World J Gastroenterol. Jan 14, 2008; 14(2): 193-199 Published online Jan 14, 2008. doi: 10.3748/wjg.14.193
Nonalcoholic fatty liver disease and mitochondrial dysfunction
Yongzhong Wei, R Scott Rector, Jamal A Ibdah, Division of Gastroenterology and Hepatology, Harry S. Truman VA Medical Center, Columbia, MO 65212, United States
John P Thyfault, Division of Gastroenterology and Hepatology, Department of Nutritional Sciences, University of Missouri-Columbia; Harry S. Truman VA Medical Center, Columbia, MO 65212, United States
ORCID number: $[AuthorORCIDs]
Correspondence to: Jamal A Ibdah, MD, PhD, Division of Gastroenterology and Hepatology, University of Missouri-Columbia, Columbia, MO 65212, United States. ibdahj@health.missouri.edu
Telephone: +1-573-8820482
Fax: +1-573-8844595
Received: July 25, 2007 Revised: September 29, 2007 Published online: January 14, 2008
Abstract
Nonalcoholic fatty liver disease (NAFLD) includes hepatic steatosis, nonalcoholic steatohepatitis (NASH), fibrosis, and cirrhosis. NAFLD is the most common liver disorder in the United States and worldwide. Due to the rapid rise of the metabolic syndrome, the prevalence of NAFLD has recently dramatically increased and will continue to increase. NAFLD has also the potential to progress to hepatocellular carcinoma (HCC) or liver failure. NAFLD is strongly linked to caloric overconsumption, physical inactivity, insulin resistance and genetic factors. Although significant progress in understanding the pathogenesis of NAFLD has been achieved in years, the primary metabolic abnormalities leading to lipid accumulation within hepatocytes has remained poorly understood. Mitochondria are critical metabolic organelles serving as “cellular power plants”. Accumulating evidence indicate that hepatic mitochondrial dysfunction is crucial to the pathogenesis of NAFLD. This review is focused on the significant role of mitochondria in the development of NAFLD.
Citation: Wei Y, Rector RS, Thyfault JP, Ibdah JA. Nonalcoholic fatty liver disease and mitochondrial dysfunction. World J Gastroenterol 2008; 14(2): 193-199
Nonalcoholic fatty liver disease (NAFLD) encompasses a disease spectrum ranging from simple hepatic steatosis to steatohepatitis (NASH), fibrosis, and cirrhosis. NAFLD has a very high prevalence in the US and worldwide[1]. It is becoming the leading cause for referral to liver clinics in most areas. In the US, it occurs in about 20%-35% of the population in over 60 million of the general adult population. Further, NAFLD occurs in about 2.6% of children and up to 53% of obese children are diagnosed with NAFLD[23]. The prevalence of NAFLD will likely continue to rise. Obesity, hyperglycemia, type 2 diabetes and hypertriglyceridemia are most important risk factors. Genetic factors undoubtedly predispose to NAFLD, as supported by higher prevalence of steatosis in Hispanics than Caucasians and African-Americans[4]. NAFLD has the potential to progress to hepatocellular carcinoma (HCC) or liver failure, both events that ultimately lead to early death.
DEFINITION AND CHARACTERISTICS OF NAFLD
NAFLD is defined as an excess of fat in the liver in which at least 5% of hepatocytes display lipid droplets[5] that exceed 5%-10% of liver weight[67] in patients who do not consume significant amounts of alcohol (140 g ethanol/week for men and 70 g ethanol/week) for women[8]. However, this definition is still controversial, because there is only 1.7% ± 0.2% of liver fat content in some healthy men[9] and liver fatty accumulation is absent in hepatic cirrhosis[10]. Morphologically, hepatic steatosis manifests as accumulation of macrovesicular or microvesicular intracytoplasmic fat droplets in hepatocytes. In macrovesicular steatosis, a large single fat vacuole fills the cytoplasm of hepatocytes and displaces the nucleus to the periphery, causing a characteristic signet ring appearance. In microvesicular steatosis, numerous small lipid droplets occupy the cytoplasm of hepatocytes and do not displace the nucleus to the periphery. Hepatic steatosis can be reversible or progress to NASH depending on the cessation or persistence of the underlying provocative cause, respectively. NASH represents steatosis, inflammation, fibrosis, ballooning hepatocytes, apoptotic cells and Mallory’s hyaline. The inflammatory extent varies considerably and does not always correlate with the degree of steatosis. Infiltrating inflammatory cells consist of lymphocytes and polymorphonuclear leukocytes. Apoptotic ballooned and Mallory hyaline hepatocytes may indicate the onset of NASH. Liver cell death and inflammation activate stellate cells leading to the development of hepatic fibrosis that often commences in zone 3 and manifests as perisinusoidal, perivenular (around terminal hepatic veins), and pericellular fibrosis. Hepatic steatosis, inflammation and aggressive fibrogenesis as well as sustained hepatocellular proliferation contribute to the development of liver cirrhosis.
DIAGNOSIS OF NAFLD
The majority of patients with NAFLD are asymptomatic or may complain of fatigue, exercise intolerance, or unspecific, vague abdominal pain in the right upper quadrant[5]. Physical examination can be unremarkable, although a palpable enlarged liver is frequent. Liver tests usually show minor nonspecific abnormalities. Alanine aminotransferases and gamma-glutamyl transpeptidase levels are elevated in most cases, but may be normal in some patients with advanced NASH and hepatic cirrhosis[11]. Imaging techniques including ultrasonography and magnetic resonance imaging are useful to detect degrees of steatosis, but can not distinguish inflammatory activity and fibrosis. Liver biopsy is still the gold standard for the diagnosis of NASH and fibrotic severity[12–14] which can be used to evaluate the degree of steatosis, inflammation, and fibrosis and also can help exclude other causes of liver disease[15].
