Splanchnic vasodilation and hyperdynamic circulatory syndrome in cirrhosis (original) (raw)
Topic Highlight Open Access
Copyright ©2014 Baishideng Publishing Group Co., Limited. All rights reserved.
World J Gastroenterol. Mar 14, 2014; 20(10): 2555-2563
Published online Mar 14, 2014. doi: 10.3748/wjg.v20.i10.2555
Splanchnic vasodilation and hyperdynamic circulatory syndrome in cirrhosis
Massimo Bolognesi, Marco Di Pascoli, Alberto Verardo, Angelo Gatta, Department of Internal Medicine-DIMED, University of Padua, Azienda Ospedaliera Università di Padova, 35128 Padova, Italy
ORCID number: $[AuthorORCIDs]
Author contributions: All authors contributed equally to this work.
Correspondence to: Massimo Bolognesi, MD, PhD, Department of Internal Medicine-DIMED, University of Padua, Azienda Ospedaliera Università di Padova, Clinica Medica 5, Via Giustiniani 2, 35128 Padova, Italy. massimo.bolognesi@unipd.it
Telephone: +39-49-8212383 Fax: +39-49-8754179
Received: October 10, 2013
Revised: November 8, 2013
Accepted: November 28, 2013
Published online: March 14, 2014
Processing time: 152 Days and 21.6 Hours
Abstract
Portal hypertension is a clinical syndrome which leads to several clinical complications, such as the formation and rupture of esophageal and/or gastric varices, ascites, hepatic encephalopathy and hepato-renal syndrome. In cirrhosis, the primary cause of the increase in portal pressure is the enhanced resistance to portal outflow. However, also an increase in splanchnic blood flow worsens and maintains portal hypertension. The vasodilatation of arterial splanchnic vessels and the opening of collateral circulation are the determinants of the increased splanchnic blood flow. Several vasoactive systems/substances, such as nitric oxide, cyclooxygenase-derivatives, carbon monoxide and endogenous cannabinoids are activated in portal hypertension and are responsible for the marked splanchnic vasodilatation. Moreover, an impaired reactivity to vasoconstrictor systems, such as the sympathetic nervous system, vasopressin, angiotensin II and endothelin-1, plays a role in this process. The opening of collateral circulation occurs through the reperfusion and dilatation of preexisting vessels, but also through the generation of new vessels. Splanchnic vasodilatation leads to the onset of the hyperdynamic circulatory syndrome, a syndrome which occurs in patients with portal hypertension and is characterized by increased cardiac output and heart rate, and decreased systemic vascular resistance with low arterial blood pressure. Understanding the pathophysiology of splanchnic vasodilatation and hyperdynamic circulatory syndrome is mandatory for the prevention and treatment of portal hypertension and its severe complications.
Core tip: In cirrhosis, portal hypertension is due to an increase in intrahepatic resistance and splanchnic blood flow. The latter is secondary to arterial splanchnic vasodilatation and the opening of collateral circulation. Though the increase in intrahepatic resistance is the earliest and most important component, at present the only treatment regimes which are available for the control of portal hypertension in cirrhosis, i.e., non-selective beta-blockers, octreotide and terlipressin, act on the splanchnic dynamic component. Thus, understanding the mechanisms that lead to splanchnic vasodilatation and to the hyperdynamic circulatory syndrome is essential for the treatment of the complications of portal hypertension.
- Citation: Bolognesi M, Di Pascoli M, Verardo A, Gatta A. Splanchnic vasodilation and hyperdynamic circulatory syndrome in cirrhosis. World J Gastroenterol 2014; 20(10): 2555-2563
- URL: https://www.wjgnet.com/1007-9327/full/v20/i10/2555.htm
- DOI: https://dx.doi.org/10.3748/wjg.v20.i10.2555
INTRODUCTION
Portal hypertension is a clinical syndrome responsible for the onset of serious clinical complications such as the formation and rupture of esophageal and/or gastric varices, ascites, and hepatic encephalopathy.
