Bile-acid-induced cell injury and protection (original) (raw)
INTRODUCTION
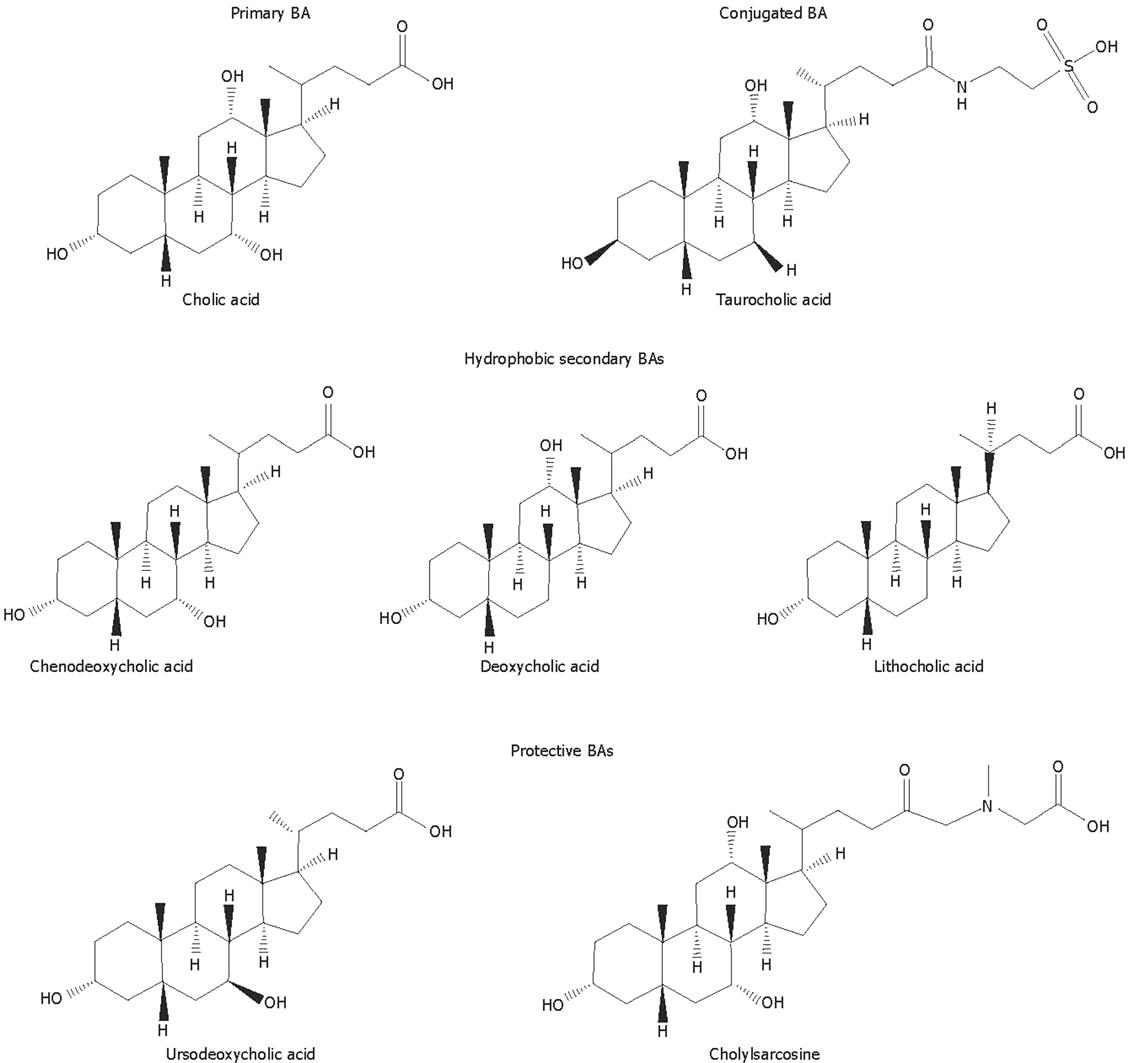
Bile acids (BAs) are the major organic solutes in bile, and are involved in several important functions in the liver and the intestine. However, the retention of hydrophobic BAs in pathophysiological conditions, such as cholestatic diseases, is believed to play an important role in liver injury by inducing apoptosis or necrosis of hepatocytes[1]. Primary BAs are directly synthesized from cholesterol by hepatocytes, by the addition of hydroxyl groups and the oxidation of its side chain to form a more water soluble end product. The hydroxylation is always on one side of the molecule resulting in an amphipathic molecule. In humans, the most abundant BA species are the primary BAs cholic acid (CA) and chenodeoxycholic acid (CDCA) (Figure 1), and the secondary BAs deoxycholic acid (DCA) and lithocholic acid (LCA) (Figure 1), generated in the intestine by bacterial biotransformation of CA and CDCA, respectively. Small amounts of other secondary BAs such as ursodeoxycholic acid (UDCA; Figure 1), currently used in the treatment of cholestatic liver diseases, are also present in the human BA pool[23].
Figure 1 Molecular structures of different potentially toxic or protective natural bile acids and synthetic bile acid analogues.
BA hydrophobicity is an important determinant of the toxicity and protection of BAs, two biological properties of these compounds which will be reviewed in this manuscript. BA hydrophobicity depends on the number, position and orientation of the hydroxyl groups, as well as amidation at the C-24 position. Regarding the magnitude of hydrophobicity of BAs the order would be UDCA < CA < CDCA < DCA < LCA[4]. Before being secreted into bile, most BA molecules are conjugated with glycine or taurine (Figure 1) within hepatocytes to more hydrophilic amidated forms. Under normal conditions, the amount of BAs undergoing further biotransformations to dianionic glucuronidated or sulfated derivatives is negligible, although these may become important in cholestasis[5].
BILE-ACID-INDUCED HEPATOCYTE INJURY
The retention and accumulation of hydrophobic BAs, such as CDCA and DCA, inside hepatocytes during cholestasis have long been implicated as a major cause of liver damage in this disease[1]. Experimentally, hydrophobic BAs are known to induce injury to isolated hepatocytes[6], cultured hepatocytes[7], and whole liver[8], but the mechanisms involved in this toxicity are not fully understood. In animal models[9] and human cholestatic disorders[10], hepatocyte swelling and the intracellular accumulation of bile pigments have been reported to occur. Moreover, swelling, pleomorphism and abnormal cristae have also been reported in mitochondria from cholestatic hepatocytes[11]. This injury to parenchymal cells, which occurs early on in the course of cholestasis, can be responsible for many of the subsequent inflammatory and fibrinogenic responses of non-parenchymal cells. Thus, altered hepatocytes may release molecules, including growth factors, cytokines, chemokines and lipid peroxide products, able to amplify the inflammatory response, stimulate fibrogenesis by hepatic stellate cells, or directly injure other nearby cells[12].
Several mechanisms may account for the cytotoxicity associated with the most hydrophobic BAs in cholestatic liver diseases[1]. BAs could disrupt cell membranes through their detergent action on lipid components[13] and promote the generation of reactive oxygen species (ROS) that, in turn, oxidatively modify lipids, proteins, and nucleic acids, and eventually cause hepatocyte apoptosis[14]. Additionally, they can activate Kupffer cells to generate ROS, which may further contribute to the liver cell insult[15].
BILE-ACID-INDUCED OXIDATIVE STRESS
Several studies have suggested that oxidative stress may play an important role in the pathogenesis of hepatic injury during cholestasis in rats[16] and humans[17]. Thus, the antioxidant α-tocopherol is able to reduce both hydrophobic BA-induced ROS generation and injury to hepatocytes _in vitro_[18] and _in vivo_[8]. Hepatic mitochondria have been proposed as a major source of the oxidative stress induced by these BAs. In this respect, it has been demonstrated that hepatic mitochondria undergo lipid peroxidation during experimental cholestasis and BA toxicity in rats[816]. Hydrophobic BAs impair respiration and electron transport in hepatic mitochondria. These compounds decrease the activities of several enzyme complexes involved in the electron transport chain, such as complexes I, III and IV, whereas complex II is not affected in isolated rat liver mitochondria[19]. Moreover, toxic BAs decrease the mitochondrial membrane potential developed upon succinate energization[20]. These compounds decrease state three and enhance state four respiration in mitochondria[20]. The decrease in state three respiration is probably related to an inhibitory action of these compounds on the phosphorylation system, although by means of a mechanism that has yet to be defined. The reported stimulation of state four by hydrophobic BAs is associated with an enhanced permeability of mitochondria to protons[20]. The observed uncoupling may be the consequence of several interactions of these compounds with the structure of mitochondria. These BAs induced membrane permeability to protons, either by acting as protonophores or by disruption of the structural organization of membrane components. Hydrophobic BAs can stimulate the generation of ROS in rat hepatic mitochondria and hepatocytes[1821] as well as in human hepatic mitochondria[22]. Intracellular ROS generation by mitochondria appears to be an early event in hydrophobic BA-induced hepatocyte toxicity[21]. As occurs in cholestatic rats, this can lead to a depletion of antioxidant defences, including total hepatic and mitochondrial glutathione contents, and the concentrations of substances involved in electron transport, such as ubiquinone-9 and ubiquinone-10[23].
Hydrophobic BAs can induce the mitochondrial permeability transition (MPT)[222425], a critical intracellular event that triggers both the apoptotic and necrotic forms of cell death in hepatocytes[26]. MPT involves a rapid increase in the permeability of the mitochondrial membrane to low-molecular-weight solutes, caused by the opening of a channel composed of the adenine nucleotide translocator of the inner membrane, the voltage-dependent anion channel of the outer membrane, and mitochondrial cyclophilin D[26]. Induction of MPT is associated with mitochondrial swelling, collapse of the mitochondrial membrane potential, reduced oxidative phosphorylation, rupture of the outer mitochondrial membrane, cytochrome c release, and generation of ROS[26]. Hydrophobic BA-induced MPT has been associated with mitochondrial ROS generation in rat hepatocytes[27] and in isolated mitochondria[14]. An elevation of the cytosolic free calcium concentration induced by hydrophobic BAs[28] may also be an important permissive factor that allows oxidant stress to open the permeability pore.
BILE-ACID-INDUCED CELL DEATH PATHWAYS
BA accumulation within the hepatocyte can result in cell injury and death through two mechanisms; lower BA concentrations induce hepatocellular apoptosis[2529–31], whereas higher concentrations induce necrosis[721]. Both types of cell death seem to play a role in cholestatic liver injury, although the contribution of each is controversial. The role of apoptosis in cholestatic injury has been demonstrated[32]. However, in certain models of BA toxicity in the mouse, it has been reported that hepatocyte necrosis is the predominant form of cell death[33].