PATHOGENESIS OF NAFLD
A currently favored hypothesis is that “two hits” are required for a subject to develop NASH[16]. The first hit leads to hepatic steatosis, and the second to hepatocyte injury and inflammation. However, the primary metabolic abnormalities leading to lipid accumulation within hepatocytes has remained poorly understood. Accumulating evidence indicates that mitochondrial dysfunction plays a central role in the pathogenesis of NAFLD, and that NAFLD is a mitochondrial disease[17].
Liver and metabolism
The liver, as a super metabolic achiever in the body, plays a critical role in metabolism of carbohydrate, protein and fat to maintain blood glucose and energy homeostasis.
Hepatic mitochondria: Mitochondria serve as the cellular powerhouse that generates ATP or heat by using substrates derived from fat and glucose. Hepatocytes are normally rich in mitochondria and each hepatocyte contains about 800 mitochondria occupying about 18% of the entire liver cell volume. Mitochondria play an important role in hepatocyte metabolism, being the primary site for the oxidation of fatty acids and oxidative phosphorylation. A mitochondrion contains inner and outer membranes composed of phospholipid bilayers and proteins. The outer mitochondrial membrane contains numerous integral proteins called porins, which contain a relatively large internal channel that is permeable to all molecules of 5000 daltons or less. Larger molecules can only traverse the outer membrane by active transport through mitochondrial membrane transport proteins. Unlike the outer membrane, the inner membrane does not contain porins, and is highly impermeable; almost all ions and molecules require special membrane transporters to enter or exit the matrix. The inner mitochondrial membrane contains proteins with four types of functions: the oxidation reactions of the respiratory chain; ATP synthase; specific transport proteins that regulate the passage of metabolites into and out of the matrix; protein import machinery. The matrix contains a highly concentrated mixture of hundreds of enzymes. Of the enzymes, the major functions include oxidation of pyruvate and fatty acids, and the citric acid cycle. The mitochondrial respiratory chain (MRC) is extremely important for energy generation and it consists of multiple polypeptides. Most of the respiratory chain polypeptides are encoded by nuclear DNA, but some are encoded by mitochondrial DNA (mtDNA). mtDNA is a circular double-stranded molecule located in the mitochondrial matrix. mtDNA is extremely sensitive to oxidative damage due to its proximity to the inner membrane, the absence of protective histones, and the incomplete DNA repair mechanisms in mitochondria. Therefore, any factors that affect mitochondrial integrate will cause reduced mitochondrial function.
Hepatic free fatty acids sources: (a) dietary triglycerides (TG) as chylomicron particles from the intestine; (b) de novo synthesis in the liver; (c) Free fatty acids (FFA). influx into the liver from lipolysis of adipose tissue; (d) diminished export of lipids from the liver; and (e) reduced oxidation of fatty acids. Imbalance of these metabolic steps will increased TG accumulation within the cytoplasm of hepatocytes.
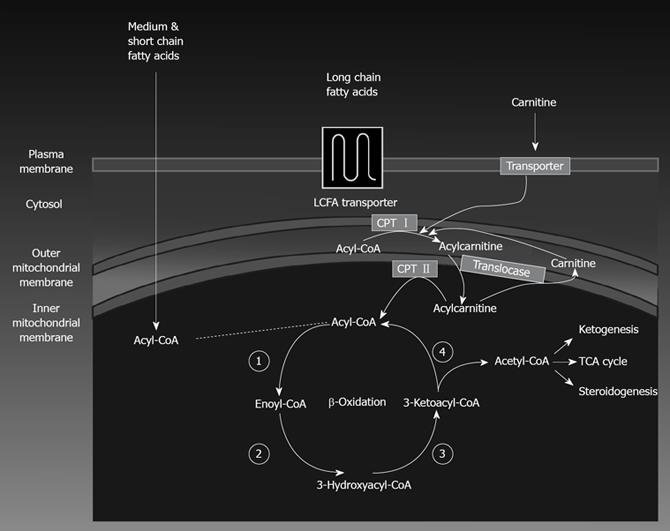
Hepatic mitochondrial fatty acid oxidation: Fatty acid oxidation (FAO) is the major source of energy for skeletal muscle and the heart, while the liver oxidizes fatty acids primarily under the conditions of prolonged fasting, during illness, and during periods of increased physical activity. FAO also plays an essential role in the intermediary metabolism of the liver. The oxidation of fatty acids in the liver fuels the synthesis of ketone bodies, 3-hydroxy butyrate and acetoacetate, which are utilized as alternative sources of energy by extrahepatic organs, like the brain when blood glucose levels are low. FAO occurs in three subcellular organelles, β-oxidation in mitochondria and peroxisomes, and ω-oxidation in the endoplasmic reticulum[1819]. Mitochondrial β-oxidation is the dominant oxidative pathway for the disposal of fatty acids under normal physiologic conditions and is primarily involved in the oxidation of short-chain (< C8), medium-chain (C8-C12), and long-chain (C12-C20) fatty acids. Short-chain and medium-chain FFAs freely enter the mitochondria, while long-chain FFAs entry into the mitochondria is regulated by the activity of the enzyme carnitine palmitoyl transferaseI(CPT-I). Glycolysis generates pyruvate, which is transformed by mitochondria into acetyl-CoA, an intermediate that goes through the citric acid cycle for the production of reducing agents and ATP. However, when glucose and energy levels are elevated, acetyl-CoA is converted to citrate which can leak out of the mitochondrial matrix into the cytosol through the tricarboxylate carrier. In the cytosol, citrate regenerates acetyl-CoA which is converted to malonyl-CoA by acetyl-CoA carboxylase. Malonyl-CoA plays important roles in both hepatic fatty acid oxidation and lipid synthesis. Malonyl-CoA is the initial component for fatty acid synthesis. High malonyl-CoA levels also actively inhibit CPT-I enzyme activity thus robustly decreasing fatty acid oxidation by reducing the rate of fatty acid entry into the mitochondria. Thus, periods of caloric overconsumption, and excessive energy supply, increase malonyl-CoA levels which promotes hepatic fatty acid synthesis (storage) and suppresses fatty acid oxidation (catabolism). Conversely, in the fasting state, hepatic malonyl-CoA levels are low, allowing extensive mitochondrial import of long-chain FFAs and high rates of β-oxidation. Figure 1 depicts the pathway for mitochondrial fatty acid oxidation. During β-oxidation in mitochondria, FFAs undergo a dehydrogenation, then hydration, followed by a second dehydrogenation, and finally thiolysis, releasing one 2-carbon acetyl-CoA molecule and a shortened fatty acid (Figure 1). The cycle is repeated to split the fatty acid into acetyl-CoA subunits. The acetyl-CoA units enter the citric acid cycle to produce reducing agents which can be converted to ATP in the electron transport chain. Under fasting conditions, acetyl-CoA moieties can be converted into ketone bodies (acetoacetate and β-hydroxybutyrate), which are released by the liver to be oxidized in peripheral tissues by the tricarboxylic acid cycle.