In cirrhosis, the main mechanism responsible for the increase in portal pressure is the increase in intrahepatic resistance to portal blood outflow.
The pivotal mechanism responsible for the increased resistance in cirrhosis is the deposition of collagen in the hepatic acinus with narrowing of the sinusoidal lumen and a consequent decrease in the total cross-sectional area of the hepatic sinusoids. A further structural change responsible for the increase in intrahepatic portal resistance is the compression of centrilobular venules by regenerating nodules, granulomas, and portal inflammation. The main role of such anatomical alterations in determining the increase in portal pressure is confirmed by the relationships between septal thickness, small nodularity, liver stiffness and portal pressure[1,2].
Apart from the structural component, a vasoactive, potentially reversible component is also involved in the increase in hepatic resistance[3]. In cirrhosis, the contractile tone of smooth muscle cells and myofibroblasts, derived from stellate cells, around the sinusoids and hepatic venules is increased[4]. Norepinephrine, substance P, thrombin, angiotensin II[5], endothelin (ET)[6] and prostanoids[7] increase the contractile tone of myofibroblasts and thus portal resistance. Nevertheless, endothelial dysfunction is the main source of the dynamic increase in intrahepatic portal resistance[8]. A decreased bioavailability of nitric oxide (NO) in the sinusoids[8-11] and an increased production of cyclooxygenase (COX)-derived prostanoids, such as prostaglandin H2 and tromboxane A2[7,12] seem to be the main players in endothelial dysfunction in cirrhosis.
Despite being crucial to the development of hemodynamic changes in cirrhosis, the mechanisms responsible for the increase in intra-hepatic resistance will not be analyzed in details in this review, as they are beyond its aim.
INCREASED SPLANCHNIC INFLOW
Due to increased intra-hepatic resistance, a reduction in portal blood flow volume may be expected in portal hypertension. However, while a dilatation of the portal vein and a decrease in portal blood velocity are detected[13], these patients are characterized by a net increase in portal, splenic and mesenteric inflow.
The opening of portal-systemic collateral circulation participates in the increase in portal inflow. However a primary splanchnic arterial vasodilation can also be observed, with increased splenic and mesenteric blood flow.
Portal pressure results from the relationship between the blood flow volume entering the portal system and the resistance to outflow of portal blood. The mathematical expression of this relationship is provided by Ohm’s formula: P = Q * S, where P represents change in pressure along the vessel, Q represents blood flow and R resistance to the flow.
The increase in resistance to portal flow is the main determinant of portal hypertension in cirrhosis[14,15], but an increase in portal inflow also plays a role. Such increase in total splanchnic inflow[16,17] has been observed in patients with cirrhosis and demonstrated in experimental models of portal hypertension[18,19].
In the liver with normal resistance, a change in portal flow does not change portal pressure[20] because of the high vascular compliance of hepatic vasculature. When outflow portal resistance is increased and vascular compliance is decreased, an increase in portal flow is responsible for an increase in portal pressure. In cirrhosis, the increase in portal inflow, which is triggered by the increase in resistance to portal flow (see below), maintains and worsens portal hypertension[21].
Several therapeutic strategies for portal hypertension aim at decreasing portal pressure by decreasing portal inflow, thus highlighting the pathogenic role of portal inflow.
PORTAL-SYSTEMIC COLLATERALS
A mechanism explaining the maintenance of a high portal inflow in portal hypertension is the opening of portal-systemic collaterals, caused by the increase in resistance to outflow from the portal system.
The opening of collateral circulation occurs through the reperfusion and dilatation of preexisting vessels, but also through the generation of new vessels, as it was demonstrated by experimental studies showing the role of angiogenetic factors such as vascular endothelial growth factor (VEGF) in the pathogenesis of collateral circulation in portal hypertensive rats[22,23].
Portal-systemic shunts are responsible for gastrointestinal hemorrhage (mostly due to the rupture of esophageal or gastric varices) and allow access to the systemic circulation of substances which are usually removed by the liver. These play a role in the pathogenesis of the hyperdynamic circulation, ascites and hepatic encephalopathy[24].
