (three xylenes, 2× 100%, 1× 95%, 1× 90%, 1× 70% ethanol, 1× PBS, for 3 min each) followed by antigen retrieval citrate buffer by using a microwave. Smooth muscle _α_-actin staining was performed as described (18). For immunofluorescent staining, sections were blocked in 2% bovine serum albumin after antigen retrieval and then incubated in anti-HMGB1 antibody overnight, followed by incubation for 60 min with a secondary antibody (Cy3). Nuclei were counterstained with Hoechst dye. Images were taken by using an Olympus Fluoview 1000 confocal microscope in the Center for Biological Imaging at the University of Pittsburgh.
Assessment of Pulmonary Vascular Remodeling
Pulmonary vascular remodeling was assessed by counting the number of partially and fully muscularized peripheral arterioles (35–100 mm) per high-power field (200× total magnification). For each mouse, at least 20 high-power fields were analyzed in multiple lung sections. Wall thickness of muscularized vessels was determined by measuring the thickness at four points on pulmonary arterioles by using the Java-based image-processing program ImageJ (National Institutes of Health, Bethesda, MD, USA).
Enzyme-Linked Immunosorbent Assay
The mouse endothelin 1 (ET-1) and mouse soluble intracellular adhesion molecule 1 (sICAM) enzyme-linked immunosorbent assays (ELISAs) were from R&D Systems (Minneapolis, MN, USA) and were performed according to the manufacturer’s instructions. The human HMGB1 ELISA was from IBL International (Hamburg, Germany) and was performed according to the manufacturer’s instructions.
Western Blot
Lung homogenate, serum or BAL (BAL was centrifuged before loading to remove contaminating cells) was separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to nitrocellulose membranes. Membranes were blocked in Tris-buffered saline, 0.1% Tween 20 (TBST), and 5% nonfat dry milk for 30 min, followed by incubation in primary antibody overnight. Membranes were washed in TBST before incubation for 1 h with horseradish peroxidase-conjugated secondary antibodies. Membranes were washed and developed by using enhanced chemiluminescence substrate (Pierce).
Cell Proliferation
Proliferation of HPASMCs was determined by measuring [3H] incorporation as previously described (18). Briefly, cells were serum-starved for 24 h in 12-well plates and treated with either 1 µg/mL recombinant human HMGB1 with or without platelet-derived growth factor (PDGF) (10 ng/mL, Sigma P4056) for 24 h. During the last 16 h, 0.2 µCi [3H]thymidine was added. After the incubation period, cells were washed twice with ice-cold PBS, and 1 mL ice-cold 10% trichloroacetic acid (Sigma T0699) was added to each well for a 30-min incubation at 4°C, after which each well was washed with 1 mL ice-cold 10% trichloroacetic acid. To each well, 0.5 mL of 0.4 N NaOH, 0.1% (wt/vol) SDS, was added, and the plates were incubated for 1 h at room temperature. The contents of each well were then transferred to 7-mL scintillation vials containing 4.5 mL Pico-Fluor-15 scintillation mixture (ICN [now MP Biomedicals, Santa Ana, CA, USA]) and counted in a liquid scintillation spectrometer.
Statistical Analysis
Statistical analyses were performed by using Graphpad Prism software. For human data, idiopathic pulmonary arterial hypertension (IPAH) versus control patient data were analyzed by Student t test with Welch correction. HMGB1 levels were correlated to mean pulmonary artery pressure by the method of Pearson. For mice, data were analyzed by one-way analysis of variance and Bonferroni post hoc tests. P values of <0.05 were considered significant.
All supplementary materials are available online at www.molmed.org.
Results
HMGB1 Release in Human and Experimental PH
To identify a role for HMGB1 in PH, we initially sought to determine whether HMGB1 is mobilized from the nucleus to the extracellular milieu, where it can act as a DAMP. We examined the localization of HMGB1 in the pulmonary arteries of IPAH patients receiving lung transplants (n = 3) or pulmonary arteries from failed donor lungs (control, non-PH). In pulmonary arteries from IPAH patients, we observed extranuclear HMGB1 in concentric and plexiform vascular lesions, whereas HMGB1 showed strict nuclear localization in pulmonary arteries of non-PH patients (Figure 1A). An increase in circulating HMGB1 was shown in several inflammatory conditions (19–26). We, therefore, measured serum HMGB1 in IPAH patients versus healthy controls. The mean serum HMGB1 concentration in the serum of IPAH patients was nearly sixfold higher than in healthy controls (33.6 versus 5.7 ng/mL HMGB1) (Figure 1B). In addition, we found that in the 16 patients for whom we had hemodynamic data available, circulating HMGB1 levels correlated with mean pulmonary artery pressures (Figure 1C). Characteristics of the patient population can be found in Table 1.
Figure 1
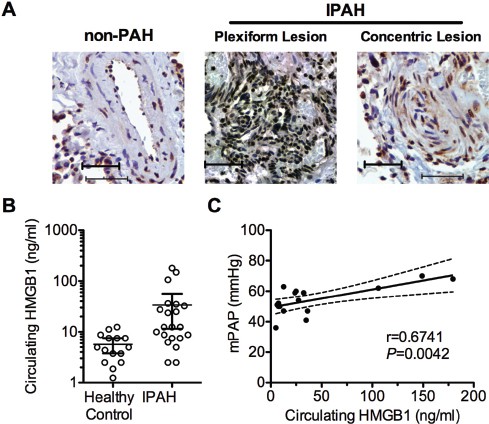
HMGB1 in human IPAH. (A) Localization of HMGB1 in lung sections of patients with and without IPAH by immunohistochemistry (brown = HMGB1, blue = hematoxylin, scale bar = 50 µm). (B) Circulating HMGB1 was measured in serum from IPAH patients (n = 21) or healthy controls (n = 15). Open circles show the levels of individual patients. The line and error bars show the mean and 95% confidence intervals. (C) Correlation of mean pulmonary artery pressure (mPAP) with circulating HMGB1 levels in IPAH patients (n = 16 patients). The r represents the Pearson correlation coefficient. Dashed lines represent the 95% confidence intervals.