Figure 1 An illustration of mitochondrial fatty acid β-oxidation. LCFA: long-chain fatty acid; TCA: tricarboxylic acid.
MITOCHONDRIAL DYSFUNCTION IN NAFLD
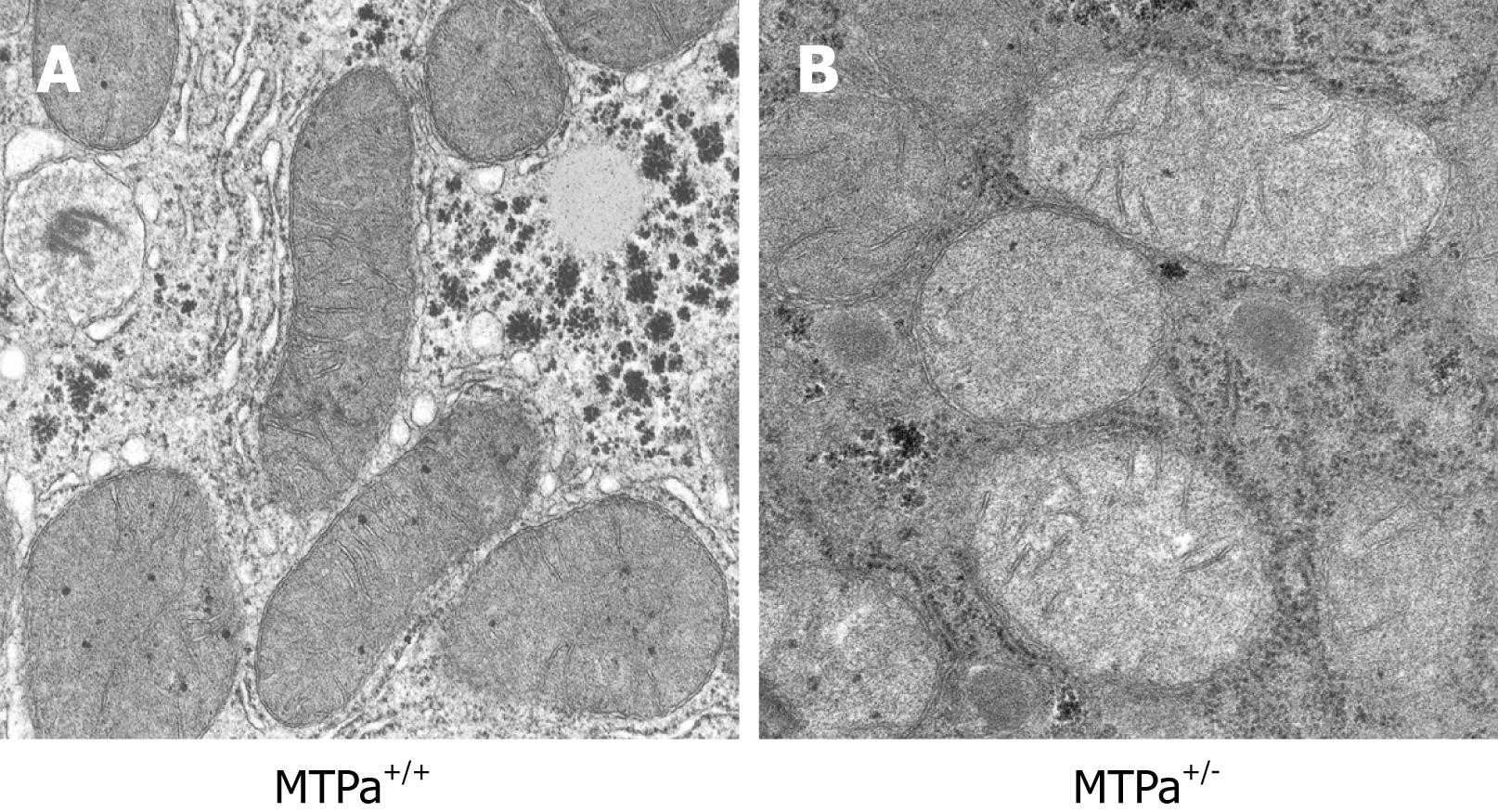
Although the mechanisms responsible for fatty liver are still not fully elucidated, decreased capacity to oxidize fatty acids, increased delivery and transport of FFAs into the liver, and augmented hepatic fatty acid synthesis are likely to play a significant role in the pathogenesis of NAFLD. We and others have shown that mitochondrial abnormalities are closely related to the pathogenesis of NAFLD which raised the possibility that NAFLD is a mitochondrial disease[1720–22]. The mitochondrial abnormalities associated with NAFLD include ultrastructural lesions, depletion of mitochondrial DNA (mtDNA), decreased activity of respiratory chain complexes, and impaired mitochondrial β-oxidation. Abnormal morphologic changes in liver mitochondria have been observed in patients and animal models with NASH[1820–24]. Electronic microscopy revealed that mitochondria in NAFLD are big and swelled, scarce in number, and that the mitochondrial matrix has paracrystalline inclusions and hypodensity. Figure 2 shows an example of the ultrastructural changes in the mitochondrial in a mouse model with a fatty acid oxidation defect and hepatic steatosis that we have generated and reported earlier[22]. We have also found similar mitochondrial lesions in other rodent models that develop hepatic steatosis/NASH (data not shown). These same mitochondrial lesions are found in liver biopsy specimens from patients treated with 4,4'-diethylaminoethoxyhexestrol, a drug that inhibits mitochondrial respiratory chain (MRC) activity and mitochondrial β-oxidation[25]. Prolonged treatment with this agent is associated with hepatic steatosis and steatohepatitis that is histologically indistinguishable from NAFLD in humans[25]. The ultrastructural mitochondrial defects in patients with NAFLD may be indicative of defective mitochondrial functions, e.g. reduced MRC activity[26] and impaired ATP synthesis[27]. NAFLD is often found in patients with insulin resistance, obesity and type 2 diabetes, the same metabolic conditions in which there is decreased oxygen consumption and ATP production, reduced total mtDNA and mtDNA transcription factor A, and reduced content of respiratory proteins in the fat, muscle and liver[28]. Many genes encoding mitochondrial proteins in skeletal muscle and fat are negatively correlated with body mass[29–33]. mtDNA depletion in hepatocytes impairs mitochondrial function and causes hepatic steatosis and other liver injury[3435]. Patients with NASH have decreased expression of mtDNA-encoded polypeptides[36] and low activity of complexes I, III, IV and V[26]. Mice with genetic deletion of NEIL1 DNA glycosylate have increased mtDNA damage and deletions and develop fatty liver disease[37].