SPLANCHNIC VASODILATION
In cirrhosis with portal hypertension, the increase in splanchnic blood flow is caused by a vasodilation of arterial splanchnic vessels, both in splenic and mesenteric vascular beds. In recent years, the mechanisms responsible for the reduction in mesenteric arterial resistance in cirrhosis have been extensively investigated. Numerous substances and systems have been proposed as possible mediators: glucagon[24-26], prostacyclin (PGI2), intestinal vasoactive peptide, histamine, substance P, estrogens, colecystokinin, ammonia, endotoxins, adenosine, biliary acids[24], NO[27-29], alpha-calcitonin gene-related peptide[30], adrenomedullin[31,32], VEGF[33], carbon monoxide (CO)[34,35], endogenous cannabinoids (ECs)[24,28]. Endothelial factors certainly play a major role[36].
The role of NO in the splanchnic vasodilatation of patients with cirrhosis and portal hypertension has been hypothesized more than 20 years ago[37] and nowadays it is considered the pivotal factor involved in the decreased mesenteric resistance of cirrhosis. NO has a very short half-life (20-30 s), it diffuses freely through the cellular membrane and acts mainly by increasing the production of cGMP by guanylate cyclase with subsequent relaxation of the smooth muscle cells. NO bioavailability is increased in patients with cirrhosis and portal hypertension[38], mostly because of an increased activity of the constitutive form of NO synthase (eNOS).
NOS activity is increased in the superior mesenteric artery and thoracic aorta of portal hypertensive rats[39]. Upregulation of eNOS can be detected even in early phases of the disease in portal hypertensive rats[35]. Several mechanisms seem to be responsible for such increase in eNOS activity. Inflammatory cytokines, VEGF and mechanical forces such as shear stress induce signaling cascades to activate Akt and heat shock protein 90 (Hsp90) which, in turn, activate eNOS[40]. Bacterial translocation from the gut into mesenteric lymph nodes is another early mechanism increasing tumor necrosis factor-alpha, eNOS cofactor tetrahydrobiopterin and eNOS-derived NO[41].
In decompensated cirrhosis, also the inducible form of NOS (iNOS) is upregulated in mesenteric arteries[42,43], even though its active role in the increased bioavailability of NO in the mesenteric vascular bed has not been clearly demonstrated. NO is also an angiogenic factor and it may play a role in the increased splanchnic angiogenesis which characterizes portal hypertensive rats[44]. In experimental cirrhosis with portal hypertension increased mesenteric angiogenesis can be reversed by chronic inhibition of NO formation[45].
However, NO/eNOS is not the only system involved in the mesenteric vasodilation of cirrhosis. Indeed, chronic administration of a NOS inhibitor in ascitic cirrhotic rats only partially corrects the mesenteric vasodilatation[35,43], thus implicating other vasoactive systems in the decrease in mesenteric resistance. PGI2, an endogenous vasodilator produced by vascular endothelial cells, is increased in patients with cirrhosis[46]. In portal hypertensive rats, an enhanced COX-1 expression within the superior mesenteric artery has been shown[47]. The inhibition of COX by indomethacin reduces portal pressure, improves hyperdynamic circulation[48,49] and reduces splanchnic blood flow[47].
It has been observed that in the superior mesenteric artery of cirrhotic rats treated with both NOS and COX inhibitors, an endothelium derived hyperpolarizing factor (EDHF) can replace NO and PGI2, inducing arterial dilation[50]. The exact nature of EDHF is controversial; however, arachidonic acid metabolites, the cation K+ and hydrogen peroxide are the main potential candidates[51]. A study conducted by our group suggested that in cirrhosis the increased NO/PGI2-indipendent vasodilation of mesenteric arteries is due, at least in part, to excess reactivity to 11,12-epoxyeicosatrienoic acid through an increased expression of myoendothelial gap junctions[52].