Table 1 Baseline characteristics of IPAH patients.
Likewise, by using the mouse model of CH-induced PH (constant exposure to 10% O2), we were able to demonstrate translocation of HMGB1 from the nucleus to the extracellular milieu. In pulmonary arteries from normoxic C57BL/6J mice, HMGB1 was localized to the nucleus. Within 48 h of exposure to hypoxia, we observed translocation of HMGB1 to extranuclear locations (Figure 2A). The translocation of HMGB1 to extranuclear compartments was associated with increased HMGB1 in the serum (Figure 2B) and BAL fluid (Figure 2C), demonstrating that HMGB1 is released into the extracellular milieu in response to hypoxia. In addition, we detected an overall increase in HMGB1 expression in lung homogenate in response to CH (Figure 2D).
Figure 2
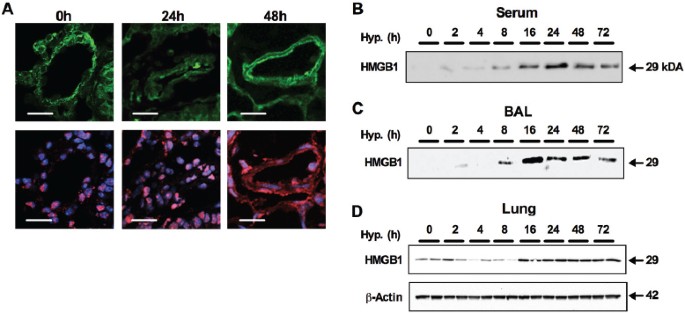
Extranuclear HMGB1 in CH-induced PH in mice. (A) Translocation of HMGB1 in lung tissue of mice exposed to 24 and 48 h of hypoxia (10% O2). Release of HMGB1 from the nucleus was assessed by confocal microscopy (red = HMGB1; blue = nuclear stain DAPI (4′,6-diamidino-2-phenylindole); green = autofluorescence to show lung morphology; scale bar = 25 µm; n = 4 mice per group). (B) Representative Western blot against HMGB1 (29 kDa) in serum of C57BL6/J mice exposed to hypoxia (10% O2 from 2 to 72 h) (n = 4 per time point; serum diluted 1:10; 30 µL loaded per well). (C) Western blot against HMGB1in bronchoalveolar lavage of C57BL6/J mice exposed to hypoxia (10% O2 from 2 to 72 h) (n = 4 per time point; loading equally 30 µL BAL per well). (D) Expression of HMGB1 in lung tissue of mice exposed to hypoxia (up to 72 h) was assessed by Western blot and normalized to _β_-actin (43 kDa) (n = 4 per time point).
HMGB1 Neutralizing Antibody Attenuates Experimental PH
To study the hemodynamic impact of extracellular HMGB1 in CH-induced PH, we treated C57BL/6J mice exposed to 3 wks of CH with an HMGB1-neutralizing antibody (_α_HMGBl) or control IgG (50 µg every other day). Untreated normoxic and CH mice served as the baseline for non-PH and CH-induced PH, respectively. As expected, CH induced an increase in RVSP and right ventricular hypertrophy (RVH) in untreated mice. Supporting the hypothesis that extracellular HMGB1 contributes to the pathogenesis of PH, treatment of mice with aHMGBl significantly attenuated the increase in RVSP and RVH in response to CH (Figures 3A, B). Treatment of mice with nonspecific IgG had no effect on the course of CH-induced PH, demonstrating the specificity of the _α_HMGB1 antibody.
Figure 3
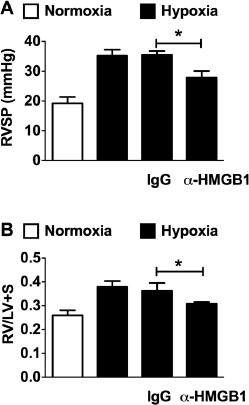
HMGB1 contributes to the pathogenesis of PH. Effect of chronic hypoxia (21 d) on RVSP (A) and right heart hypertrophy (B) (Fulton index; weight of RV/weight of LV + septum) in C57B16/J mice treated with aHMGB1 antibody or control IgG (50 µg every other day, IP) during hypoxia (n = 6 per group). Untreated normoxic and hypoxic C57BL6/J mice served as additional controls.
We then investigated the effect of CH on vascular remodeling in these mice by assessing the degree of muscularization of peripheral pulmonary arteries. Increased muscularization was observed in the CH wild-type (WT) mice, as reflected by enhanced immunoreactivity for a-smooth muscle cell actin (SMA). Treatment with aHMGBl, but not control IgG, attenuated pulmonary vascular remodeling. Figures 4A-C show representative photomicrographs of SMA-stained lung sections from hypoxic WT mice and hypoxic WT mice treated with control IgG or aHMGBl, respectively. Morphometric analysis reveals that treating mice with aHMGB1 had no effect on the number of partially muscularized arterioles (Figure 4D), but led to a decreased number of fully muscularized arterioles (<150 µmol/L) (Figure 4E) as well as decreased vessel wall thickness (Figure 4F) when compared with untreated or IgG-treated CH mice. In vitro experiments reveal that HMGB1 had no effect on human pulmonary artery smooth muscle cell proliferation, suggesting that the effects of HMGB1 on vascular remodeling were indirect (Figure 4G).