Figure 2 Representative electron micrograph of hepatocytes from control wild-type mice (MTPa+/+) (A) and mice heterozygous for a mitochondrial trifunctional protein defect (MTPa+/-) (B) at 11 700 × magnification. The MTPa+/- mice develop hepatic steatosis (see Figure 3). The mitochondria from the MTPa+/- mice were swollen with hypodense matrix and disrupted cristae. Reproduced with permission from Gastroenterology. 2005; 128: 1381-1390.
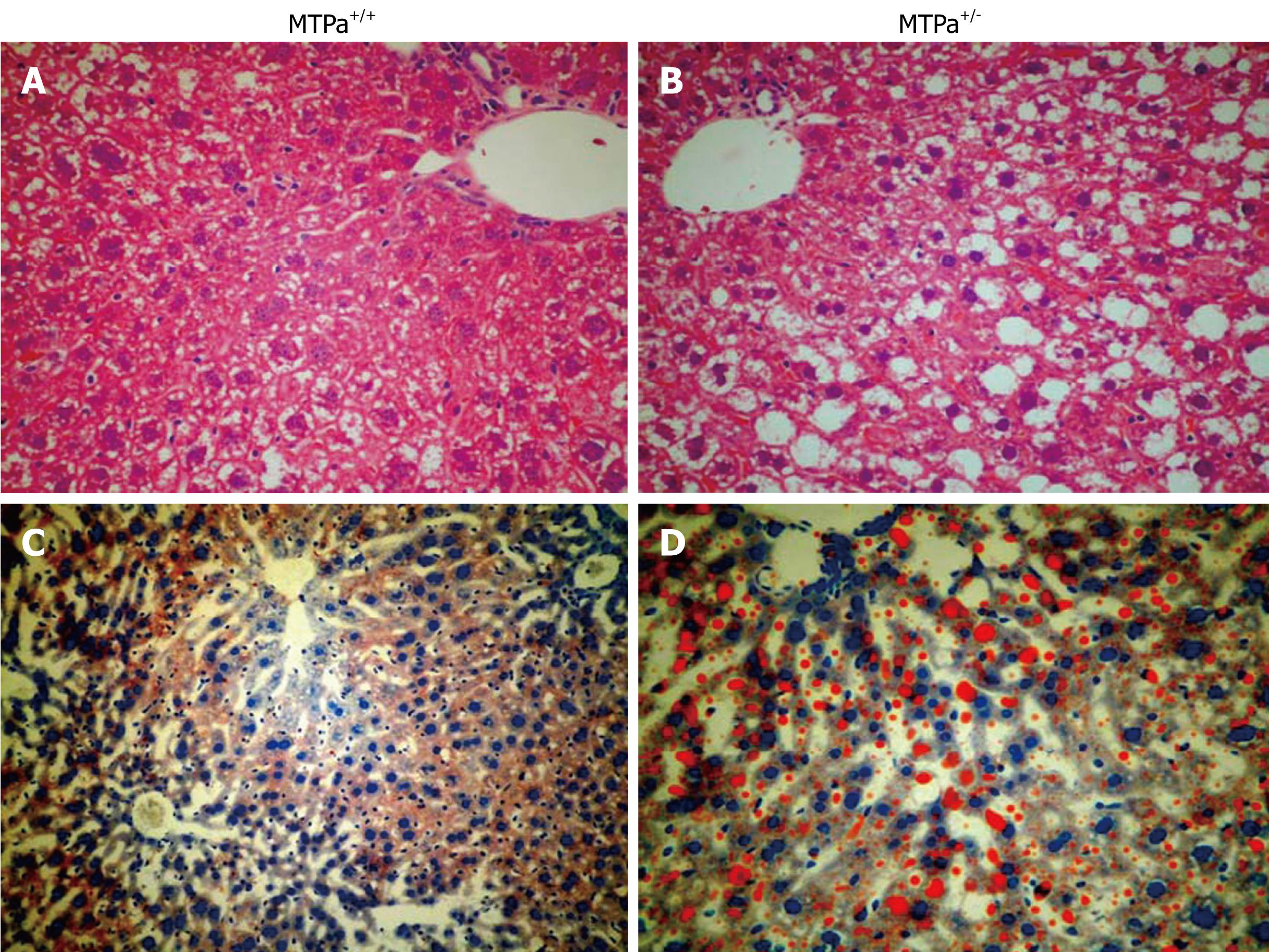
Figure 3 Representative liver sections obtained from control wild-type mice (MTPa+/+) (A and C) and mice with a defect in mitochondrial trifunctional protein (MTPa+/-) (B and D) littermates stained with hematoxylin-eosin (A and B) and oil red O (C and D) (20 ×). Reproduced with permission from Gastroenterology. 2005; 128: 1381-1390.