Also CO, an end product of heme catabolism by heme oxygenase (HO), is involved in the splanchnic vasodilatation in cirrhosis. CO, like NO, is an activator of guanyl cyclase and of large-conductance calcium-activated potassium channels (BKCa). It has been hypothesized that NO and CO may act in a coordinated fashion to maintain the patency of the sinusoids as a reaction to the upregulation of sinusoidal constrictors such as ET[53]. In a cirrhotic liver, the downregulation of NOS activity may not be compensated by a sufficient upregulation of other dilators, such as CO, with an increase in sinusoidal resistance as a result. On the other hand, in the aorta[54] and in the mesenteric arteries[35,54-56] of cirrhotic rats, the expression of HO-1 is increased. Moreover, the inhibition of HO improves the hyperdynamic circulatory syndrome[54] and normalizes the response of mesenteric arteries to phenylephrine in cirrhotic rats with ascites[35]. These data suggest that in cirrhosis the HO/CO system may play a role in the splanchnic hemodynamic alterations, especially in the later stages of the disease. In mesenteric arteries of ascitic cirrhotic rats, the increased levels of CO due to HO upregulation induce the expression of BKCa[56]. In cirrhotic rats with ascites, the upregulation of BKCa may explain the hypersensitivity of mesenteric arteries to CO[56].
ECs are ubiquitous lipid signaling molecules that determine central and peripheral effects through specific receptors CB1 and CB2. Experimental data suggest that ECs contribute to the development of splanchnic vasodilatation and portal hypertension by overactivating CB1 receptors within the mesenteric vasculature[51,57-59]. The effects of ECs on the splanchnic vessels seems to be due both to an increase in NO production[57] and to a NO-independent mechanism, since in isolated mesenteric arteries from cirrhotic rats endothelial denudation did not abolish the vasodilating effect[60].
In patients with chronic liver disease, autonomic dysfunction has also been observed and peripheral vasodilatation is probably also a consequence of a decreased reactivity to vasoconstrictor systems, such as the sympathetic nervous system[61], vasopressin, angiotensin II and ET-1. Autonomic dysfunction has been often attributed to alcohol-related neuropathy, but other mechanisms may also be involved as neuropathy is more common in in chronic alcohol misusers with liver damage than in their conterparts without liver damage[62]. A downregulation or a decreased affinity of the receptors for vasoconstrictors may explain the sustained systemic and splanchnic vasodilatation that occurs despite the activation of the vasoconstrictor systems. Finally, a downregulation or an impaired vasoconstrictor-induced activation of Rho kinase may represent a further mechanism contributing to defective contractility in cirrhosis[63].
SPLENIC CIRCULATION AND PORTAL HYPERTENSION
In cirrhosis with portal hypertension, the increase in mesenteric blood flow is not the only factor determining an increase in portal blood inflow, since splenic blood flow is also enhanced[64]. Indeed, it has been observed that in patients with cirrhosis and portal hypertension splenic blood flow increases to a higher extent than mesenteric flow[65,66]. While in healthy subjects the spleen contributes to portal blood flow for only about 40%[24], in patients with cirrhosis the splenic component represents more than 50% of portal inflow[67]. This increase in splenic blood flow has been observed in patients with cirrhosis and splenomegaly[64,68]. Moreover, a correlation exists between spleen size and portal vein diameter[69,70], splenic blood flow[71,72] and portal blood flow[69,73].
These data suggest that in cirrhosis the splenic congestion is not a passive phenomenon and the spleen actively contributes in maintaining portal hypertension by congesting the portal system[17].
HYPERDYNAMIC CIRCULATORY SYNDROME
In cirrhosis, the hyperdynamic circulation is characterized by increased cardiac output and heart rate, and decreased systemic vascular resistance with low arterial blood pressure[74,75]. The main cause of the onset of the syndrome is the systemic and splanchnic vasodilatation, which eventually leads to abnormalities in the cardiovascular system, and several regional vascular beds. Chronic administration of a NOS inhibitor in ascitic cirrhotic rats completely normalized the parameters of hyperdynamic circulation[76], but only partially corrected the mesenteric vasodilatation[35,43]. Neuropeptide Y, a sympathetic co-transmitter of norepinephrine, exerted marked portal hypotensive effects and ameliorated the hyperdynamic circulation through a pronounced splanchnic vasoconstriction and a reduction in splanchnic blood flow in cirrhotic rats with ascites[77].