Figure 4
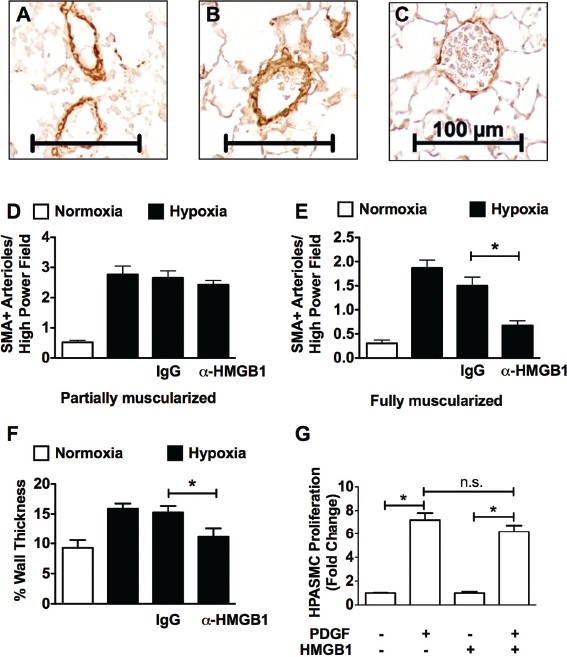
_α_HMGB1 attenuates pulmonary vascular remodeling in mice. Effect of chronic hypoxia (21 d) on vascular remodeling in C57Bl6/J mice treated with aHMGB1 antibody or control IgG (50 µg every other day, IP) during hypoxia (n = 6 per group). Untreated normoxic and hypoxic C57BL6/J mice served as additional controls. (A–C) Representative photomicrographs of SMA stained lung sections from hypoxic control mice, IgG-treated mice and aHMGB1-treated mice, respectively (scale bar = 100 µm). Morphometric analyses of the number of partially and fully muscularized arterioles (D, E) and vessel wall thickness in normoxic WT mice, hypoxic WT mice, and hypoxic WT mice treated with control IgG antibody or µHGMB1 (F) are shown. (G) Effect of HMGB1 (1,000 ng/mL) on PDGF (10 ng/mL)-stimulated HPASMC proliferation by using (3H)thymidine incorporation. Data represent the mean ± standard error of the mean (SEM) of three independent experiments. Analysis of variance: *P < 0.05. n.s., Not significant.
Endothelial dysfunction and inflammation contributes to the development of PH by promoting vasoconstriction and pulmonary vascular remodeling. Several markers of endothelial dysfunction and inflammation are increased in human and experimental PH, including intracellular adhesion molecule 1 (ICAM-1) and ET-1 (27,28), and drugs that target ET-1 receptors are used to treat PH (29). We observed increased lung ICAM-1 and plasma ET-1 in untreated or IgG-treated mice exposed to CH. In CH mice treated with µHMGB1 antibody, the increase in ICAM-1 and ET-1 was abrogated (Figures 5A, B), suggesting that HMGB1 contributes to endothelial dysfunction and inflammation in PH.
Figure 5
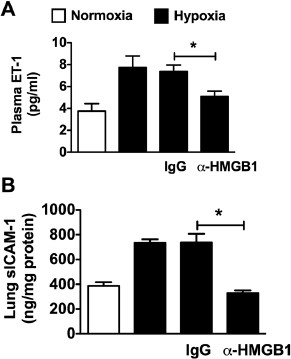
HMGB1 promotes endothelial dysfunction and inflammation in hypoxia-induced PH. (A) Plasma ET-1 in WT mice treated with aHMGB1 antibody or control IgG (50 µg every other day, IP) during hypoxia (n = 6 per group). (B) ICAM levels of lung homogenate from WT mice treated with _α_HMGB1 antibody or control IgG (50 µg every other day, IP) during hypoxia (n = 6 per group). Untreated normoxic and hypoxic WT mice served as controls (A and B). Data represent the mean ± SEM of six mice per group. Analysis of variance: *P < 0.05.
TLR4 Contributes to the Pathogenesis of PH
HMGB1 is known to bind to and activate both TLR4 and RAGE. There are no data on the role of RAGE in PH, whereas a recent study demonstrated that mice expressing mutant TLR4 (C3H/HEJ mice) are protected from PH. We first examined whether TLR4 or RAGE is up-regulated in response to CH. RNA isolated from the lungs of age-matched male C57BL/6J mice exposed to normoxia or 3 wks of CH was reverse-transcribed, and RAGE or TLR4 message was assessed by quantitative polymerase chain reaction (Figure 6A). TLR4 expression was approximately threefold higher in the lungs of CH mice compared with normoxic mice. RAGE expression was nearly twofold greater; however, the difference was not statistically significant.
Figure 6
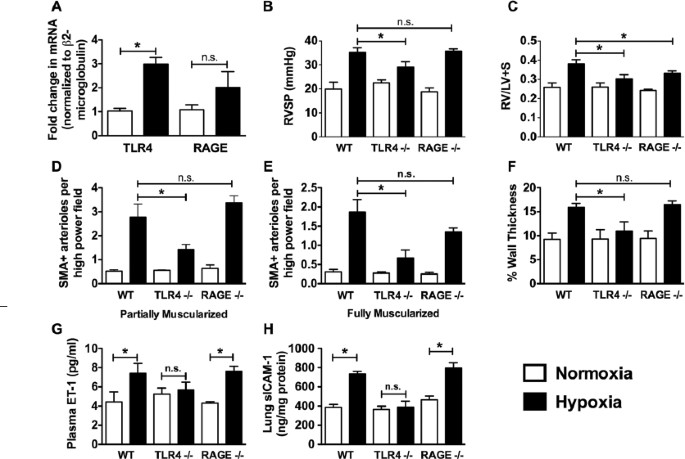
TLR4 contributes to the pathogenesis of experimental PH. (A) The effect of chronic hypoxia (21 d) on TLR4 and RAGE mRNA expression in the lungs of WT mice. Data represent the mean ± SEM of four mice per group. Analysis of variance: *P < 0.05. Effect of chronic hypoxia (21 d) on RVSP (B), RV hypertrophy (C), muscularization of pulmonary arterioles (D, E) and pulmonary arteriolar wall thickness (F) in WT, Tlr4 −/− and Rage −/− mice are shown. Effect of chronic hypoxia on plasma ET-1 (G) and lung ICAM levels (H) in WT, Tlr4 −/− and Rage −/− mice. Data represent the mean ± SEM of six mice per group. Analysis of variance: *P < 0.05. n.s., Not significant.