Interestingly, at the level of apoptosis induction, cytotoxicity of BAs does not always correlate with their hydrophobicity, which is not the case in necrosis. Apoptosis induction is indeed dependent on the BA, its concentration, or its conjugation state[29].
Necrosis
In rat hepatocytes, cellular necrosis is characterized by cell swelling and disruption of the intracellular and plasma membranes. BA-induced lethal hepatocellular injury has been attributed to direct membrane damage due to the detergent-like properties of hydrophobic BAs[13], as well as to depletion of ATP, ion dysregulation, mitochondrial and cellular swelling, plasma membrane failure, and cell lysis, releasing intracellular contents[6]. In the liver of humans with cholestatic disorders, the histological features of hepatocyte necrosis, such as massive swelling of hepatocytes containing accumulated bile and elevated serum hepatocellular aminotransferase enzymes, have been observed[10]. Antioxidants have been shown to prevent hepatocellular necrosis and reduce oxidant stress in isolated hepatocytes exposed to hydrophobic BAs[1821] and to inhibit the dissipation of mitochondrial membrane potential[14]. Thus, it has been proposed that MPT induction by ROS generated in hepatocyte mitochondria[27] is a critical event promoting BA-induced hepatocyte necrosis. In contrast to BA-induced apoptosis, only high concentrations of BAs are able to induce hepatocyte necrosis[721]. The generation of ROS can result from a direct detergent effect of BAs on membrane enzymes, such as phospholipase A2, which upon activation release arachidonic acid. Mitochondrial damage due to BAs can arise from several causes, including the endogenous generation of arachidonic acid[34].
Apoptosis
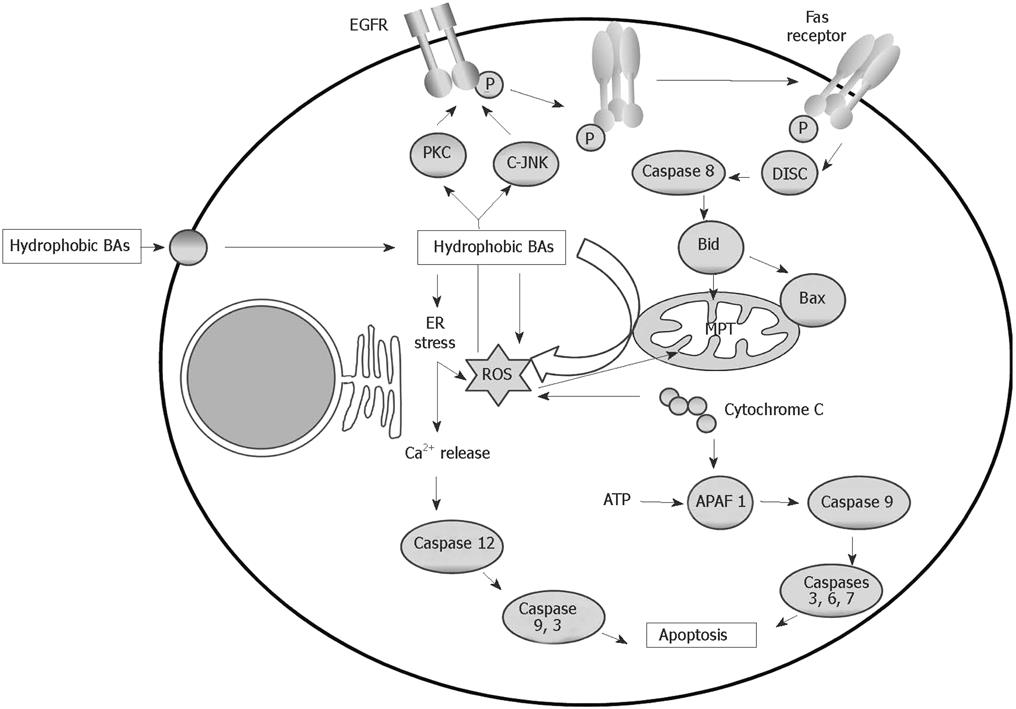
The impairment of apoptosis in hepatocytes and bile duct epithelial cells has been implicated in the pathogenesis of many liver diseases[32]. It is well known that hydrophobic BAs can induce apoptosis by activating the death receptor or extrinsic pathway[3536], and through the mitochondrial or intrinsic pathway[252730] (Figure 2). More recently, it has been demonstrated that hydrophobic BAs can also induce apoptosis in hepatocytes by causing endoplasmic reticulum (ER) stress (Figure 2). In contrast to BA-induced cell necrosis, apoptosis is characterized by the maintenance of cellular ATP content[29].
Figure 2 Intracellular mechanisms of bile acid-induced hepatocyte apoptosis. In this schema, further transduction after activation of death receptors and formation of the DISC, direct mitochondrial toxicity and ER stress are implicated.
Extrinsic pathway of apoptosis: BA-associated hepatocyte apoptosis has been shown to occur through the death receptors Fas[35] and TRAIL-R2. In contrast, BAs do not appear to enhance tumor necrosis factor (TNF)-α/TNF-R1 cytotoxicity[36]. Toxic BAs cause cell death, partly due to the activation of a protease cascade. The proximal signaling protease caspase 8 appears to be activated by toxic BAs in a Fas-receptor-dependent manner. After caspase 8 activation, cathepsin B activity also increases. Inhibition of either protease attenuates apoptosis in vitro, suggesting that they both play a critical role in BA-induced apoptosis[35]. Moreover, toxic BAs can induce Fas aggregation on the plasma membrane via a Fas-ligand-independent mechanism[37]. These compounds appear to promote Fas activation by altering the cellular trafficking of this death receptor. The shuttling of Fas and the resulting apoptosis can be inhibited by Golgi-disrupting agents and microtubule poisons. By inference, a Golgi-associated and microtubule-dependent pathway appears to be involved in the trafficking of Fas to the cell surface during BA cytotoxicity[38].
Oxidative stress has been implicated in the stimulation of Fas translocation induced by BAs. It has been demonstrated that BA-induced oxidative stress may trigger the activation of c-Jun-N-terminal kinases (JNKs) and protein kinase C (PKC). These are responsible for activating the epidermal growth factor receptor (EGFR), which associates with Fas in a JNK-dependent manner. The resulting phosphorylation of Fas induces its mobilization to the plasma membrane[39]. The increased density of Fas on the cell surface also sensitizes hepatocytes to cell death induced by Fas agonists[38]. Thus, toxic BAs also promote Fas-ligand-dependent hepatocyte apoptosis.
After death receptor activation and death-inducing signaling complex (DISC) formation, caspase 8 is activated and the pro-apoptotic protein Bid is cleaved and translocated to the mitochondria, which results in opening of the MPT pore, and the release of cytochrome c and other proapoptotic intermembrane space small molecules. Cytosolic cytochrome c leads to the binding of apoptosis activating factor-1 (APAF 1) with procaspase 9, resulting in the activation of caspase 9. A caspase cascade in then initiated and, finally, activation of the effector caspases, which leads to irreversible hepatocyte death[40].
The death receptor TRAIL-R2 has been also suggested to be involved in the apoptosis induced by BA. Enhanced expression and oligomerization of this death receptor has been described in glycochenodeoxycholic acid (GCDCA)-induced apoptosis in Fas-deficient cell lines[36].
Intrinsic pathway of apoptosis: BAs are able also to induce apoptosis through the mitochondrial or intrinsic pathway, in which intracellular stress causes mitochondrial dysfunction and the subsequent release of proapoptotic factors[252730]. As mentioned above, BAs can directly induce the generation of ROS by mitochondria[1821] and mitochondrial membrane potential depolarization[273031] in rat hepatocytes. MPT induction in apoptosis induced by several BAs has also been demonstrated[2425]. Antioxidants prevented GCDCA-induced apoptosis in rat hepatocytes through a mechanism involving the MPT[25]. It has also been demonstrated that DCA-triggered apoptosis, involved in decreased mitochondrial membrane potential and alterations in Bax subcellular distribution, exhibits increased mitochondrial-associated Bax protein levels[31]. These results suggest that BA-induced MPT is the initial event, and the initial cytochrome c release stimulates Bax translocation to the mitochondria, accomplishing further cytochrome c release[31]. In vivo studies show that 3 d after bile-duct-ligation, Bax expression is increased but then decreases over time. Bax translocation to the mitochondria and cytochrome c release are also found in these conditions[41].
Mitochondrial dysfunction can also occur in death-receptor-mediated apoptosis, especially the so called “type II cells”, such as hepatocytes. Mitochondrial cytochrome c release is (Fas-associated death domain) FADD/caspase 8-dependent during the death receptor-mediated apoptosis of type II cells[42].
ER stress-induced apoptosis: More recently, it has been demonstrated that hydrophobic BAs can also induce apoptosis in hepatocytes through another intracellular pathway of cell death involving ER stress. Thus, GCDCA induces ER stress which, in turn, induces apoptotic signalling in a time-dependent manner in isolated rat hepatocytes. This BA caused Ca2+ release from ER, which in turn induced extracellular Ca2+ influx followed by the activation of calpain and caspase-12. In this study it is suggested that ER stress induced by GCDCA may trigger the activation of both ER mediated apoptosis and mitochondria-mediated apoptosis in isolated rat hepatocytes by cross-talk between ER and mitochondria using calcium ions as signal substances[4344]. Recent studies have suggested that ER stress might be involved in hepatocyte cell death caused by cholestasis or BAs[4546]. It has been demonstrated that cholestasis induces ER stress mediated by CHOP, a key component in ER stress mediated apoptosis, and triggers hepatocyte cell death. Moreover, CHOP deficiency attenuates this cell death and subsequent liver fibrosis. The results demonstrate an essential role of CHOP in development of liver fibrosis due to cholestatic liver damage[46].