Collateral circulation contributes to the development of hyperdynamic syndrome, both directly, by reducing peripheral resistance, and indirectly, by allowing intestinal vasoactive substances to bypass the liver and reach the systemic circulation. Neurogenic, biochemical and local mechanisms are also involved in the progression of the syndrome[21]. Even if overall peripheral and splanchnic vascular resistance is markedly reduced, a decrease in resistance is not present in all vascular beds[78]. A blood flow reduction has been observed in the kidney[79-82], in the brain[83-86] and in muscles[87]. The more the liver disease and the splanchnic vasodilatation worsen, the more the blood flow to other organs decreases[78]. Renal vasoconstriction is a consequence of effective hypovolemia and the activation of neurohumoral systems, providing the rationale for improving renal blood flow not by renal vasodilators, but by albumin infusion and splanchnic vasoconstrictors such as terlipressin or octreotide[88-91].
The hyperdynamic circulatory syndrome is the pathogenetic basis for the development of several complications of cirrhosis, such as hepato-renal syndrome, hepato-pulmonary syndrome, shock susceptibility and tissue hypoxemia. Moreover, since the gut and liver receive a third of the entire cardiac output, hyperdynamic circulation directly or indirectly contributes to two of the most serious complications of cirrhosis: ascites and variceal bleeding. Also, several symptoms of portal hypertension (i.e., tachycardia, subcyanotic warm skin, systemic arterial hypotension) are a direct consequence of this syndrome. Another main finding in the hyperdynamic circulatory syndrome is the increase in circulating blood volume[92-95]. According to the ‘‘peripheral arterial vasodilatation theory’’[96], splanchnic vasodilatation leads to renal sodium retention and, as a consequence, hypervolemia which contributes to the hyperdynamic syndrome by increasing cardiac pre-load. The hyperdynamic syndrome, in turn, maintains and enhances portal hypertension, while anti-aldosteronic drugs reduce portal pressure in cirrhotic patients without ascites by decreasing blood volume[97,98] (Figure 1). The hyperdynamic circulatory syndrome only develops in the supine position, in which a portion of blood volume is moved towards the ‘‘central’’ area[99,100]. In cirrhosis, the increase in cardiac output with compensatory reduction in peripheral vascular resistance is a consequence of the enhanced cardiac pre-load due to the supine posture[99,100].
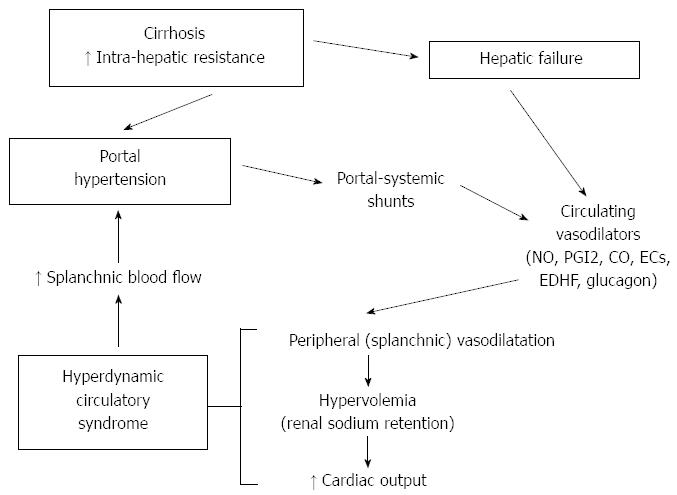
Figure 1 Schema of the relationships among splanchnic vasodilation, hyperdynamic circulatory syndrome and portal hypertension in liver cirrhosis. NO: Nitric oxide; PGI2: Prostacyclin; CO: Carbon monoxide; ECs: Endogenous cannabinoids; EDHF: Endothelium derived hyperpolarizing factor.