Multiple enzymes are involved in mitochondrial β-oxidation and deficiency of these enzymes may lead to the development of hepatic steatosis, e.g. mice with disrupted medium-chain and very-long-chain acyl-CoA dehydrogenase genes manifest defects in fatty acid oxidation that likely lead to the witnessed micro- and macrovascular hepatic steatosis found in these mice. Mitochondrial trifunctional protein (MTP) is a heterotrimeric protein that consists of four α-subunits and four β-subunits and catalyzes long-chain fatty acid oxidation. MTP defects in humans are recessively inherited, and children with defects of any of the three enzymatic activities exhibit mostly microvesicular hepatic steatosis. We have generated a mouse model for a null mutation causing complete MTP deficiency and demonstrated that homozygous mice develop hepatic steatosis immediately after birth[38]. In subsequent report, we have documented that aging mice heterozygous for the MTP defect also become insulin resistant and develop hepatic steatosis as shown in Figure 3[22]. Further, the activity of mitochondrial respiratory chain complex is decreased in liver tissue of patients and animal models with NAFLD[2639].
A number of mechanisms can be considered to explain the mitochondrial dysfunction found in NAFLD patients and animal models. Possible mechanisms include (a) excessive reactive oxygen species (ROS) production, (b) increased TNF-α expression, and (c) altered PGC-1 expression.
Reactive oxygen species
MRC dysfunction can directly lead to the production of ROS. If electron flow is interrupted at any point in the respiratory chain, the preceding respiratory intermediates can transfer electrons to molecular oxygen to produce superoxide anions and hydrogen peroxide[4041]. Malondialdehyde and 4-hydroxynonenal, two byproducts of lipid peroxidation, can inhibit mitochondrial cytochrome c oxidase by forming adducts with this enzyme. ROS-induced depletion in mtDNA can severely lower mitochondrial number and function leading to steatosis and liver lesions[34]. Such depletion can impair the synthesis of complexes I, III, IV and V of the MRC, because mtDNA encodes for 13 of the MRC polypeptides. Evidence of oxidative stress has been found in patients with NASH[21]. Decreased activity of the MRC in ob/ob mice is in part attributable to the oxidative stress, because treatment of ob/ob mice with antioxidant MnTBAP, a mimic of manganese superoxide dismutase, improved the activity of several complexes of the MRC. Surprisingly, activity of complex II, which polypeptides are only encoded by nuclear DNA, decreased even more in ob/ob mice treated with MnTBAP. Furthermore, liver histology improved markedly in mice treated with MnTBAP. MRC complexes’ activity was reduced by 30% to 50% versus control activity. These complexes’ activity was inversely correlated to blood TNF-α levels, body mass index, and HOMA index[26].
TNF-α
Another important factor to consider in the pathogenesis of mitochondrial dysfunction is TNF-α. High blood TNF-α levels have been found in patients with NASH[264243]. In ob/ob mice TNF-α concentrations in liver tissue were some 20-fold higher than in normal mice[39]. The likely sources of the hepatic TNF-α are hepatocytes and Kupffer cells[44]. TNF-α induces mitochondrial swelling with a lighter matrix and a loss of septa. In addition, TNF-α induced swelling of the mitochondria causes a bursting of the mitochondrial membrane leading to an interference between MRC complexes I and III[4546]. Anti-TNF-α treatment in ob/ob mice improve complex I, II, III and V activity, β-oxidation activity, and liver histology[39].
PGC-1
Nuclear receptors are pleiotropic regulators of glycolytic and oxidative metabolism[47]. Mitochondrial activity is transcriptionally controlled, in part, by the nuclear receptors and the peroxisome proliferator-activated receptor-γ coactivator 1 (PGC-1)-related protein family such as PGC-1α and PGC-1β[4849]. PGC-1α and PGC-1β are preferentially expressed in tissues with high oxidative capacity such as heart, skeletal muscle and brown adipose tissue, where they serve critical roles in the regulation of mitochondrial functional capacity and cellular energy metabolism[50–53] PGC-1α potently induces the expression of genes implicated in energy homeostasis in almost every cell type through known mitochondrial regulators such as the estrogen-related receptors (ERRs), peroxisome proliferator-activated receptor δ, or nuclear respiratory factor (NRF-1, 2)[48495455]. Overexpression of PGC-1α in skeletal muscle cells results in an increased energy expenditure, mitochondrial biogenesis[5253], whereas loss of PGC-1α results in reduced muscle performance, cardiac defects, and other metabolic and behavioral defects[5657]. Liver expresses low levels of PGC-1α and PGC-1β at normal condition, however, their expression is upregulated at fasting[58–60]. PGC-1α and PGC-1β activate hepatic fatty oxidation by inducing expression of PPARα[5960]. Hepatocytes from PGC-1α deficient mice have diminished FAO activity and mitochondrial respiration rates[49].
CONCLUSION
Genetic, environmental and nutritional factors are involved in the pathogenesis of NAFLD, which is nowadays a major public health problem. In the absence of proven pharmacological therapy of NAFLD, it is critical to explore its pathogenesis and novel therapeutic pathways. In this Editorial, we have reviewed evidence that implicates mitochondrial dysfunction as a primary mechanism for development of NAFLD. Mitochondrial dysfunction may not only cause fat accumulation, but also may lead to the generation of ROS and cytokine production contributing to progression of NAFLD by inducing hepatic inflammation and fibrosis.