BILE-ACID-INDUCED DAMAGE IN THE PLASMA MEMBRANE
In hepatocytes, high concentrations of BAs can cause damage to the basolateral membrane, cell organelle membranes, and because they are more concentrated in bile, these compounds might be particularly harmful to the outside layer of the canalicular membrane. Hydrophobic BA-induced structural and functional injury of hepatocyte membranes plays an important role in the pathogenesis of cholestatic liver diseases in humans[47]. Conjugated-BAs are more hydrophilic and not usually cytotoxic until their concentrations approach their critical micellar concentration[48]. The effect of BAs on cell membranes is induced by binding to the membrane components[49]. The authors carried out experiments using large unilamellar vesicles and showed that at very low BA concentrations, BA/lipid aggregates are formed in the outer membrane monolayer, with a size and BA-binding strength that depended on the species of BA and lipids involved. As BA concentrations increased, binding to membranes also rose until a certain threshold was reached and the BA-membrane interaction then resulted in the formation of transient membrane holes[49], which caused the disruption of plasma membrane integrity and subsequently lysis of hepatocytes[1]. The efficiency of BAs to solubilise membrane lipids, such as phospholipids, cholesterol or fatty acids, is generally enhanced with increasing BA hydrophobicity[113]. CDCA and DCA have critical micellar concentrations lower than that of CA; therefore at any given concentration they are more cytotoxic[48]. In the hepatocyte, when the concentration of hydrophobic BAs exceeds the binding capacity of the cytosolic proteins, these compounds possibly damage organelle membranes, especially in the case of mitochondria, which leads to mitochondrial damage and ultimately to apoptosis or necrosis[48].
The amount of BAs present in bile as monomers depends on their degree of association with phospholipids and cholesterol to form mixed micelles. Such mixed micelles are formed at concentration values well below cytotoxic levels, which accounts for the lack of BA-induced injury in the biliary system and small intestine under physiological circumstances. In _Mdr2_-knockout mice and patients lacking the canalicular phosphatidylcholine transporter MDR3 (Mdr2 in rodents), phospholipid secretion is markedly impaired, leading to an increase in the concentration of monomeric BAs, which causes damage to biliary epithelial cells[48].
BILE-ACID-INDUCED INJURY IN OTHER CELL TYPES
Biliary cells
In addition to the cytoprotective role of biliary phospholipids against the detergent activity of BAs, other mechanisms prevent cell damage, necrosis and apoptosis in the biliary epithelium even through this is exposed to high concentrations of these compounds. In immortalized mouse cholangiocytes it has been demonstrated that hydrophobic BAs cause apoptosis when they are taken up and accumulate inside the cell. This is, in part, inhibited by the activity of Mrp3, an export pump able to reduce intracellular BA concentrations[50]. However, when the accumulation of hydrophobic BAs occurs in chronic or subchronic cholestasis, this triggers cholangiocyte proliferation[51]. This stimulation capacity has been observed both in normal cholangiocytes and in cholangiocarcinoma cells[5253]. Hydrophobic BA-induced cholangiocellular proliferation occurs by transactivation of EGFR[3754], which stimulates a phosphatidylinositol 3-kinase-dependent pathway[55].
Gastrointestinal cells
In humans, an increased incidence of cancer of the laryngopharyngeal tract, esophagus, stomach, pancreas, small intestine and colon is associated with high levels of hydrophobic BAs[34]. DCA has been proposed to be a cancer promoter in these organs[56]. These BAs cause DNA damage in several human colon cancer cell lines[5758], probably through an indirect mechanism involving the induction of oxidative stress and the production of ROS. Repeated DNA damage is likely to increase the mutation rate in several genes, including those coding for tumour suppressor factors and oncogenes. Moreover, hydrophobic BAs may behave as selection agents for apoptosis-resistant cells. The sub-population of cells with a reduced apoptosis capability may be selected to survive and proliferate versus the more fragile normal cells. Indeed, long-term exposure of a human colonic epithelial cell line to sublethal concentrations of DCA has been reported to result in the selection of a population of cells partially resistant to DCA-induced apoptosis[59].
Kidney and lung cells
Regarding extrahepatic tissues, BA accumulation in the systemic circulation may contribute to endothelial injury in the kidney and lungs[60]. Hydrophobic BAs cause oxidative damage to tubular cell membranes by stimulating the generation of ROS from mitochondria, as well as promoting their release from neutrophils and macrophages. Oxidative stress can promote the formation of a variety of vasoactive mediators including endothelin-1, cysteinyl leukotrienes, F2-isoprostanes and endogenous products of lipid peroxidation. These mediators can affect renal function directly by causing renal vasoconstriction or by decreasing the coefficient of glomerular capillary ultrafiltration, and thus reducing the glomerular filtration rate. Collectively, these factors contribute to the onset of renal failure in patients with biliary obstruction[61]. Disturbances in the biochemical functions of the kidney glomeruli and tubules have been reported in patients with intrahepatic cholestasis during pregnancy[62].
BAs may reach the lung accidentally by aspiration of gastrointestinal contents or by direct uptake from blood. Bile aspiration produces a severe chemical pneumonitis in a porcine lung model[63] and intratracheally injected BAs have been shown to produce severe pulmonary edema in rabbits[64]. When intratracheal instillation of taurocholic acid (TCA, Figure 1) in rabbits was studied, microscopic evidence of widespread atelectasis, the pooling of eosinophilic substances in the intra-alveolar spaces, and the formation of hyaline membranes were found. The authors speculated that surfactant activity might be inhibited by BAs[65].
Placental and fetal cells
The accumulation of BAs may also have deleterious effects on the fetal-placental unit. The excretion of BAs produced by the fetus is performed by the placenta and the maternal liver, therefore impairment in biliary excretion may lead to the accumulation of BAs, in particular in the fetal liver-placenta-maternal liver trio[66]. An interesting issue that has recently been addressed is how this accumulation of BAs can affect fetal and placental tissues. In cholestatic pregnant rats, hydrophobic BA accumulation induces impairment of the placental antioxidant system and oxidative damage. These alterations are accompanied by enhanced activation of the mitochondrial pathway of apoptosis[67]. Moreover, the accumulation of hydrophobic BAs in the fetal compartment also causes marked oxidative damage and apoptosis in the fetal liver[68]. This is evidenced by enhanced lipid peroxidation and protein carbonylation, a pro-apoptotic imbalance in the Bax-α/Bcl-2 ratio, caspase-3 activation, and DNA fragmentation. Different sensitivities to BAs have been reported in fetal and maternal hepatocytes in short-term primary culture[69]. Although the basal production of ROS by fetal hepatocytes has been found to be higher than by maternal hepatocytes, ROS production is higher in maternal hepatocytes in response to exposure to relatively high concentrations of GCDCA[69].
BA accumulation can cause other complications in extrahepatic fetal tissues. TCA (Figure 1) impairs neonatal rat cardiomyocyte function by altering calcium dynamics and impairing the function of gap junctions in these cells, resulting in dysrhythmia. This has been proposed as a mechanism for intrauterine fetal death in obstetric cholestasis[70]. Recently, it has been reported that BAs can induce lung injury in newborn infants because these compounds are detectable in the bronchoalveolar lavage fluid of newborns from mothers with intrahepatic cholestasis during pregnancy affected by respiratory distress syndrome. Elevated serum BA levels in these infants can reach the lung after uptake from the circulation. These findings support the concept of a role of BA in the etiopathogenesis of some types of pneumonia[71].
BILE-ACID-INDUCED CELL PROTECTION
UDCA: protective effects and mechanisms of action
UDCA (Figure 1) and its taurine-conjugated derivative, tauroursodeoxycholic acid (TUDCA) are hydrophilic BAs that have become highly popular owing to their low toxicity and efficiency in the treatment of several cholestatic liver diseases, such as cholelithiasis, primary biliary cirrhosis, primary sclerosing cholangitis, cystic fibrosis and intrahepatic cholestasis of pregnancy[23]. These BAs may be also useful to protect organs other than the liver, such as the brain[72] or placenta[6773–75].
UDCA is a major primary BA in some species of bears[76]. In fact, for centuries dried bear bile has been used in traditional Chinese medicine as a remedy for liver disorders[76]. In humans, UDCA is considered as a minor secondary BA because it is formed by 7β-epimerization of CDCA in the gut by intestinal bacteria. The abundance of UDCA in the total BA pool is less than 3%[77]. Despite its clinical efficacy, the precise mechanism by which UDCA improves liver function during cholestasis is still under study. It is considered that the choleretic effect of UDCA, together with its ability to cause a marked shift in the composition of the BA pool towards hydrophilicity, accounts for the beneficial properties of UDCA in the treatment of liver disorders[278]. Moreover, it has recently become evident that UDCA and TUDCA are capable of exerting direct protective effects at the cellular and molecular level, including the stabilization of hepatocyte membranes, the enhancement of defences against oxidative stress and the inhibition of apoptosis induced by several agents[2]. Other proposed mechanisms of action for UDCA, such as the immunomodulation and stimulation of bile secretion by hepatocytes and bile duct epithelial cells, may also contribute to the cytoprotective effects of this hydrophilic BA[78]. Nevertheless, the predominant mechanism of action of UDCA may vary, depending on the pathophysiology of the underlying liver disease.
Protection against oxidative stress: UDCA has shown to play an important role in the prevention of the oxidative injury induced by several agents, either through a direct antioxidant effect or an increase in antioxidant defences. The enhancement of glutathione levels, which has been demonstrated using isolated rat hepatocytes[79] and rats with bile-duct-ligation-induced secondary biliary cirrhosis[80], can be included among the beneficial effects of UDCA treatment, and may be due to a higher expression of the enzymes involved in glutathione synthesis. In this sense, an enhancement of γ-glutamylcysteine synthetase at the transcriptional level has been found in isolated rat hepatocytes treated with UDCA, which allows these cells to be more resistant to cadmium- or hydrogen-peroxide-induced oxidative injury[79]. UDCA treatment of chronic bile-duct-ligated rats also leads to an up-regulation of γ-glutamylcysteine synthetase and prevents the marked increase in the production of mitochondrial peroxide and of hydroxynonenal-protein adducts observed during chronic cholestasis[80]. The activity of methionine S-adenosyltransferase, another enzyme involved in glutathione (GSH) biosynthesis, has also been found to be increased by UDCA in rat livers[81].
Pre-treatment of hepatocytes with UDCA also increases the amount of thiol-containing proteins such as metallothioneins, which efficiently scavenge hydroxyl radicals (OH•), the most highly reactive ROS[79]. In human hepatoblastoma HepG2 cells, UDCA has been shown to be able to activate the metallothionein IIA promoter[82].