To confirm a role for these receptors in the development of PH, WT (C57BL/6J), Rage −/− and Tlr4 −/− mice were exposed to CH for 3 wks. Age-matched mice kept in room air served as controls. The loss of TLR4, but not RAGE, significantly attenuated the development of PH in hypoxic Tlr4 −/− mice when compared with hypoxic WT mice as determined by measurement of RVSP (Figure 6B). On the other hand, both hypoxic Rage_−/−_ mice and hypoxic Tlr4 −/− mice had decreased RV hypertrophy when compared with WT hypoxic mice (Figure 6C).
SMA staining of lung sections from WT mice revealed an increase in muscularization of arterioles (<150 _µ_mol/L) in hypoxic versus normoxic WT mice. There were significantly less partially and fully muscularized arterioles in the lungs of Tlr4 −/− hypoxic versus WT hypoxic mice (Figures 6D, E). In contrast, we observed no difference in partially muscularized vessels in hypoxic Rage −/− versus hypoxic WT mice. The number of fully muscularized vessels in Rage −/− mice tended to be fewer when compared to WT mice, but the difference did not reach statistical significance. Vessel wall thickness was also increased in WT mice in response to CH, and there was no difference between WT and Rage −/− mice. When compared with WT mice, however, there was significantly less thickening of pulmonary arterioles in Tlr4 −/− mice (Figure 6F). Similarly, genetic deletion of TLR4, but not RAGE, abrogated CH-induced increases in ET-1 and sICAM-1 (Figures 6G, H). These data suggest an important role for TLR4, but not RAGE, in the development of PH.
Exogenous HMGB1 Exacerbates CH-Induced PH in a TLR4-Dependent Manner
On the basis of our results demonstrating a role for HMGB1 and TLR4 in the development of PH, we next tested whether the effect of HMGB1 in PH is TLR4 dependent. WT or Tlr4 −/− mice were exposed to CH for 10 d with or without daily injections of recombinant human HMGB1 (10 _µ_g/d IP). In normoxia, HMGB1 had no effect on RVSP, RVH or pulmonary vascular remodeling in WT or Tlr4 −/− mice (Supplementary Figures S1A-E). Ten-day CH caused a statistically significant increase in RVSP in WT mice (19.9 ± 2.8 mmHg in normoxia versus 27.4 ± 2.2 mmHg in hypoxia) but not Tlr4 −/− mice (22.4 ± 1.3 mmHg in normoxia versus 26.4 ± 1.7 mmHg in hypoxia). Similarly, RVH, as measured by Fulton index [RV/(LV + S)], was increased in WT mice (0.259 ± 0.021 in normoxia versus 0.303 ± 0.013 in hypoxia) but not Tlr4 −/− mice (0.260 ± 0.020 in normoxia versus 0.287 ± 0.025 in hypoxia). Treatment of WT mice, but not Tlr4 −/− mice, with HMGB1 exacerbated CH-induced increases in RVSP and RVH (Figures 7A, B). Likewise, pulmonary vascular remodeling worsened in WT mice treated with HMGB1, whereas Tlr4 −/− mice were unaffected (Figures 7C-E). HMGB1 treatment of hypoxic WT mice also increased plasma ET-1 levels over hypoxia alone without affecting sICAM levels (Figure 8A). HMGB1 had no effect on ET-1 or sICAM in Tlr4 −/− mice (Figure 8B). Finally, treatment of human pulmonary artery endothelial cells with HMGB1 resulted in a significant increase in ET-1 released into the cell culture media, as determined by ELISA (Figure 8C). The effect of HMGB1 on ET-1 release in HPAECs was abrogated by a TLR4-neutralizing antibody demonstrating that the effect was mediated by TLR4. Taken together, these data demonstrate that HMGB1 promotes experimental PH via TLR4.
Figure 7
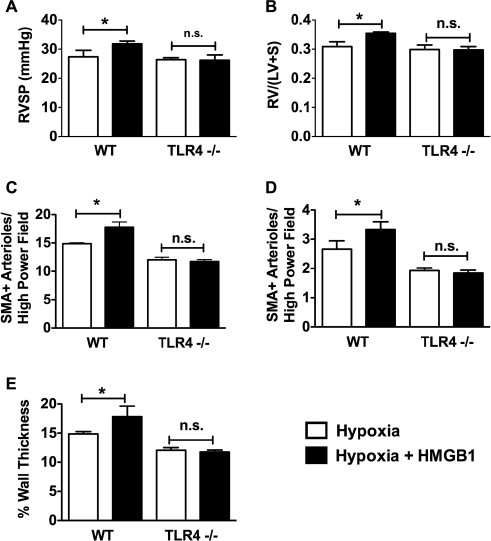
Exogenous HMGB1 exacerbates hypoxia-induced PH via TLR4. WT and Tlr4 −/− mice were given daily injections of HMGB1 (10 µg IP) during 10 d of chronic hypoxia and assessed for changes in RVSP (A), RV hypertrophy (B), muscularization of pulmonary arterioles (C, D) and pulmonary arteriolar wall thickness (E). Data represent the mean ± SEM of six mice per group. Analysis of variance: *P < 0.05. n.s., Not significant.