Footnotes
S- Editor Liu Y L- Editor Alpini GD E- Editor Liu Y
References
1.
Clark JM, Brancati FL, Diehl AM. Nonalcoholic fatty liver disease. Gastroenterology. 2002;122:1649-1657. [PubMed] [DOI] [Cited in This Article: ]
2.
Franzese A, Vajro P, Argenziano A, Puzziello A, Iannucci MP, Saviano MC, Brunetti F, Rubino A. Liver involvement in obese children. Ultrasonography and liver enzyme levels at diagnosis and during follow-up in an Italian population. Dig Dis Sci. 1997;42:1428-1432. [PubMed] [DOI] [Cited in This Article: ]
3.
Tominaga K, Kurata JH, Chen YK, Fujimoto E, Miyagawa S, Abe I, Kusano Y. Prevalence of fatty liver in Japanese children and relationship to obesity. An epidemiological ultrasonographic survey. Dig Dis Sci. 1995;40:2002-2009. [PubMed] [DOI] [Cited in This Article: ]
4.
Browning JD, Szczepaniak LS, Dobbins R, Nuremberg P, Horton JD, Cohen JC, Grundy SM, Hobbs HH. Prevalence of hepatic steatosis in an urban population in the United States: impact of ethnicity. Hepatology. 2004;40:1387-1395. [PubMed] [DOI] [Cited in This Article: ]
5.
Neuschwander-Tetri BA. Nonalcoholic steatohepatitis and the metabolic syndrome. Am J Med Sci. 2005;330:326-335. [PubMed] [DOI] [Cited in This Article: ]
6.
Adams LA, Lymp JF, St Sauver J, Sanderson SO, Lindor KD, Feldstein A, Angulo P. The natural history of nonalcoholic fatty liver disease: a population-based cohort study. Gastroenterology. 2005;129:113-121. [PubMed] [DOI] [Cited in This Article: ]
7.
Browning JD, Horton JD. Molecular mediators of hepatic steatosis and liver injury. J Clin Invest. 2004;114:147-152. [PubMed] [DOI] [Cited in This Article: ]
9.
Seppala-Lindroos A, Vehkavaara S, Hakkinen AM, Goto T, Westerbacka J, Sovijarvi A, Halavaara J, Yki-Jarvinen H. Fat accumulation in the liver is associated with defects in insulin suppression of glucose production and serum free fatty acids independent of obesity in normal men. J Clin Endocrinol Metab. 2002;87:3023-3028. [PubMed] [DOI] [Cited in This Article: ]
10.
Loria P, Lonardo A, Carulli N. Should nonalcoholic fatty liver disease be renamed? Dig Dis. 2005;23:72-82. [PubMed] [DOI] [Cited in This Article: ]
11.
Ong JP, Elariny H, Collantes R, Younoszai A, Chandhoke V, Reines HD, Goodman Z, Younossi ZM. Predictors of nonalcoholic steatohepatitis and advanced fibrosis in morbidly obese patients. Obes Surg. 2005;15:310-315. [PubMed] [DOI] [Cited in This Article: ]
12.
Ahmed MH, Osman KA, Osman MM. Invasive and non-invasive investigations for tamoxifen-induced non-alcoholic steatohepatitis (NASH): the benefit of computed tomography scan guided liver biopsy. Pathology. 2006;38:270-271. [PubMed] [DOI] [Cited in This Article: ]
13.
Brunt EM, Janney CG, Di Bisceglie AM, Neuschwander-Tetri BA, Bacon BR. Nonalcoholic steatohepatitis: a proposal for grading and staging the histological lesions. Am J Gastroenterol. 1999;94:2467-2474. [PubMed] [DOI] [Cited in This Article: ]
14.
Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, Ferrell LD, Liu YC, Torbenson MS, Unalp-Arida A. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41:1313-1321. [PubMed] [DOI] [Cited in This Article: ]
15.
Skelly MM, James PD, Ryder SD. Findings on liver biopsy to investigate abnormal liver function tests in the absence of diagnostic serology. J Hepatol. 2001;35:195-199. [PubMed] [DOI] [Cited in This Article: ]
17.
Pessayre D, Fromenty B. NASH: a mitochondrial disease. J Hepatol. 2005;42:928-940. [PubMed] [DOI] [Cited in This Article: ]
18.
Rao MS, Reddy JK. PPARalpha in the pathogenesis of fatty liver disease. Hepatology. 2004;40:783-786. [PubMed] [DOI] [Cited in This Article: ]
19.
Reddy JK, Hashimoto T. Peroxisomal beta-oxidation and peroxisome proliferator-activated receptor alpha: an adaptive metabolic system. Annu Rev Nutr. 2001;21:193-230. [PubMed] [DOI] [Cited in This Article: ]
20.
Caldwell SH, Swerdlow RH, Khan EM, Iezzoni JC, Hespenheide EE, Parks JK, Parker WD Jr. Mitochondrial abnormalities in non-alcoholic steatohepatitis. J Hepatol. 1999;31:430-434. [PubMed] [DOI] [Cited in This Article: ]
21.
Sanyal AJ, Campbell-Sargent C, Mirshahi F, Rizzo WB, Contos MJ, Sterling RK, Luketic VA, Shiffman ML, Clore JN. Nonalcoholic steatohepatitis: association of insulin resistance and mitochondrial abnormalities. Gastroenterology. 2001;120:1183-1192. [PubMed] [DOI] [Cited in This Article: ]
22.
Ibdah JA, Perlegas P, Zhao Y, Angdisen J, Borgerink H, Shadoan MK, Wagner JD, Matern D, Rinaldo P, Cline JM. Mice heterozygous for a defect in mitochondrial trifunctional protein develop hepatic steatosis and insulin resistance. Gastroenterology. 2005;128:1381-1390. [PubMed] [DOI] [Cited in This Article: ]
23.