During rat development, there is an excessive mitochondrial response to pro-oxidant stimuli, together with less well developed antioxidant protection mechanisms. This may account for the particularly high sensitivity of the foetal liver to lipid peroxidation[83]. In rats, obstructive cholestasis during pregnancy (OCP) increases the degree of lipid peroxidation and protein carbonylation in placenta[67] and in foetal liver[68]. Treatment of rats with UDCA during pregnancy prevents oxidative injury in the placenta[67] and foetal liver[68]. The drop in the activities of the enzymes involved in the mechanisms of resistance against oxidative stress, such as catalase, glutathione peroxidase and glutathione-S-transferase[67], increases GSH content and the GSH/glutathione disulfide ratio. The impairment in liver structure and function in 4-wk-old pups born from rats with OCP is also partially prevented if the mothers are treated with UDCA[75].
Hydrophobic BAs stimulate Kupffer cells, increasing their capacity to generate ROS, which in turn attack nucleic acids, thiol proteins or membrane lipids, causing lipid peroxidation. UDCA can block hydrophobic-BA-induced cellular phenomena, therefore, it could also antagonise macrophage activation by hydrophobic BAs to blunt their capacity to generate ROS[15].
UDCA has direct antioxidant properties, which are evident at therapeutically relevant drug concentrations and are especially relevant against Fe3+- and OH•-induced oxidative damage[84]. The OH•-scavenging efficiency of UDCA appears remarkable, considering that its rate constant for reactions with this radical species is about 10-fold higher than that of the typical pharmacological scavenger (mannitol) and of the physiological scavengers glucose and histidine[84]. Thus, together with the high therapeutic concentrations of UDCA reached in human bile, the drug could readily act as an effective OH• scavenger, especially in the biliary milieu[84]. It is therefore possible that UDCA could act not only as a site-specific OH• scavenger, but also as an antioxidant against iron (IV)-induced oxidative damage.
Inhibition of apoptosis: UDCA and TUDCA have been shown to prevent in vitro apoptosis induced by several agents in both hepatic and non-hepatic cells[27]. In rats with OCP, UDCA also prevents apoptosis in the placenta[67] and fetal liver[68]. In patients with primary biliary cirrhosis treated with UDCA, this drug shows a potential effect in reducing nuclear DNA fragmentation in biliary epithelial cells[85]. It has been suggested that UDCA could function as a therapeutic agent in the treatment of neurodegenerative disorders associated with increased levels of apoptosis[72]. Indeed, UDCA is neuroprotective in pharmacological and transgenic animal models of Huntington’s disease[86]; it inhibits unconjugated bilirubin-induced apoptosis in both glial and neuronal rat cells in culture[87], improves graft survival in Parkinsonian rats[86], and protects against neurological injury after acute ischemic and hemorrhagic stroke[86]. The ability of UDCA to prevent apoptosis induced by a wide variety of compounds, such as hydrophobic BAs, ethanol, transforming growth factor beta 1 (TGFβ1), Fas ligand, and okadaic acid, suggests a mechanism that is common to each of the different apoptotic pathways[27]. A possible explanation for this ubiquitous antiapoptotic effect of UDCA seems to involve a blockage of mitochondrial dysfunction, mitochondria having a role as integrators of apoptosis signalling pathways[2430]. UDCA is involved in both short- and long-term mechanisms in the prevention of the MPT responsible for hepatocyte cell death[2430]. UDCA is able to reduce BA-induced disruption of the mitochondrial membrane potential, ROS production, and Bax protein abundance in mitochondria[30]. UDCA treatment also prevents the marked decrease in cardiolipin levels, which modulate apoptotic processes by inhibiting the MPT of damaged and primarily apoptotic hepatocytes during biliary cirrhosis induced by chronic cholestasis in rats[88]. Additionally, UDCA prevents the release of cytochrome c from mitochondria to the cytoplasm after mitochondrial injury and the subsequent cytosolic caspase activation and cleavage of the nuclear enzyme poly(ADP-ribose) polymerase[31].
Nevertheless, the mechanisms by which UDCA prevents cell death involve molecular targets other than mitochondria, such as the ER. In an ER stress model in the liver-derived cell line Huh7, it has been shown that stress in this organelle activates caspase-12 and triggers apoptosis without the involvement of mitochondria[89]. TUDCA is able to abolish the typical morphological changes of ER stress preceding apoptosis, including activation of caspase-3 and -7, DNA fragmentation, and cleavage of poly(ADP-ribose) polymerase. TUDCA also blocks one of the calcium-mediated apoptotic pathways by a reduction in calcium efflux and the inhibition of caspase-12 activation[89].
Apoptosis can be inhibited, not only by blocking pro-apoptotic pathways, but also by promoting survival signals, including the cAMP, Akt, nuclear factor kappaB (NF-κB), mitogen-activated protein kinases (MAPK), and phosphatidylinositol 3-kinase (PI3K) signalling pathways[9091]. The antiapoptotic effect of TUDCA against hydrophobic BA-induced apoptosis in rat hepatocytes is independent of caspase-8 inhibition, but results from the activation of the p38 MAPK, extracellular signal regulated-kinase (ERK) MAPK, and PI3K survival pathways[91]. UDCA stimulates the activation of the intracellular MAPK pathway through the activation of the epidermal growth factor receptor (EGFR)[90]. TGFβ1-induced hepatocyte apoptosis is associated with the activation of E2F transcription factors and p53 stabilization through its inhibitor Mdm-2. UDCA interferes in the E2F-1 transcriptional activation of apoptosis, thus modulating p53 stabilization, NF-κB activation, and the expression of Bcl-2 family members[92], through a mechanism that appears to involve nuclear glucocorticoid and mineralocorticoid receptors[93].
Stimulation of bile flow and detoxification of cholephilic compounds: The impairment of bile formation in cholestasis results in the accumulation of potentially toxic BAs and other biliary constituents in the liver, which can result in necrosis, apoptosis, fibrosis, and ultimately liver cirrhosis[94]. UDCA stimulates the biliary excretion of BAs and other cholephilic organic anions, even in situations of impaired bile secretion, such as primary biliary cirrhosis and primary sclerosing cholangitis, two disorders in which the effects of UDCA have mostly been studied[3]. In patients with these diseases, UDCA treatment decreases the serum levels of bilirubin and the accumulation of hydrophobic BAs[95], probably due to stimulation of the expression of the export pumps involved in the detoxification process of cholephilic organic compounds by hepatocytes[96], which are down-regulated in cholestatic liver diseases[9496].
Transient latent cholestasis in young rats born from mothers with OCP has been reported[97]. UDCA treatment of rats with OCP has long-term beneficial effects on their offspring by partially preventing the congenital impairment in hepatobiliary function of the pups that affects their biliary lipid secretion[75]. UDCA also has a beneficial effect on BA transport mechanisms in placentas from patients with intrahepatic cholestasis of pregnancy[73] and rats with OCP[74]. UDCA partially prevents OCP-induced impairment in the placenta-maternal liver tandem excretory pathway, by preserving trophoblast structure and function[74].
In rodents fed for several weeks with diets supplemented with CA, CDCA and LCA, a down-regulation of the expression of xenobiotic-metabolizing enzymes, such as hepatic glutathione S-transferase in mice[98] and intestinal UDP-glucuronyltransferase in rats[99], has been found. The prevention by UDCA of the decrease in the activity of phase-II-metabolizing enzymes by hydrophobic BAs helps to maintain detoxification processes of cholephilic compounds[9899].
The secretory capacity of hepatocytes is mainly determined by the number and activity of carrier proteins in the apical membrane. In mouse liver, UDCA stimulates the gene expression of both the canalicular bile salt export pump (Bsep) and the canalicular multidrug resistance associated protein 2 (Mrp2)[100]. UDCA also stimulates the alternative basolateral ATP-binding cassette proteins Mrp3[101] and Mrp4[102], which represent a compensatory overflow system under cholestatic conditions when the function of canalicular export pumps is impaired. UDCA also stimulates murine renal (Mrp2 and Mrp4) and intestinal (Mrp2 and Mrp3) efflux transport proteins, resulting in an increased overall elimination capacity for potentially toxic biliary compounds[101102].
Besides the long-term effects of UDCA on the regulation of gene transcription, post-transcriptional events such as increased targeting of transport proteins to the canalicular membrane via stimulation of vesicular exocytosis, can be accounted for by the increased expression of hepatobiliary transporters under treatment with UDCA or its conjugated derivatives[78]. Calcium-[103], protein kinase C (PKC)-[104] and MAPK-dependent mechanisms[105] mediate the enhancement of the secretory capacity of cholestatic hepatocytes by TUDCA. Indeed, TUDCA favors the insertion of rat Mrp2 into apical membranes via PKC-dependent mechanisms[104]. Furthermore, p38 MAPK signalling pathways are involved in the enhanced insertion of both rat Bsep[105] and human BSEP[106] into the canalicular membrane of hepatocytes by TUDCA. Since the Golgi apparatus may serve as a BSEP pool and since p38 MAPK regulates BSEP trafficking from the Golgi apparatus to the plasma membrane, the activation of p38 MAPK by TUDCA can recruit Golgi-associated BSEP and insert it into the canalicular membrane[106].
Besides the up-regulation of synthesis, apical targeting and insertion, direct activation of key canalicular transporters through modification of their phosphorylation status may contribute to the anticholestatic action of UDCA. This has been shown for mouse Bsep, whose transport capacity is increased by TUDCA via PKCα-mediated phosphorylation[107].
Protection of cholangiocytes against toxic effects of hydrophobic BAs: The enrichment of bile with UDCA contributes to the prevention of toxic effects of more hydrophobic BAs, because this compound renders bile more hydrophilic, modifying the structure and composition of mixed phospholipid-rich micelles[108]. In addition, UDCA and its conjugates exert a direct effect on membranes by stabilizing their structure. This effect appears to be due to the incorporation of UDCA into the non-polar domain of the lipid bilayer and of its conjugates, into the interface[109].
An experimental model in which the in vivo effects of UDCA on the protection of cholangiocytes have been studied is the _Mdr2_-knockout mouse. These animals lack the ability to secrete phospholipids into bile and develop a chronic cholangitis resembling human chronic cholestatic liver disease[110]. The feeding of mice with UDCA or TUDCA decreases the degree of cholangiocellular injury, portal inflammation, and ductular proliferation[110]. 24-norursodeoxycholic acid seems be superior to UDCA in the treatment of cholangitis in _Mdr2_-knockout mice[111]. Similar beneficial effects of UDCA have been found in bile-duct-ligated rats[112]. Likewise, in patients with primary cholestatic liver diseases under treatment with UDCA, the inflammatory reaction around bile ducts has been reported to be less severe[113].