Figure 8
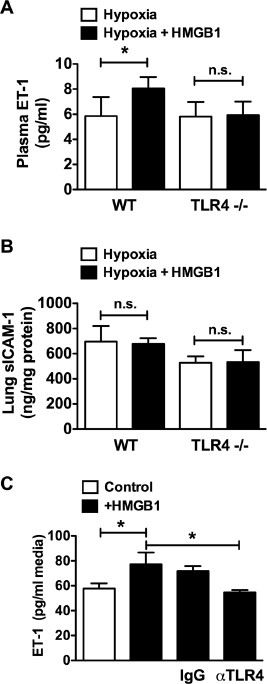
Exogenous HMGB1 exacerbates hypoxia-induced endothelial dysfunction and inflammation. WT and Tlr4 −/− mice were treated with recombinant human HMGB1 (10 µg/day IP) during 10 d of chronic hypoxia and were assessed for plasma ET-1 (A) and sICAM levels (B) in lung homogenates. WT and Tlr4 −/− mice injected with sterile saline served as controls (A, B). Data represent the mean ± SEM of six mice per group. Analysis of variance: *P < 0.05. (C) Effect of TLR4-neutralizing antibody (20 µg/mL) on HMGB1 induced ET-1 release in human pulmonary artery endothelial cells. Data represent the mean ± SEM of three independent experiments. Analysis of variance: *P < 0.05. n.s., Not significant.
Discussion
PH is classically described as a disease of endothelial dysfunction involving pulmonary vasoconstriction and pulmonary vascular remodeling, leading to increased pulmonary vascular resistance. However, clinical data demonstrate that despite advances in therapeutics aimed at promoting pulmonary vasodilation, survival remains poor (30). These data highlight the necessity of novel approaches to treating PH. There is mounting evidence of a role for the immune system in the pathogenesis of PH. A role for inflammation in human PH is based on the identification of inflammatory cells in vascular lesions of PH patients (31), elevated circulating levels of inflammatory cytokines in PH patients (32) and case studies in PH patients demonstrating improvement with antiinflammatory drugs (33,34). In the mouse model of CH-induced PH, there is an influx of inflammatory cells within the first 48 h that diminishes by 2 wks. This increase in inflammatory cells coincides with increased secretion of proinflammatory cytokines, including TNF_α_, interleukin (IL)-1_β_, IL-6 and MCP-1 (35). In addition, IL-6 overexpression promotes, and genetic deletion of IL-6 attenuates, PH in this model (36,37).
A key facet of the innate immune response is self-/nonself recognition. Through the use of PRRs, the innate immune system is capable of detecting conserved pathogen motifs, termed pathogen-associated molecular patterns (PAMPs), thereby stimulating an immune response (38). In recent years, however, it was acknowledged that the self-/nonself paradigm fails to explain innate immune activation in diseases involving sterile inflammation (39). The discovery of DAMPs, which have a similar function to PAMPs, has provided a context for understanding innate immune activation in the absence of foreign pathogens (9). HMGB1 fulfills the functions of a DAMP, being involved in inflammation caused by both infectious and noninfectious stimuli (40). Once released, HMGB1 signals through various PRRs to activate immune and parenchymal cells involved in the immune process (11–13).
Our studies clearly establish a role for extracellular HMGB1 in the pathogenesis of CH-induced PH in mice. In particular, data showing translocation of HMGB1 into the extracellular milieu and that HMGB1-neutralizing antibody attenuates CH-induced PH show the necessity of extracellular HMGB1 to fully develop the disease. HMGB1 may have additional intracellular roles in PH; however, the strategies used in this study likely do not address these roles. The clinical relevance of these data is indicated by the observations that (a) there is a diffuse extranuclear staining pattern of HMGB1 in pulmonary vascular lesions of PH patients and (b) there is increased circulating HMGB1 in PH patients, which correlates with mean pulmonary arterial pressure.
In addition to our findings in mice and humans, it was also reported, in the form of an a published abstract, that there is increased circulating HMGB1 in monocrotaline rats and that HMGB1 is released from smooth muscle cells and alveolar macrophages in this model (41). The consistency of this phenomenon (increased circulating HMGB1) across mice, rats and humans suggests that HMGB1 is an important mediator of PH across species. Future studies using similar strategies (that is, HMGB1-neutralizing antibodies) in other experimental PH models, such as monocrotaline and the SU5416/hypoxia model in rats, will be important to support this hypothesis.
A role for HMGB1-TLR4 interaction in driving immunopathology was initially described in 2005. In that study, it was found that an HMGB1-neutralizing antibody protects WT mice (C3H/HeouJ) but not _Tlr4_−/− mice (C3H/HEJ) from liver ischemia reperfusion injury (42). Since then, several studies have demonstrated a role for HMGB1-TLR4 interactions in the immunopathology of diseases, including inflammation-induced seizures (43), inflammation-induced skin cancer (44), end organ injury after tissue trauma (45) and ischemic kidney injury (46). Our study establishes a role for HMGB1 and TLR4 interaction in the pathogenesis of PH. Genetic deletion of TLR4 attenuated CH-induced PH, as assessed by measuring RVSP, RVH and pulmonary vascular remodeling. Furthermore, exogenous HMGB1 exacerbated CH-induced PH in WT but not Tlr4 −/− mice, establishing a link between HMGB1 and TLR4 in PH. Consistent with a role for HMGB1 and TLR4 in endothelial activation and inflammation, HMGB1 neutralizing antibody or loss of TLR4 also prevented hypoxia-induced increases in endothelin-1 and soluble ICAM-1. Interestingly, however, treating mice with exogenous HMGB1 during hypoxia exacerbated the increase in ET-1 but not sICAM-1 in WT mice. This result suggests that HMGB1 may directly contribute to the elevation of ET-1 in PH. Indeed, human pulmonary artery endothelial cells treated with HMGB1 secreted more ET-1 than untreated cells, which was attenuated by TLR4-neutralizing antibody. TLR4 agonists have also been shown to induce production of ET-1 by dendritic cells (47). The effect of the HMGB1-neutralizing antibody or genetic deletion of TLR4 on sICAM levels would appear to be indirect, since exogenous HMGB1 did not further increase sICAM. Alternatively, it is possible that sICAM expression is maximally induced by endogenous HMGB1 and cannot be further induced by addition of the exogenous protein.