Sobaniec-Lotowska ME, Lebensztejn DM. Ultrastructure of hepatocyte mitochondria in nonalcoholic steatohepatitis in pediatric patients: usefulness of electron microscopy in the diagnosis of the disease. Am J Gastroenterol. 2003;98:1664-1665. [PubMed] [DOI] [Cited in This Article: ]
24.
Begriche K, Igoudjil A, Pessayre D, Fromenty B. Mitochondrial dysfunction in NASH: causes, consequences and possible means to prevent it. Mitochondrion. 2006;6:1-28. [PubMed] [DOI] [Cited in This Article: ]
25.
Berson A, De Beco V, Letteron P, Robin MA, Moreau C, El Kahwaji J, Verthier N, Feldmann G, Fromenty B, Pessayre D. Steatohepatitis-inducing drugs cause mitochondrial dysfunction and lipid peroxidation in rat hepatocytes. Gastroenterology. 1998;114:764-774. [PubMed] [DOI] [Cited in This Article: ]
26.
Perez-Carreras M, Del Hoyo P, Martin MA, Rubio JC, Martin A, Castellano G, Colina F, Arenas J, Solis-Herruzo JA. Defective hepatic mitochondrial respiratory chain in patients with nonalcoholic steatohepatitis. Hepatology. 2003;38:999-1007. [PubMed] [DOI] [Cited in This Article: ]
27.
Cortez-Pinto H, Zhi Lin H, Qi Yang S, Odwin Da Costa S, Diehl AM. Lipids up-regulate uncoupling protein 2 expression in rat hepatocytes. Gastroenterology. 1999;116:1184-1193. [PubMed] [DOI] [Cited in This Article: ]
28.
Valerio A, Cardile A, Cozzi V, Bracale R, Tedesco L, Pisconti A, Palomba L, Cantoni O, Clementi E, Moncada S. TNF-alpha downregulates eNOS expression and mitochondrial biogenesis in fat and muscle of obese rodents. J Clin Invest. 2006;116:2791-2798. [PubMed] [DOI] [Cited in This Article: ]
29.
Sparks LM, Xie H, Koza RA, Mynatt R, Hulver MW, Bray GA, Smith SR. A high-fat diet coordinately downregulates genes required for mitochondrial oxidative phosphorylation in skeletal muscle. Diabetes. 2005;54:1926-1933. [PubMed] [DOI] [Cited in This Article: ]
30.
Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, Sole J, Nichols A, Ross JS, Tartaglia LA. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. 2003;112:1821-1830. [PubMed] [DOI] [Cited in This Article: ]
31.
Wilson-Fritch L, Nicoloro S, Chouinard M, Lazar MA, Chui PC, Leszyk J, Straubhaar J, Czech MP, Corvera S. Mitochondrial remodeling in adipose tissue associated with obesity and treatment with rosiglitazone. J Clin Invest. 2004;114:1281-1289. [PubMed] [DOI] [Cited in This Article: ]
32.
Patti ME, Butte AJ, Crunkhorn S, Cusi K, Berria R, Kashyap S, Miyazaki Y, Kohane I, Costello M, Saccone R. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: Potential role of PGC1 and NRF1. Proc Natl Acad Sci USA. 2003;100:8466-8671. [PubMed] [DOI] [Cited in This Article: ]
33.
Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstrale M, Laurila E. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34:267-273. [PubMed] [DOI] [Cited in This Article: ]
34.
Demeilliers C, Maisonneuve C, Grodet A, Mansouri A, Nguyen R, Tinel M, Letteron P, Degott C, Feldmann G, Pessayre D. Impaired adaptive resynthesis and prolonged depletion of hepatic mitochondrial DNA after repeated alcohol binges in mice. Gastroenterology. 2002;123:1278-1290. [PubMed] [DOI] [Cited in This Article: ]
35.
Fromenty B, Pessayre D. Inhibition of mitochondrial beta-oxidation as a mechanism of hepatotoxicity. Pharmacol Ther. 1995;67:101-154. [PubMed] [DOI] [Cited in This Article: ]
36.
Santamaria E, Avila MA, Latasa MU, Rubio A, Martin-Duce A, Lu SC, Mato JM, Corrales FJ. Functional proteomics of nonalcoholic steatohepatitis: mitochondrial proteins as targets of S-adenosylmethionine. Proc Natl Acad Sci USA. 2003;100:3065-3070. [PubMed] [DOI] [Cited in This Article: ]
37.
Vartanian V, Lowell B, Minko IG, Wood TG, Ceci JD, George S, Ballinger SW, Corless CL, McCullough AK, Lloyd RS. The metabolic syndrome resulting from a knockout of the NEIL1 DNA glycosylase. Proc Natl Acad Sci USA. 2006;103:1864-1869. [PubMed] [DOI] [Cited in This Article: ]
38.
Ibdah JA, Paul H, Zhao Y, Binford S, Salleng K, Cline M, Matern D, Bennett MJ, Rinaldo P, Strauss AW. Lack of mitochondrial trifunctional protein in mice causes neonatal hypoglycemia and sudden death. J Clin Invest. 2001;107:1403-1409. [PubMed] [DOI] [Cited in This Article: ]
39.
Garcia-Ruiz I, Rodriguez-Juan C, Diaz-Sanjuan T, del Hoyo P, Colina F, Munoz-Yague T, Solis-Herruzo JA. Uric acid and anti-TNF antibody improve mitochondrial dysfunction in ob/ob mice. Hepatology. 2006;44:581-591. [PubMed] [DOI] [Cited in This Article: ]
40.
Garcia-Ruiz C, Colell A, Morales A, Kaplowitz N, Fernandez-Checa JC. Role of oxidative stress generated from the mitochondrial electron transport chain and mitochondrial glutathione status in loss of mitochondrial function and activation of transcription factor nuclear factor-kappa B: studies with isolated mitochondria and rat hepatocytes. Mol Pharmacol. 1995;48:825-834. [PubMed] [DOI] [Cited in This Article: ]
41.