In addition to stabilizing cell membranes, other molecular mechanisms responsible for the protective effects of UDCA on cholangiocytes have been described recently. BAs are taken up by cholangiocytes mainly via the apical sodium-dependent BA transporter (ASBT). The accumulation of BAs in chronic cholestasis triggers cholangiocyte proliferation and secretion through a PI3K-dependent pathway[55]. UDCA and TUDCA reduce ASBT expression in cholangiocytes isolated from bile-duct-ligated rats, and they inhibit cellular growth and secretion[114] through Ca2+- and PKCα-dependent mechanisms[115]. TUDCA also inhibits the growth of the cholangiocarcinoma cell line Mz-ChA-1 through a signal-transduction pathway involving MAPK p42/44 and PKCα, which suggests that this compound may be a candidate for the treatment of cholangiocarcinoma[116].
Immunomodulation: The immunomodulatory mechanism of action of UDCA accounts for the beneficial properties of treatment with this BA against several autoimmune liver diseases, such as primary biliary cirrhosis and chronic viral hepatitis. Among the in vitro experimental evidence of the immunomodulatory activity of UDCA reported is its ability to decrease the secretion of interleukins 2 and 4, TNF-α and interferon-γfrom activated T lymphocytes, and immunoglobulin production from B lymphocytes[117]. The suppression of cytokine and immunoglobulin production and T-cell-mediated cytotoxicity in mice by the treatment not only with UDCA[118] but also with CDCA[119] has been observed. In patients with primary biliary cirrhosis, the administration of UDCA down-regulates the expression of abnormal major histocompatibility complex (MHC) class I molecules in periportal hepatocytes, whereas the expression of abnormal MHC class II molecules in bile-duct epithelial cells does not change[120]. However, UDCA suppresses the interferon-γ-mediated induction of MHC class II gene expression via the glucocorticoid-receptor-mediated pathway[121].
Protective effects of other bile acids
Other natural BA molecules or their derivatives have also aroused pharmacological interest owing to their protective properties. Besides the efficiency of CDCA in cholesterol cholecystolithiasis[122] and cerebrotendinous xanthomatosis[48123], BA replacement therapy with a mixture of this BA together with CA is used in the treatment of inborn errors of BA biosynthesis involving the A ring[48]. This treatment suppresses the synthesis of cytotoxic BA precursors and restores the input of primary BAs into the enterohepatic circulation[48]. Cholyl-N-methylglycine or cholylsarcosine (Figure 1) is a synthetic BA analog that has been shown to prevent the severe fat malabsorption seen in patients with short-bowel syndrome due to a BA deficiency, which leads to impaired micellar solubilization in the proximal intestine[124].
CONCLUSION
Rapid advances in the understanding of the cellular and molecular pathophysiology of BAs have led to a better knowledge of hepatocyte injury caused by the retention of hydrophobic BAs in cholestatic diseases. These BAs can damage cell membranes and promote the generation of ROS, and eventually cause necrosis and apoptosis. Hydrophobic BAs can also trigger hepatocyte apoptosis by the activation of death receptors, through the mitochondrial pathways, and by the induction of ER stress. The accumulation of hydrophobic BAs in the systemic circulation may have also deleterious effects on extrahepatic tissues such as kidney, lung, placenta, and foetal cells.
UDCA is a hydrophilic BA useful for the treatment of cholestatic liver diseases by its ability to modulate hydrophobic-BA-induced injury in hepatocytes. Underlying mechanisms of their beneficial effects are only now being clarified, and include protection against cytotoxicity due to more toxic BAs, stimulation of bile secretion, immunomodulation, protection against oxidative stress, and the inhibition of apoptosis. Other natural BAs or their derivatives, such as CA, CDCA and cholylsarcosine, have also aroused pharmacological interest owing to their protective properties in several diseases.
Supported by Instituto de Salud Carlos III, FIS, Spain (Grants PI070517 and PI080151); Fundacion Investigacion Medica Mutua Madrileña, Spain (Conv-III, 2006); Junta de Castilla y Leon, Spain (Grants GR75-2008, SA033A08, SA03508 and SA03608); and Ministerio de Ciencia y Tecnologia, Plan Nacional de Investigacion Cientifica, Desarrollo e Innovacion Tecnologica, Spain (Grant BFU2006-12577). The group is member of the Network for Cooperative Research on Membrane Transport Proteins (REIT), co-funded by the Ministerio de Educacion y Ciencia, Spain, and the European Regional Development Fund (ERDF) (Grant BFU2007-30688-E/BFI); and belongs to the CIBERehd (Centro de Investigacion Biomedica en Red para el Estudio de Enfermedades Hepaticas y Digestivas), Instituto de Salud Carlos III
1.
Attili AF, Angelico M, Cantafora A, Alvaro D, Capocaccia L. Bile acid-induced liver toxicity: relation to the hydrophobic-hydrophilic balance of bile acids. Med Hypotheses. 1986;19:57-69. [PubMed] [DOI] [Cited in This Article: ]
2.
Paumgartner G, Beuers U. Ursodeoxycholic acid in cholestatic liver disease: mechanisms of action and therapeutic use revisited. Hepatology. 2002;36:525-531. [PubMed] [DOI] [Cited in This Article: ]
3.
Pusl T, Beuers U. Ursodeoxycholic acid treatment of vanishing bile duct syndromes. World J Gastroenterol. 2006;12:3487-3495. [PubMed] [DOI] [Cited in This Article: ]
4.
Thomas C, Pellicciari R, Pruzanski M, Auwerx J, Schoonjans K. Targeting bile-acid signalling for metabolic diseases. Nat Rev Drug Discov. 2008;7:678-693. [PubMed] [DOI] [Cited in This Article: ]
6.
Spivey JR, Bronk SF, Gores GJ. Glycochenodeoxycholate-induced lethal hepatocellular injury in rat hepatocytes. Role of ATP depletion and cytosolic free calcium. J Clin Invest. 1993;92:17-24. [PubMed] [DOI] [Cited in This Article: ]
7.
Galle PR, Theilmann L, Raedsch R, Otto G, Stiehl A. Ursodeoxycholate reduces hepatotoxicity of bile salts in primary human hepatocytes. Hepatology. 1990;12:486-491. [PubMed] [DOI] [Cited in This Article: ]
8.
Sokol RJ, McKim JM Jr, Goff MC, Ruyle SZ, Devereaux MW, Han D, Packer L, Everson G. Vitamin E reduces oxidant injury to mitochondria and the hepatotoxicity of taurochenodeoxycholic acid in the rat. Gastroenterology. 1998;114:164-174. [PubMed] [DOI] [Cited in This Article: ]
9.
Kountouras J, Billing BH, Scheuer PJ. Prolonged bile duct obstruction: a new experimental model for cirrhosis in the rat. Br J Exp Pathol. 1984;65:305-311. [PubMed] [DOI] [Cited in This Article: ]
12.
Maher JJ, Friedman SL. Parenchymal and nonparenchymal cell interactions in the liver. Semin Liver Dis. 1993;13:13-20. [PubMed] [DOI] [Cited in This Article: ]
13.
Billington D, Evans CE, Godfrey PP, Coleman R. Effects of bile salts on the plasma membranes of isolated rat hepatocytes. Biochem J. 1980;188:321-327. [PubMed] [DOI] [Cited in This Article: ]
14.
Sokol RJ, Straka MS, Dahl R, Devereaux MW, Yerushalmi B, Gumpricht E, Elkins N, Everson G. Role of oxidant stress in the permeability transition induced in rat hepatic mitochondria by hydrophobic bile acids. Pediatr Res. 2001;49:519-531. [PubMed] [DOI] [Cited in This Article: ]
15.
Ljubuncic P, Fuhrman B, Oiknine J, Aviram M, Bomzon A. Effect of deoxycholic acid and ursodeoxycholic acid on lipid peroxidation in cultured macrophages. Gut. 1996;39:475-478. [PubMed] [DOI] [Cited in This Article: ]
16.
Sokol RJ, Devereaux M, Khandwala RA. Effect of dietary lipid and vitamin E on mitochondrial lipid peroxidation and hepatic injury in the bile duct-ligated rat. J Lipid Res. 1991;32:1349-1357. [PubMed] [DOI] [Cited in This Article: ]
17.
Togashi H, Shinzawa H, Wakabayashi H, Nakamura T, Yamada N, Takahashi T, Ishikawa M. Activities of free oxygen radical scavenger enzymes in human liver. J Hepatol. 1990;11:200-205. [PubMed] [DOI] [Cited in This Article: ]
18.
Sokol RJ, Devereaux M, Khandwala R, O'Brien K. Evidence for involvement of oxygen free radicals in bile acid toxicity to isolated rat hepatocytes. Hepatology. 1993;17:869-881. [PubMed] [DOI] [Cited in This Article: ]
19.
Krähenbühl S, Talos C, Fischer S, Reichen J. Toxicity of bile acids on the electron transport chain of isolated rat liver mitochondria. Hepatology. 1994;19:471-479. [PubMed] [DOI] [Cited in This Article: ]
20.
Rolo AP, Oliveira PJ, Moreno AJ, Palmeira CM. Bile acids affect liver mitochondrial bioenergetics: possible relevance for cholestasis therapy. Toxicol Sci. 2000;57:177-185. [PubMed] [DOI] [Cited in This Article: ]
21.
Sokol RJ, Winklhofer-Roob BM, Devereaux MW, McKim JM Jr. Generation of hydroperoxides in isolated rat hepatocytes and hepatic mitochondria exposed to hydrophobic bile acids. Gastroenterology. 1995;109:1249-1256. [PubMed] [DOI] [Cited in This Article: ]
22.
Sokol RJ, Dahl R, Devereaux MW, Yerushalmi B, Kobak GE, Gumpricht E. Human hepatic mitochondria generate reactive oxygen species and undergo the permeability transition in response to hydrophobic bile acids. J Pediatr Gastroenterol Nutr. 2005;41:235-243. [PubMed] [DOI] [Cited in This Article: ]
23.
Krähenbühl S, Talos C, Lauterburg BH, Reichen J. Reduced antioxidative capacity in liver mitochondria from bile duct ligated rats. Hepatology. 1995;22:607-612. [PubMed] [DOI] [Cited in This Article: ]
24.
Botla R, Spivey JR, Aguilar H, Bronk SF, Gores GJ. Ursodeoxycholate (UDCA) inhibits the mitochondrial membrane permeability transition induced by glycochenodeoxycholate: a mechanism of UDCA cytoprotection. J Pharmacol Exp Ther. 1995;272:930-938. [PubMed] [DOI] [Cited in This Article: ]
25.