In addition to its effects on ET-1 and ICAM-1 expression, recent data from our laboratory demonstrate that HMGB1 is a potent inhibitor of endothelial cell migration, suggesting one mechanism may be the inhibition of endothelial repair mechanisms (17). In addition, in that study, we show that hypoxia induces the release of HMGB1 from endothelial cells, suggesting that endothelial cells could be one source of increased circulating HMGB1 in PH. The HMGB1/TLR4 signaling axis has been shown to stimulate neutrophil NADPH oxidase (NOX2) in both neutrophils and lung microvascular endothelial cells, and NOX2 is thought to be important in the pathogenesis of PH (48). TLR4 is also expressed on multiple other cell types important in the pathogenesis of PH. HMGB1 induces macrophages to secrete proinflammatory cytokines in a TLR4-dependent manner (49). Studies also demonstrate that TLR4 is found on platelets and that activation of platelet TLR4 promotes platelet aggregation (50), which may promote in situ thrombosis, a feature of human IPAH. Future experiments using bone marrow chimeric mice and tissue-specific knockouts will help reveal cell-specific mechanisms by which TLR4/HMGB1 signaling promotes PH.
We also explored the possibility that RAGE contributes to the development of CH-induced PH. RAGE is highly enriched in the lung (51,52) and has been reported to be a important receptor for HMGB1 (53–55). Interestingly Rage −/− mice developed the same degree of PH (as determined by RVSP) as WT mice when exposed to CH, suggesting that RAGE does not contribute to increased pulmonary vascular resistance in CH-induced PH. This was corroborated by data showing that CH induced a similar degree of pulmonary vascular remodeling in WT and Rage −/− mice. Despite these observations, CH-induced RVH was significantly attenuated in Rage −/− mice when compared with WT mice. These data suggest that RAGE is involved in the hypertrophic response of the right heart to increased pulmonary arterial pressure. This result is consistent with a previous study demonstrating a role for RAGE in mediating cardiac hypertrophy in mice fed a Western diet (56). Whereas the attenuated RVH in Rage −/− mice is an important observation, experiments interrogating a role for RAGE in RVH are beyond the scope of the current study.
Conclusion
The data presented herein demonstrate that HMGB1-mediated activation of TLR4 promotes experimental PH and identify HMGB1 and/or TLR4 as potential therapeutic targets for the treatment of this devastating disease.
Disclosure
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
References
- Simonneau G, et al. (2004) Clinical classification of pulmonary hypertension. J. Am. Coll. Cardiol. 43:5S–12S.
Article Google Scholar - Humbert M, et al. (2004) Cellular and molecular pathobiology of pulmonary arterial hypertension. J. Am. Coll. Cardiol. 43:13S–24S.
Article CAS Google Scholar - Rabinovitch M. (2008) Molecular pathogenesis of pulmonary arterial hypertension. J. Clin. Invest. 118:2372–9.
Article CAS Google Scholar - Tuder RM, et al. (2009) Development and pathology of pulmonary hypertension. J. Am. Coll. Cardiol. 54:S3–9.
Article CAS Google Scholar - Tuder RM, Marecki JC, Richter A, Fijalkowska I, Flores S. (2007) Pathology of pulmonary hypertension. Clin. Chest Med. 28:23–42, vii.
Article Google Scholar - Michelakis ED, Wilkins MR, Rabinovitch M. (2008) Emerging concepts and translational priorities in pulmonary arterial hypertension. Circulation. 118:1486–95.
Article Google Scholar - Galie N, et al. (2009) A meta-analysis of randomized controlled trials in pulmonary arterial hypertension. Eur. Heart J. 30:394–403.
Article Google Scholar - Macchia A, et al. (2007) A meta-analysis of trials of pulmonary hypertension: a clinical condition looking for drugs and research methodology. Am. Heart J. 153:1037–47.
Article Google Scholar - Bianchi ME. (2007) DAMPs, PAMPs and alarmins: all we need to know about danger. J. Leukoc. Biol. 81:1–5.
Article CAS Google Scholar - Bonaldi T, Langst G, Strohner R, Becker PB, Bianchi ME. (2002) The DNA chaperone HMGB1 facilitates ACF/CHRAC-dependent nucleosome sliding. EMBO J. 21:6865–73.
Article CAS Google Scholar - Park JS, et al. (2006) High mobility group box 1 protein interacts with multiple Toll-like receptors. Am. J. Physiol. Cell Physiol. 290:C917–24.
Article CAS Google Scholar - Park JS, et al. (2004) Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J. Biol. Chem. 279:7370–7.
Article CAS Google Scholar - Hori O, et al. (1995) The receptor for advanced glycation end products (RAGE) is a cellular binding site for amphoterin: mediation of neurite outgrowth and co-expression of rage and amphoterin in the developing nervous system. J. Biol. Chem. 270:25752–61.
Article CAS Google Scholar - Sodhi CP, et al. (2010) Toll-like receptor-4 inhibits enterocyte proliferation via impaired beta-catenin signaling in necrotizing enterocolitis. Gastroenterology. 138:185–96.
Article CAS Google Scholar - Liliensiek B, et al. (2004) Receptor for advanced glycation end products (RAGE) regulates sepsis but not the adaptive immune response. J. Clin. Invest. 113:1641–50.
Article CAS Google Scholar - Yang H, et al. (2004) Reversing established sepsis with antagonists of endogenous high-mobility group box 1. Proc. Natl. Acad. Sci. U. S. A. 101:296–301.
Article CAS Google Scholar - Bauer EM, Shapiro R, Billiar TR, Bauer PM. (2013) High mobility group box 1 inhibits human pulmonary artery endothelial cell migration via a toll-like receptor 4- and interferon response factor 3-dependent mechanism(s). J. Biol. Chem. 288:1365–73.