Hensley K, Kotake Y, Sang H, Pye QN, Wallis GL, Kolker LM, Tabatabaie T, Stewart CA, Konishi Y, Nakae D. Dietary choline restriction causes complex I dysfunction and increased H(2)O(2) generation in liver mitochondria. Carcinogenesis. 2000;21:983-989. [PubMed] [DOI] [Cited in This Article: ]
42.
Kugelmas M, Hill DB, Vivian B, Marsano L, McClain CJ. Cytokines and NASH: a pilot study of the effects of lifestyle modification and vitamin E. Hepatology. 2003;38:413-419. [PubMed] [DOI] [Cited in This Article: ]
43.
Hui JM, Hodge A, Farrell GC, Kench JG, Kriketos A, George J. Beyond insulin resistance in NASH: TNF-alpha or adiponectin? Hepatology. 2004;40:46-54. [PubMed] [DOI] [Cited in This Article: ]
44.
Crespo J, Cayon A, Fernandez-Gil P, Hernandez-Guerra M, Mayorga M, Dominguez-Diez A, Fernandez-Escalante JC, Pons-Romero F. Gene expression of tumor necrosis factor alpha and TNF-receptors, p55 and p75, in nonalcoholic steatohepatitis patients. Hepatology. 2001;34:1158-1163. [PubMed] [DOI] [Cited in This Article: ]
45.
Sanchez-Alcazar JA, Schneider E, Martinez MA, Carmona P, Hernandez-Munoz I, Siles E, De La Torre P, Ruiz-Cabello J, Garcia I, Solis-Herruzo JA. Tumor necrosis factor-alpha increases the steady-state reduction of cytochrome b of the mitochondrial respiratory chain in metabolically inhibited L929 cells. J Biol Chem. 2000;275:13353-13361. [PubMed] [DOI] [Cited in This Article: ]
46.
Higuchi M, Proske RJ, Yeh ET. Inhibition of mitochondrial respiratory chain complex I by TNF results in cytochrome c release, membrane permeability transition, and apoptosis. Oncogene. 1998;17:2515-2524. [PubMed] [DOI] [Cited in This Article: ]
47.
Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schutz G, Umesono K, Blumberg B, Kastner P, Mark M, Chambon P. The nuclear receptor superfamily: the second decade. Cell. 1995;83:835-839. [PubMed] [DOI] [Cited in This Article: ]
48.
Lin J, Handschin C, Spiegelman BM. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 2005;1:361-370. [PubMed] [DOI] [Cited in This Article: ]
49.
Finck BN, Kelly DP. PGC-1 coactivators: inducible regulators of energy metabolism in health and disease. J Clin Invest. 2006;116:615-622. [PubMed] [DOI] [Cited in This Article: ]
50.
Kamei Y, Ohizumi H, Fujitani Y, Nemoto T, Tanaka T, Takahashi N, Kawada T, Miyoshi M, Ezaki O, Kakizuka A. PPARgamma coactivator 1beta/ERR ligand 1 is an ERR protein ligand, whose expression induces a high-energy expenditure and antagonizes obesity. Proc Natl Acad Sci USA. 2003;100:12378-12383. [PubMed] [DOI] [Cited in This Article: ]
51.
Lin J, Puigserver P, Donovan J, Tarr P, Spiegelman BM. Peroxisome proliferator-activated receptor gamma coactivator 1beta (PGC-1beta ), a novel PGC-1-related transcription coactivator associated with host cell factor. J Biol Chem. 2002;277:1645-1648. [PubMed] [DOI] [Cited in This Article: ]
52.
St-Pierre J, Lin J, Krauss S, Tarr PT, Yang R, Newgard CB, Spiegelman BM. Bioenergetic analysis of peroxisome proliferator-activated receptor gamma coactivators 1alpha and 1beta (PGC-1alpha and PGC-1beta) in muscle cells. J Biol Chem. 2003;278:26597-26603. [PubMed] [DOI] [Cited in This Article: ]
53.
Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. 1999;98:115-124. [PubMed] [DOI] [Cited in This Article: ]
54.
Wang YX, Lee CH, Tiep S, Yu RT, Ham J, Kang H, Evans RM. Peroxisome-proliferator-activated receptor delta activates fat metabolism to prevent obesity. Cell. 2003;113:159-170. [PubMed] [DOI] [Cited in This Article: ]
56.
Lin J, Wu H, Tarr PT, Zhang CY, Wu Z, Boss O, Michael LF, Puigserver P, Isotani E, Olson EN. Transcriptional co-activator PGC-1 alpha drives the formation of slow-twitch muscle fibres. Nature. 2002;418:797-801. [PubMed] [DOI] [Cited in This Article: ]
57.
Lin J, Wu PH, Tarr PT, Lindenberg KS, St-Pierre J, Zhang CY, Mootha VK, Jager S, Vianna CR, Reznick RM. Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1alpha null mice. Cell. 2004;119:121-135. [PubMed] [DOI] [Cited in This Article: ]
58.
Herzig S, Long F, Jhala US, Hedrick S, Quinn R, Bauer A, Rudolph D, Schutz G, Yoon C, Puigserver P, Spiegelman B, Montminy M. CREB regulates hepatic gluconeogenesis through the coactivator PGC-1. Nature. 2001;413:179-183. [PubMed] [DOI] [Cited in This Article: ]
59.
Lin J, Tarr PT, Yang R, Rhee J, Puigserver P, Newgard CB, Spiegelman BM. PGC-1beta in the regulation of hepatic glucose and energy metabolism. J Biol Chem. 2003;278:30843-30848. [PubMed] [DOI] [Cited in This Article: ]
60.
Yoon JC, Puigserver P, Chen G, Donovan J, Wu Z, Rhee J, Adelmant G, Stafford J, Kahn CR, Granner DK. Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature. 2001;413:131-138. [PubMed] [DOI] [Cited in This Article: ]