Yerushalmi B, Dahl R, Devereaux MW, Gumpricht E, Sokol RJ. Bile acid-induced rat hepatocyte apoptosis is inhibited by antioxidants and blockers of the mitochondrial permeability transition. Hepatology. 2001;33:616-626. [PubMed] [DOI] [Cited in This Article: ]
26.
Lemasters JJ, Nieminen AL, Qian T, Trost LC, Elmore SP, Nishimura Y, Crowe RA, Cascio WE, Bradham CA, Brenner DA. The mitochondrial permeability transition in cell death: a common mechanism in necrosis, apoptosis and autophagy. Biochim Biophys Acta. 1998;1366:177-196. [PubMed] [DOI] [Cited in This Article: ]
27.
Rodrigues CM, Fan G, Wong PY, Kren BT, Steer CJ. Ursodeoxycholic acid may inhibit deoxycholic acid-induced apoptosis by modulating mitochondrial transmembrane potential and reactive oxygen species production. Mol Med. 1998;4:165-178. [PubMed] [DOI] [Cited in This Article: ]
28.
Anwer MS, Engelking LR, Nolan K, Sullivan D, Zimniak P, Lester R. Hepatotoxic bile acids increase cytosolic Ca++ activity of isolated rat hepatocytes. Hepatology. 1988;8:887-891. [PubMed] [DOI] [Cited in This Article: ]
29.
Patel T, Bronk SF, Gores GJ. Increases of intracellular magnesium promote glycodeoxycholate-induced apoptosis in rat hepatocytes. J Clin Invest. 1994;94:2183-2192. [PubMed] [DOI] [Cited in This Article: ]
30.
Rodrigues CM, Fan G, Ma X, Kren BT, Steer CJ. A novel role for ursodeoxycholic acid in inhibiting apoptosis by modulating mitochondrial membrane perturbation. J Clin Invest. 1998;101:2790-2799. [PubMed] [DOI] [Cited in This Article: ]
31.
Rodrigues CM, Ma X, Linehan-Stieers C, Fan G, Kren BT, Steer CJ. Ursodeoxycholic acid prevents cytochrome c release in apoptosis by inhibiting mitochondrial membrane depolarization and channel formation. Cell Death Differ. 1999;6:842-854. [PubMed] [DOI] [Cited in This Article: ]
32.
Guicciardi ME, Gores GJ. Apoptosis: a mechanism of acute and chronic liver injury. Gut. 2005;54:1024-1033. [PubMed] [DOI] [Cited in This Article: ]
33.
Fickert P, Trauner M, Fuchsbichler A, Zollner G, Wagner M, Marschall HU, Zatloukal K, Denk H. Oncosis represents the main type of cell death in mouse models of cholestasis. J Hepatol. 2005;42:378-385. [PubMed] [DOI] [Cited in This Article: ]
34.
Bernstein H, Bernstein C, Payne CM, Dvorakova K, Garewal H. Bile acids as carcinogens in human gastrointestinal cancers. Mutat Res. 2005;589:47-65. [PubMed] [DOI] [Cited in This Article: ]
35.
Faubion WA, Guicciardi ME, Miyoshi H, Bronk SF, Roberts PJ, Svingen PA, Kaufmann SH, Gores GJ. Toxic bile salts induce rodent hepatocyte apoptosis via direct activation of Fas. J Clin Invest. 1999;103:137-145. [PubMed] [DOI] [Cited in This Article: ]
36.
Higuchi H, Bronk SF, Takikawa Y, Werneburg N, Takimoto R, El-Deiry W, Gores GJ. The bile acid glycochenodeoxycholate induces trail-receptor 2/DR5 expression and apoptosis. J Biol Chem. 2001;276:38610-38618. [PubMed] [DOI] [Cited in This Article: ]
37.
Qiao L, Studer E, Leach K, McKinstry R, Gupta S, Decker R, Kukreja R, Valerie K, Nagarkatti P, El Deiry W. Deoxycholic acid (DCA) causes ligand-independent activation of epidermal growth factor receptor (EGFR) and FAS receptor in primary hepatocytes: inhibition of EGFR/mitogen-activated protein kinase-signaling module enhances DCA-induced apoptosis. Mol Biol Cell. 2001;12:2629-2645. [PubMed] [DOI] [Cited in This Article: ]
39.
Reinehr R, Becker S, Eberle A, Grether-Beck S, Häussinger D. Involvement of NADPH oxidase isoforms and Src family kinases in CD95-dependent hepatocyte apoptosis. J Biol Chem. 2005;280:27179-27194. [PubMed] [DOI] [Cited in This Article: ]
40.
Yin XM, Ding WX. Death receptor activation-induced hepatocyte apoptosis and liver injury. Curr Mol Med. 2003;3:491-508. [PubMed] [DOI] [Cited in This Article: ]
41.
Oh SH, Yun KJ, Nan JX, Sohn DH, Lee BH. Changes in expression and immunolocalization of protein associated with toxic bile salts-induced apoptosis in rat hepatocytes. Arch Toxicol. 2003;77:110-115. [PubMed] [DOI] [Cited in This Article: ]
42.
Scaffidi C, Fulda S, Srinivasan A, Friesen C, Li F, Tomaselli KJ, Debatin KM, Krammer PH, Peter ME. Two CD95 (APO-1/Fas) signaling pathways. EMBO J. 1998;17:1675-1687. [PubMed] [DOI] [Cited in This Article: ]
43.
Tsuchiya S, Tsuji M, Morio Y, Oguchi K. Involvement of endoplasmic reticulum in glycochenodeoxycholic acid-induced apoptosis in rat hepatocytes. Toxicol Lett. 2006;166:140-149. [PubMed] [DOI] [Cited in This Article: ]
44.
Iizaka T, Tsuji M, Oyamada H, Morio Y, Oguchi K. Interaction between caspase-8 activation and endoplasmic reticulum stress in glycochenodeoxycholic acid-induced apoptotic HepG2 cells. Toxicology. 2007;241:146-156. [PubMed] [DOI] [Cited in This Article: ]
45.
Mencin A, Seki E, Osawa Y, Kodama Y, De Minicis S, Knowles M, Brenner DA. Alpha-1 antitrypsin Z protein (PiZ) increases hepatic fibrosis in a murine model of cholestasis. Hepatology. 2007;46:1443-1452. [PubMed] [DOI] [Cited in This Article: ]
46.
Tamaki N, Hatano E, Taura K, Tada M, Kodama Y, Nitta T, Iwaisako K, Seo S, Nakajima A, Ikai I. CHOP deficiency attenuates cholestasis-induced liver fibrosis by reduction of hepatocyte injury. Am J Physiol Gastrointest Liver Physiol. 2008;294:G498-G505. [PubMed] [DOI] [Cited in This Article: ]
47.
Greim H, Trülzsch D, Czygan P, Rudick J, Hutterer F, Schaffner F, Popper H. Mechanism of cholestasis. 6. Bile acids in human livers with or without biliary obstruction. Gastroenterology. 1972;63:846-850. [PubMed] [DOI] [Cited in This Article: ]
48.
Hofmann AF. The continuing importance of bile acids in liver and intestinal disease. Arch Intern Med. 1999;159:2647-2658. [PubMed] [DOI] [Cited in This Article: ]
49.
Schubert R, Schmidt KH. Structural changes in vesicle membranes and mixed micelles of various lipid compositions after binding of different bile salts. Biochemistry. 1988;27:8787-8794. [PubMed] [DOI] [Cited in This Article: ]
50.
Komichi D, Tazuma S, Nishioka T, Hyogo H, Une M, Chayama K. Unique inhibition of bile salt-induced apoptosis by lecithins and cytoprotective bile salts in immortalized mouse cholangiocytes. Dig Dis Sci. 2003;48:2315-2322. [PubMed] [DOI] [Cited in This Article: ]
51.
Strazzabosco M, Spirlí C, Okolicsanyi L. Pathophysiology of the intrahepatic biliary epithelium. J Gastroenterol Hepatol. 2000;15:244-253. [PubMed] [DOI] [Cited in This Article: ]
52.
Alpini G, Glaser S, Robertson W, Phinizy JL, Rodgers RE, Caligiuri A, LeSage G. Bile acids stimulate proliferative and secretory events in large but not small cholangiocytes. Am J Physiol. 1997;273:G518-G529. [PubMed] [DOI] [Cited in This Article: ]
53.
Alpini G, Glaser SS, Ueno Y, Rodgers R, Phinizy JL, Francis H, Baiocchi L, Holcomb LA, Caligiuri A, LeSage GD. Bile acid feeding induces cholangiocyte proliferation and secretion: evidence for bile acid-regulated ductal secretion. Gastroenterology. 1999;116:179-186. [PubMed] [DOI] [Cited in This Article: ]
54.
Yoon JH, Higuchi H, Werneburg NW, Kaufmann SH, Gores GJ. Bile acids induce cyclooxygenase-2 expression via the epidermal growth factor receptor in a human cholangiocarcinoma cell line. Gastroenterology. 2002;122:985-993. [PubMed] [DOI] [Cited in This Article: ]
55.
Alpini G, Glaser S, Alvaro D, Ueno Y, Marzioni M, Francis H, Baiocchi L, Stati T, Barbaro B, Phinizy JL. Bile acid depletion and repletion regulate cholangiocyte growth and secretion by a phosphatidylinositol 3-kinase-dependent pathway in rats. Gastroenterology. 2002;123:1226-1237. [PubMed] [DOI] [Cited in This Article: ]
56.
Cook JW, Kennaway EL, Kennaway NM. Production of tumours in mice by deoxycholic acid. Nature. 1940;145:627. [PubMed] [DOI] [Cited in This Article: ]
57.
Payne CM, Crowley C, Washo-Stultz D, Briehl M, Bernstein H, Bernstein C, Beard S, Holubec H, Warneke J. The stress-response proteins poly(ADP-ribose) polymerase and NF-kappaB protect against bile salt-induced apoptosis. Cell Death Differ. 1998;5:623-636. [PubMed] [DOI] [Cited in This Article: ]
58.
Venturi M, Hambly RJ, Glinghammar B, Rafter JJ, Rowland IR. Genotoxic activity in human faecal water and the role of bile acids: a study using the alkaline comet assay. Carcinogenesis. 1997;18:2353-2359. [PubMed] [DOI] [Cited in This Article: ]
59.
Crowley-Weber CL, Payne CM, Gleason-Guzman M, Watts GS, Futscher B, Waltmire CN, Crowley C, Dvorakova K, Bernstein C, Craven M. Development and molecular characterization of HCT-116 cell lines resistant to the tumor promoter and multiple stress-inducer, deoxycholate. Carcinogenesis. 2002;23:2063-2080. [PubMed] [DOI] [Cited in This Article: ]
60.