Article CAS Google Scholar - Bauer EM, et al. (2011) Complement C3 deficiency attenuates chronic hypoxia-induced pulmonary hypertension in mice. PLoS One. 6:e28578.
Article CAS Google Scholar - Troseid M, et al. (2012) Circulating levels of HMGB1 are correlated strongly with MD2 in HIV-infection: possible implication for TLR4-signalling and chronic immune activation. Innate Immun. 2012, Oct 15 [Epub ahead of print].
- Stoetzer OJ, et al. (2012) Circulating immunogenic cell death biomarkers HMGB1 and RAGE in breast cancer patients during neoadjuvant chemotherapy. Tumour Biol. 2012, Sep 15 [Epub ahead of print].
- Hashimoto T, et al. (2012) Circulating high-mobility group box 1 and cardiovascular mortality in unstable angina and non-ST-segment elevation myocardial infarction. Atherosclerosis. 221:490–5.
Article CAS Google Scholar - Oktayoglu P, et al. (2012) Elevated serum levels of high mobility group box protein 1 (HMGB1) in patients with ankylosing spondylitis and its association with disease activity and quality of life. Rheumatol. Int. 2012, Nov 10 [Epub ahead of print].
- Allonso D, et al. (2012) Elevated serum levels of high mobility group box 1 (HMGB1) protein in dengue-infected patients are associated with disease symptoms and secondary infection. J. Clin. Virol. 55:214–9.
Article CAS Google Scholar - Chung HW, et al. (2012) Serum high mobility group box-1 is a powerful diagnostic and prognostic biomarker for pancreatic ductal adenocarcinoma. Cancer Sci. 103:1714–21.
Article CAS Google Scholar - Zickert A, et al. (2012) Renal expression and serum levels of high mobility group box 1 protein in lupus nephritis. Arthritis Res. Ther. 14:R36.
Article CAS Google Scholar - Dupire G, Nicaise C, Gangji V, Soyfoo MS. (2012) Increased serum levels of high-mobility group box 1 (HMGB1) in primary Sjogren’s syndrome. Scand. J. Rheumatol. 41:120–3.
Article CAS Google Scholar - Iannone F, et al. (2008) Bosentan regulates the expression of adhesion molecules on circulating T cells and serum soluble adhesion molecules in systemic sclerosis-associated pulmonary arterial hypertension. Ann. Rheum. Dis. 67:1121–6.
Article CAS Google Scholar - Giaid A, et al. (1993) Expression of endothelin-1 in the lungs of patients with pulmonary hypertension. N. Engl. J. Med. 328:1732–9.
Article CAS Google Scholar - Channick RN, et al. (2001) Effects of the dual endothelin-receptor antagonist bosentan in patients with pulmonary hypertension: a randomised placebo-controlled study. Lancet. 358:1119–23.
Article CAS Google Scholar - Humbert M, et al. (2010) Survival in patients with idiopathic, familial, and anorexigen-associated pulmonary arterial hypertension in the modern management era. Circulation. 122:156–63.
Article Google Scholar - Tuder RM, Groves B, Badesch DB, Voelkel NF. (1994) Exuberant endothelial cell growth and elements of inflammation are present in plexiform lesions of pulmonary hypertension. Am. J. Pathol. 144:275–85.
CAS PubMed PubMed Central Google Scholar - Hassoun PM, et al. (2009) Inflammation, growth factors, and pulmonary vascular remodeling. J. Am. Coll. Cardiol. 54:S10–9.
Article CAS Google Scholar - Dahl M, Chalmers A, Wade J, Calverley D, Munt B. (1992) Ten year survival of a patient with advanced pulmonary hypertension and mixed connective tissue disease treated with immunosuppressive therapy. J. Rheumatol. 19:1807–9.
CAS PubMed Google Scholar - Friedman DM, Mitnick HJ, Danilowicz D. (1992) Recovery from pulmonary hypertension in an adolescent with mixed connective tissue disease. Ann. Rheum. Dis. 51:1001–4.
Article CAS Google Scholar - Minamino T, et al. (2001) Targeted expression of heme oxygenase-1 prevents the pulmonary inflammatory and vascular responses to hypoxia. Proc. Natl. Acad. Sci. U. S. A. 98:8798–803.
Article CAS Google Scholar - Savale L, et al. (2009) Impact of interleukin-6 on hypoxia-induced pulmonary hypertension and lung inflammation in mice. Respir. Res. 10:6.
Article Google Scholar - Steiner MK, et al. (2009) Interleukin-6 overexpression induces pulmonary hypertension. Circ. Res. 104:236–44.
Article CAS Google Scholar - Kumar H, Kawai T, Akira S. (2011) Pathogen recognition by the innate immune system. Int. Rev. Immunol. 30:16–34.
Article CAS Google Scholar - Matzinger P. (1994) Tolerance, danger, and the extended family. Annu. Rev. Immunol. 12:991–1045.
Article CAS Google Scholar - Klune JR, Dhupar R, Cardinal J, Billiar TR, Tsung A. (2008) HMGB1: endogenous danger signaling. Mol. Med. 14:476–84.
Article CAS Google Scholar - Sadamura Y, et al. (2011) The role of high mobility group box1 in monocrotaline-induced pulmonary hypertension in rats. Am. J. Respir. Crit. Care Med. 183:A3413.
Google Scholar - Tsung A, et al. (2005) The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia-reperfusion. J. Exp. Med. 201:1135–43.
Article CAS Google Scholar - Maroso M, et al. (2010) Toll-like receptor 4 and high-mobility group box-1 are involved in ictogenesis and can be targeted to reduce seizures. Nat. Med. 16:413–9.
Article CAS Google Scholar - Mittal D, et al. (2010) TLR4-mediated skin carcinogenesis is dependent on immune and radioresistant cells. EMBO J. 29:2242–52.