Hofmann AF. Cholestatic liver disease: pathophysiology and therapeutic options. Liver. 2002;22 Suppl 2:14-19. [PubMed] [DOI] [Cited in This Article: ]
61.
Bomzon A, Holt S, Moore K. Bile acids, oxidative stress, and renal function in biliary obstruction. Semin Nephrol. 1997;17:549-562. [PubMed] [DOI] [Cited in This Article: ]
62.
Smolarczyk R, Wójcicka-Jagodzińska J, Piekarski P, Romejko E, Czajkowski K. The biochemical functions of the renal tubules and glomeruli in the course of intrahepatic cholestasis in pregnancy. Eur J Obstet Gynecol Reprod Biol. 2000;89:35-39. [PubMed] [DOI] [Cited in This Article: ]
63.
Porembka DT, Kier A, Sehlhorst S, Boyce S, Orlowski JP, Davis K Jr. The pathophysiologic changes following bile aspiration in a porcine lung model. Chest. 1993;104:919-924. [PubMed] [DOI] [Cited in This Article: ]
65.
Kaneko T, Sato T, Katsuya H, Miyauchi Y. Surfactant therapy for pulmonary edema due to intratracheally injected bile acid. Crit Care Med. 1990;18:77-83. [PubMed] [DOI] [Cited in This Article: ]
66.
Marín JJ, Macías RI, Briz O, Pérez MJ, Serrano MA. Molecular bases of the excretion of fetal bile acids and pigments through the fetal liver-placenta-maternal liver pathway. Ann Hepatol. 2005;4:70-76. [PubMed] [DOI] [Cited in This Article: ]
67.
Perez MJ, Velasco E, Monte MJ, Gonzalez-Buitrago JM, Marin JJ. Maternal ethanol consumption during pregnancy enhances bile acid-induced oxidative stress and apoptosis in fetal rat liver. Toxicology. 2006;225:183-194. [PubMed] [DOI] [Cited in This Article: ]
68.
Perez MJ, Macias RI, Duran C, Monte MJ, Gonzalez-Buitrago JM, Marin JJ. Oxidative stress and apoptosis in fetal rat liver induced by maternal cholestasis. Protective effect of ursodeoxycholic acid. J Hepatol. 2005;43:324-332. [PubMed] [DOI] [Cited in This Article: ]
69.
Perez MJ, Macias RI, Marin JJ. Maternal cholestasis induces placental oxidative stress and apoptosis. Protective effect of ursodeoxycholic acid. Placenta. 2006;27:34-41. [PubMed] [DOI] [Cited in This Article: ]
70.
Williamson C, Gorelik J, Eaton BM, Lab M, de Swiet M, Korchev Y. The bile acid taurocholate impairs rat cardiomyocyte function: a proposed mechanism for intra-uterine fetal death in obstetric cholestasis. Clin Sci (Lond). 2001;100:363-369. [PubMed] [DOI] [Cited in This Article: ]
71.
Zecca E, De Luca D, Baroni S, Vento G, Tiberi E, Romagnoli C. Bile acid-induced lung injury in newborn infants: a bronchoalveolar lavage fluid study. Pediatrics. 2008;121:e146-e149. [PubMed] [DOI] [Cited in This Article: ]
72.
Keene CD, Rodrigues CM, Eich T, Linehan-Stieers C, Abt A, Kren BT, Steer CJ, Low WC. A bile acid protects against motor and cognitive deficits and reduces striatal degeneration in the 3-nitropropionic acid model of Huntington's disease. Exp Neurol. 2001;171:351-360. [PubMed] [DOI] [Cited in This Article: ]
73.
Serrano MA, Brites D, Larena MG, Monte MJ, Bravo MP, Oliveira N, Marin JJ. Beneficial effect of ursodeoxycholic acid on alterations induced by cholestasis of pregnancy in bile acid transport across the human placenta. J Hepatol. 1998;28:829-839. [PubMed] [DOI] [Cited in This Article: ]
74.
Serrano MA, Macias RI, Vallejo M, Briz O, Bravo A, Pascual MJ, St-Pierre MV, Stieger B, Meier PJ, Marin JJ. Effect of ursodeoxycholic acid on the impairment induced by maternal cholestasis in the rat placenta-maternal liver tandem excretory pathway. J Pharmacol Exp Ther. 2003;305:515-524. [PubMed] [DOI] [Cited in This Article: ]
75.
Macias RI, Serrano MA, Monte MJ, Jimenez S, Hernandez B, Marin JJ. Long-term effect of treating pregnant rats with ursodeoxycholic acid on the congenital impairment of bile secretion induced in the pups by maternal cholestasis. J Pharmacol Exp Ther. 2005;312:751-758. [PubMed] [DOI] [Cited in This Article: ]
76.
Hagey LR, Crombie DL, Espinosa E, Carey MC, Igimi H, Hofmann AF. Ursodeoxycholic acid in the Ursidae: biliary bile acids of bears, pandas, and related carnivores. J Lipid Res. 1993;34:1911-1917. [PubMed] [DOI] [Cited in This Article: ]
78.
Trauner M, Graziadei IW. Review article: mechanisms of action and therapeutic applications of ursodeoxycholic acid in chronic liver diseases. Aliment Pharmacol Ther. 1999;13:979-996. [PubMed] [DOI] [Cited in This Article: ]
79.
Mitsuyoshi H, Nakashima T, Sumida Y, Yoh T, Nakajima Y, Ishikawa H, Inaba K, Sakamoto Y, Okanoue T, Kashima K. Ursodeoxycholic acid protects hepatocytes against oxidative injury via induction of antioxidants. Biochem Biophys Res Commun. 1999;263:537-542. [PubMed] [DOI] [Cited in This Article: ]
80.
Ljubuncic P, Tanne Z, Bomzon A. Ursodeoxycholic acid suppresses extent of lipid peroxidation in diseased liver in experimental cholestatic liver disease. Dig Dis Sci. 2000;45:1921-1928. [PubMed] [DOI] [Cited in This Article: ]
81.
Rodríguez-Ortigosa CM, Cincu RN, Sanz S, Ruiz F, Quiroga J, Prieto J. Effect of ursodeoxycholic acid on methionine adenosyltransferase activity and hepatic glutathione metabolism in rats. Gut. 2002;50:701-706. [PubMed] [DOI] [Cited in This Article: ]
82.
Bernstein C, Payne CM, Bernstein H, Garewal H. Activation of the metallothionein IIA promoter and other key stress response elements by ursodeoxycholate in HepG2 cells: relevance to the cytoprotective function of ursodeoxycholate. Pharmacology. 2002;65:2-9. [PubMed] [DOI] [Cited in This Article: ]
83.
Gonzalez MM, Madrid R, Arahuetes RM. Physiological changes in antioxidant defences in fetal and neonatal rat liver. Reprod Fertil Dev. 1995;7:1375-1380. [PubMed] [DOI] [Cited in This Article: ]
84.
Lapenna D, Ciofani G, Festi D, Neri M, Pierdomenico SD, Giamberardino MA, Cuccurullo F. Antioxidant properties of ursodeoxycholic acid. Biochem Pharmacol. 2002;64:1661-1667. [PubMed] [DOI] [Cited in This Article: ]
85.
Koga H, Sakisaka S, Ohishi M, Sata M, Tanikawa K. Nuclear DNA fragmentation and expression of Bcl-2 in primary biliary cirrhosis. Hepatology. 1997;25:1077-1084. [PubMed] [DOI] [Cited in This Article: ]
86.
Ramalho RM, Viana RJ, Low WC, Steer CJ, Rodrigues CM. Bile acids and apoptosis modulation: an emerging role in experimental Alzheimer's disease. Trends Mol Med. 2008;14:54-62. [PubMed] [DOI] [Cited in This Article: ]
87.
Silva RF, Rodrigues CM, Brites D. Bilirubin-induced apoptosis in cultured rat neural cells is aggravated by chenodeoxycholic acid but prevented by ursodeoxycholic acid. J Hepatol. 2001;34:402-408. [PubMed] [DOI] [Cited in This Article: ]
88.
Serviddio G, Pereda J, Pallardó FV, Carretero J, Borras C, Cutrin J, Vendemiale G, Poli G, Viña J, Sastre J. Ursodeoxycholic acid protects against secondary biliary cirrhosis in rats by preventing mitochondrial oxidative stress. Hepatology. 2004;39:711-720. [PubMed] [DOI] [Cited in This Article: ]
89.
Xie Q, Khaoustov VI, Chung CC, Sohn J, Krishnan B, Lewis DE, Yoffe B. Effect of tauroursodeoxycholic acid on endoplasmic reticulum stress-induced caspase-12 activation. Hepatology. 2002;36:592-601. [PubMed] [DOI] [Cited in This Article: ]
90.
Qiao L, Yacoub A, Studer E, Gupta S, Pei XY, Grant S, Hylemon PB, Dent P. Inhibition of the MAPK and PI3K pathways enhances UDCA-induced apoptosis in primary rodent hepatocytes. Hepatology. 2002;35:779-789. [PubMed] [DOI] [Cited in This Article: ]
91.
Schoemaker MH, Conde de la Rosa L, Buist-Homan M, Vrenken TE, Havinga R, Poelstra K, Haisma HJ, Jansen PL, Moshage H. Tauroursodeoxycholic acid protects rat hepatocytes from bile acid-induced apoptosis via activation of survival pathways. Hepatology. 2004;39:1563-1573. [PubMed] [DOI] [Cited in This Article: ]
92.
Sola S, Ma X, Castro RE, Kren BT, Steer CJ, Rodrigues CM. Ursodeoxycholic acid modulates E2F-1 and p53 expression through a caspase-independent mechanism in transforming growth factor beta1-induced apoptosis of rat hepatocytes. J Biol Chem. 2003;278:48831-48838. [PubMed] [DOI] [Cited in This Article: ]
93.
Solá S, Castro RE, Kren BT, Steer CJ, Rodrigues CM. Modulation of nuclear steroid receptors by ursodeoxycholic acid inhibits TGF-beta1-induced E2F-1/p53-mediated apoptosis of rat hepatocytes. Biochemistry. 2004;43:8429-8438. [PubMed] [DOI] [Cited in This Article: ]
94.
Trauner M, Meier PJ, Boyer JL. Molecular pathogenesis of cholestasis. N Engl J Med. 1998;339:1217-1227. [PubMed] [DOI] [Cited in This Article: ]
95.
Combes B, Carithers RL Jr, Maddrey WC, Lin D, McDonald MF, Wheeler DE, Eigenbrodt EH, Muñoz SJ, Rubin R, Garcia-Tsao G. A randomized, double-blind, placebo-controlled trial of ursodeoxycholic acid in primary biliary cirrhosis. Hepatology. 1995;22:759-766. [PubMed] [DOI] [Cited in This Article: ]
96.
Jazrawi RP, de Caestecker JS, Goggin PM, Britten AJ, Joseph AE, Maxwell JD, Northfield TC. Kinetics of hepatic bile acid handling in cholestatic liver disease: effect of ursodeoxycholic acid. Gastroenterology. 1994;106:134-142. [PubMed] [DOI] [Cited in This Article: ]
97.
Monte MJ, Morales AI, Arevalo M, Alvaro I, Macias RI, Marin JJ. Reversible impairment of neonatal hepatobiliary function by maternal cholestasis. Hepatology. 1996;23:1208-1217. [PubMed] [DOI] [Cited in This Article: ]
98.
Kitani K, Kanai S, Sato Y, Ohta M, Nokubo M. Ursodeoxycholic acid reduces the systemic toxicity of 1,2-dichloro,4-nitrobenzene by stimulating hepatic glutathione S-transferase in mice. Life Sci. 1994;54:983-989. [PubMed] [DOI] [Cited in This Article: ]
99.
Baijal PK, Fitzpatrick DW, Bird RP. Modulation of colonic xenobiotic metabolizing enzymes by feeding bile acids: comparative effects of cholic, deoxycholic, lithocholic and ursodeoxycholic acids. Food Chem Toxicol. 1998;36:601-607. [PubMed] [DOI] [Cited in This Article: ]
100.
Fickert P, Zollner G, Fuchsbichler A, Stumptner C, Pojer C, Zenz R, Lammert F, Stieger B, Meier PJ, Zatloukal K. Effects of ursodeoxycholic and cholic acid feeding on hepatocellular transporter expression in mouse liver. Gastroenterology. 2001;121:170-183. [PubMed] [DOI] [Cited in This Article: ]
101.
Zollner G, Fickert P, Fuchsbichler A, Silbert D, Wagner M, Arbeiter S, Gonzalez FJ, Marschall HU, Zatloukal K, Denk H. Role of nuclear bile acid receptor, FXR, in adaptive ABC transporter regulation by cholic and ursodeoxycholic acid in mouse liver, kidney and intestine. J Hepatol. 2003;39:480-488. [PubMed] [DOI] [Cited in This Article: ]
102.
Zollner G, Wagner M, Moustafa T, Fickert P, Silbert D, Gumhold J, Fuchsbichler A, Halilbasic E, Denk H, Marschall HU. Coordinated induction of bile acid detoxification and alternative elimination in mice: role of FXR-regulated organic solute transporter-alpha/beta in the adaptive response to bile acids. Am J Physiol Gastrointest Liver Physiol. 2006;290:G923-G932. [PubMed] [DOI] [Cited in This Article: ]
103.
Beuers U, Nathanson MH, Isales CM, Boyer JL. Tauroursodeoxycholic acid stimulates hepatocellular exocytosis and mobilizes extracellular Ca++ mechanisms defective in cholestasis. J Clin Invest. 1993;92:2984-2993. [PubMed] [DOI] [Cited in This Article: ]
104.
Beuers U, Bilzer M, Chittattu A, Kullak-Ublick GA, Keppler D, Paumgartner G, Dombrowski F. Tauroursodeoxycholic acid inserts the apical conjugate export pump, Mrp2, into canalicular membranes and stimulates organic anion secretion by protein kinase C-dependent mechanisms in cholestatic rat liver. Hepatology. 2001;33:1206-1216. [PubMed] [DOI] [Cited in This Article: ]
105.
Kurz AK, Graf D, Schmitt M, Vom Dahl S, Häussinger D. Tauroursodesoxycholate-induced choleresis involves p38(MAPK) activation and translocation of the bile salt export pump in rats. Gastroenterology. 2001;121:407-419. [PubMed] [DOI] [Cited in This Article: ]
106.
Kubitz R, Sütfels G, Kühlkamp T, Kölling R, Häussinger D. Trafficking of the bile salt export pump from the Golgi to the canalicular membrane is regulated by the p38 MAP kinase. Gastroenterology. 2004;126:541-553. [PubMed] [DOI] [Cited in This Article: ]
107.
Noe J, Hagenbuch B, Meier PJ, St-Pierre MV. Characterization of the mouse bile salt export pump overexpressed in the baculovirus system. Hepatology. 2001;33:1223-1231. [PubMed] [DOI] [Cited in This Article: ]
108.
Heuman DM, Bajaj RS, Lin Q. Adsorption of mixtures of bile salt taurine conjugates to lecithin-cholesterol membranes: implications for bile salt toxicity and cytoprotection. J Lipid Res. 1996;37:562-573. [PubMed] [DOI] [Cited in This Article: ]
109.
Leuschner U, Guldutuna S, Bhatti S, Elze A, Imhof M, You T, Zimmer G. TUDCA and UDCA are incorporated into hepatocyte membranes: different sites, but similar effects. Ital J Gastroenterol. 1995;27:376-377. [PubMed] [DOI] [Cited in This Article: ]
110.
Van Nieuwkerk CM, Elferink RP, Groen AK, Ottenhoff R, Tytgat GN, Dingemans KP, Van Den Bergh Weerman MA, Offerhaus GJ. Effects of Ursodeoxycholate and cholate feeding on liver disease in FVB mice with a disrupted mdr2 P-glycoprotein gene. Gastroenterology. 1996;111:165-171. [PubMed] [DOI] [Cited in This Article: ]
111.
Fickert P, Wagner M, Marschall HU, Fuchsbichler A, Zollner G, Tsybrovskyy O, Zatloukal K, Liu J, Waalkes MP, Cover C. 24-norUrsodeoxycholic acid is superior to ursodeoxycholic acid in the treatment of sclerosing cholangitis in Mdr2 (Abcb4) knockout mice. Gastroenterology. 2006;130:465-481. [PubMed] [DOI] [Cited in This Article: ]
112.
Frezza EE, Gerunda GE, Plebani M, Galligioni A, Giacomini A, Neri D, Faccioli AM, Tiribelli C. Effect of ursodeoxycholic acid administration on bile duct proliferation and cholestasis in bile duct ligated rat. Dig Dis Sci. 1993;38:1291-1296. [PubMed] [DOI] [Cited in This Article: ]
114.
Alpini G, Baiocchi L, Glaser S, Ueno Y, Marzioni M, Francis H, Phinizy JL, Angelico M, Lesage G. Ursodeoxycholate and tauroursodeoxycholate inhibit cholangiocyte growth and secretion of BDL rats through activation of PKC alpha. Hepatology. 2002;35:1041-1052. [PubMed] [DOI] [Cited in This Article: ]
115.
Marzioni M, Francis H, Benedetti A, Ueno Y, Fava G, Venter J, Reichenbach R, Mancino MG, Summers R, Alpini G. Ca2+-dependent cytoprotective effects of ursodeoxycholic and tauroursodeoxycholic acid on the biliary epithelium in a rat model of cholestasis and loss of bile ducts. Am J Pathol. 2006;168:398-409. [PubMed] [DOI] [Cited in This Article: ]
116.
Alpini G, Kanno N, Phinizy JL, Glaser S, Francis H, Taffetani S, LeSage G. Tauroursodeoxycholate inhibits human cholangiocarcinoma growth via Ca2+-, PKC-, and MAPK-dependent pathways. Am J Physiol Gastrointest Liver Physiol. 2004;286:G973-G982. [PubMed] [DOI] [Cited in This Article: ]
117.
Calmus Y, Guechot J, Podevin P, Bonnefis MT, Giboudeau J, Poupon R. Differential effects of chenodeoxycholic and ursodeoxycholic acids on interleukin 1, interleukin 6 and tumor necrosis factor-alpha production by monocytes. Hepatology. 1992;16:719-723. [PubMed] [DOI] [Cited in This Article: ]
118.
Yoshikawa M, Matsui Y, Kawamoto H, Toyohara M, Matsumura K, Yamao J, Kuriyama S, Fukui H, Ishizaka S. Intragastric administration of ursodeoxycholic acid suppresses immunoglobulin secretion by lymphocytes from liver, but not from peripheral blood, spleen or Peyer's patches in mice. Int J Immunopharmacol. 1998;20:29-38. [PubMed] [DOI] [Cited in This Article: ]
119.
Calmus Y, Weill B, Ozier Y, Chéreau C, Houssin D, Poupon R. Immunosuppressive properties of chenodeoxycholic and ursodeoxycholic acids in the mouse. Gastroenterology. 1992;103:617-621. [PubMed] [DOI] [Cited in This Article: ]
120.
Calmus Y, Gane P, Rouger P, Poupon R. Hepatic expression of class I and class II major histocompatibility complex molecules in primary biliary cirrhosis: effect of ursodeoxycholic acid. Hepatology. 1990;11:12-15. [PubMed] [DOI] [Cited in This Article: ]
121.
Tanaka H, Makino Y, Miura T, Hirano F, Okamoto K, Komura K, Sato Y, Makino I. Ligand-independent activation of the glucocorticoid receptor by ursodeoxycholic acid. Repression of IFN-gamma-induced MHC class II gene expression via a glucocorticoid receptor-dependent pathway. J Immunol. 1996;156:1601-1608. [PubMed] [DOI] [Cited in This Article: ]
122.
Danzinger RG, Hofmann AF, Schoenfield LJ, Thistle JL. Dissolution of cholesterol gallstones by chenodeoxycholic acid. N Engl J Med. 1972;286:1-8. [PubMed] [DOI] [Cited in This Article: ]
123.
Oftebro H, Björkhem I, Størmer FC, Pedersen JI. Cerebrotendinous xanthomatosis: defective liver mitochondrial hydroxylation of chenodeoxycholic acid precursors. J Lipid Res. 1981;22:632-40. [PubMed] [DOI] [Cited in This Article: ]
124.
Gruy-Kapral C, Little KH, Fordtran JS, Meziere TL, Hagey LR, Hofmann AF. Conjugated bile acid replacement therapy for short-bowel syndrome. Gastroenterology. 1999;116:15-21. [PubMed] [DOI] [Cited in This Article: ]