Article CAS Google Scholar - Levy RM, et al. (2007) Systemic inflammation and remote organ injury following trauma require HMGB1. Am. J. Physiol. Regul. Integr. Comp. Physiol. 293:R1538–44.
Article CAS Google Scholar - Chen J, et al. (2011) Early interleukin 6 production by leukocytes during ischemic acute kidney injury is regulated by TLR4. Kidney Int. 80:504–15.
Article CAS Google Scholar - Spirig R, et al. (2009) TLR2 and TLR4 agonists induce production of the vasoactive peptide endothelin-1 by human dendritic cells. Mol. Immunol. 46:3178–82.
Article CAS Google Scholar - Liu JQ, Zelko IN, Erbynn EM, Sham JS, Folz RJ. (2006) Hypoxic pulmonary hypertension: role of superoxide and NADPH oxidase (gp91phox). Am. J. Physiol. Lung Cell Mol. Physiol. 290:L2–10.
Article CAS Google Scholar - Yang H, et al. (2010) A critical cysteine is required for HMGB1 binding to Toll-like receptor 4 and activation of macrophage cytokine release. Proc. Natl. Acad. Sci. U. S. A. 107:11942–7.
Article CAS Google Scholar - Andonegui G, et al. (2005) Platelets express functional Toll-like receptor-4. Blood. 106:2417–23.
Article CAS Google Scholar - Katsuoka F, et al. (1997) Type II alveolar epithelial cells in lung express receptor for advanced glycation end products (RAGE) gene. Biochem. Biophys. Res. Commun. 238:512–6.
Article CAS Google Scholar - Buckley ST, Ehrhardt C. (2010) The receptor for advanced glycation end products (RAGE) and the lung. J. Biomed. Biotechnol. 2010:917108.
Article Google Scholar - Lotze MT, DeMarco RA. (2003) Dealing with death: HMGB1 as a novel target for cancer therapy. Curr. Opin. Investig. Drugs. 4:1405–9.
CAS PubMed Google Scholar - Volz HC, Kaya Z, Katus HA, Andrassy M. (2010) The role of HMGB1/RAGE in inflammatory cardiomyopathy. Semin. Thromb. Hemost. 36:185–94.
Article CAS Google Scholar - Nogueira-Machado JA, Volpe CM, Veloso CA, Chaves MM. (2011) HMGB1, TLR and RAGE: a functional tripod that leads to diabetic inflammation. Expert Opin. Ther. Targets. 15:1023–35.
Article CAS Google Scholar - Tikellis C, et al. (2008) Cardiac inflammation associated with a Western diet is mediated via activation of RAGE by AGEs. Am. J. Physiol. Endocrinol. Metab. 295:E323–30.
Article CAS Google Scholar
Acknowledgments
We thank Kevin Tracey (North Shore-LIJ Health System, Feinstein Institute for Medical Research) for providing the HMGB1 neutralizing antibody. We thank Suchitra Barge, Michael Lotze and Lisa Butterfield (University of Pittsburgh) for assistance in obtaining control patient samples. Some of the IPAH patient samples were provided by Cooperative Human Tissue Network, Southern Division, The University of Alabama at Birmingham, under the Pulmonary Hypertension Breakthrough Initiative (PHBI). Funding for the PHBI was provided by the Cardiovascular Medical Research and Education Fund. This work was supported by grants to PM Bauer (R01-HL085134 and NIH R03-HL110794), SC Erzurum (R01-HL60917), TR Billiar (P50-GM53789) and EM Bauer (T32-098036).
Author information
Authors and Affiliations
- Department of Surgery, University of Pittsburgh School of Medicine, 200 Lothrop Street, Starzl Biomedical Science Tower W1147, Pittsburgh, Pennsylvania, 15261, USA
Eileen M Bauer, Richard Shapiro, Han Zheng, Timothy R Billiar & Philip M Bauer - Department of Medicine, University of Pittsburgh School of Medicine, Pittsburgh, Pennsylvania, USA
Ferhaan Ahmad & David Ishizawar - Department of Human Genetics, University of Pittsburgh School of Medicine, Pittsburgh, Pennsylvania, USA
Ferhaan Ahmad - Vascular Medicine Institute, University of Pittsburgh School of Medicine, Pittsburgh, Pennsylvania, USA
Ferhaan Ahmad & Philip M Bauer - Department of Pathobiology, Lerner Research Institute, and the Respiratory Institute, Cleveland Clinic, Cleveland, Ohio, USA
Suzy A Comhair & Serpil C Erzurum - Department of Pharmacology and Chemical Biology, University of Pittsburgh School of Medicine, Pittsburgh, Pennsylvania, USA
Philip M Bauer
Authors
- Eileen M Bauer
You can also search for this author inPubMed Google Scholar - Richard Shapiro
You can also search for this author inPubMed Google Scholar - Han Zheng
You can also search for this author inPubMed Google Scholar - Ferhaan Ahmad
You can also search for this author inPubMed Google Scholar - David Ishizawar
You can also search for this author inPubMed Google Scholar - Suzy A Comhair
You can also search for this author inPubMed Google Scholar - Serpil C Erzurum
You can also search for this author inPubMed Google Scholar - Timothy R Billiar
You can also search for this author inPubMed Google Scholar - Philip M Bauer
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toPhilip M Bauer.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, and provide a link to the Creative Commons license. You do not have permission under this license to share adapted material derived from this article or parts of it.
The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this license, visit (http://creativecommons.org/licenses/by-nc-nd/4.0/)
About this article
Cite this article
Bauer, E.M., Shapiro, R., Zheng, H. et al. High Mobility Group Box 1 Contributes to the Pathogenesis of Experimental Pulmonary Hypertension via Activation of Toll-like Receptor 4.Mol Med 18, 1509–1518 (2012). https://doi.org/10.2119/molmed.2012.00283
- Received: 19 December 2012
- Published: 20 December 2012
- Issue Date: December 2012
- DOI: https://doi.org/10.2119/molmed.2012.00283