In patients with cirrhosis, a higher cardiac output is independently associated with higher hepatic venous pressure gradient and hepatic blood flow[101]. Despite the increase in the baseline cardiac output, the systolic and diastolic ventricular response to stress is blunted in conjunction with ventricular hypertrophy or dilatation and electrophysiological abnormalities such as prolonged QT interval. The following mechanisms seem to be involved in the development of cirrhotic cardiomyopathy: an altered β-adrenergic signalling[102,103], a dysfunction of the cardiomyocytes membrane, and an activation of cardiodepressant substances such as NO, cytokines and ECs[104].
PHARMACOLOGICAL APPROACHES TO SPLANCHNIC VASODILATATION AND HYPERDYNAMIC CIRCULATORY SYNDROME
Several therapeutic strategies for portal hypertension complications aim at decreasing portal pressure by decreasing portal inflow, thus highlighting the pathogenic role of splanchnic vasodilatation and hyperdynamic circulatory syndrome.
In the latest Baveno conference (May 2010), the therapeutic recommendations for non-selective β-blockers included primary prophylaxis of bleeding from gastro-esophageal varices and the prevention of recurrent hemorrhage[105]. The efficacy of non selective beta-blockers in patients with portal hypertension is due to the fact that they increase splanchnic resistance and decrease cardiac output, thus decreasing portal inflow[106].
Vasopressin analogues and somatostatin are the most widely utilized vasoactive drugs during an acute variceal bleeding. Terlipressin, a synthetic vasopressin analogue, causes splanchnic and systemic vasoconstriction by acting on the V1a receptors within the arterial smooth muscle. Somatostatin and its analogues cause splanchnic vasoconstriction both by inhibiting the release of the vasodilator glucagon, and through a direct mesenteric vasoconstrictive effect. Vasopressin analogues and somatostatin also blunt the increase in portal pressure induced by volume expansion[107,108]. Splanchnic vasodilation is responsible for the hypoperfusion of the renal system which leads to the activation of the renin-angiotensin-aldosterone system and to fluid retention. Diuretics are the pivotal drugs in the control of ascites. The standard combination includes spironolactone, an aldosterone antagonist, and furosemide[109]. In patients with ascites, albumin is used to prevent paracentesis-induced circulatory dysfunction, or an acute worsening of the hyperdynamic circulatory syndrome with a marked increase in plasma renin activity and plasma norepinephrine that occurs after a large volume paracentesis[110]. Also terlipressin and midodrine, an alpha-1 agonist upon arterial and venous vessels, seem to be effective in reducing the manifestations of paracentesis-related circulatory dysfunction[111,112]. Moreover, in refractory or recurrent ascites, midodrine significantly decreased cardiac output and increased systemic vascular resistance, thus being superior to standard medical therapy alone in controlling ascites[113]. In patients with hepatorenal syndrome, the rationale for the use of terlipressin, norepinephrine or midodrine in association with albumin is to counteract the splanchnic arterial vasodilation, thus increasing the effective circulating volume and, in turn, renal perfusion[114,115].
CONCLUSION
In cirrhosis, the increase in portal pressure which is responsible for the onset of complications such as gastrointestinal hemorrhage, ascites, hepatic encephalopathy, hepato-renal syndrome, hepato-pulmonary syndrome and spontaneous bacterial peritonitis, is due not only to an enhanced intrahepatic resistance to portal blood outflow, but also to an increase in splanchnic blood inflow into the portal vascular system.
The increase in splanchnic blood flow is determined by a vasodilatation of the arterial splanchnic vessels, both in the splenic and mesenteric vascular beds, and by the development of collateral circulation. The increase in NO production in the splanchnic vascular bed is considered the main contributor to splanchnic vasodilatation. However, several others molecules, such as PGI2, EDHF, CO and ECs also play a role. Splanchnic vasodilatation leads to the development of hyperdynamic circulatory syndrome, which is characterized by increased cardiac output and heart rate, and decreased systemic vascular resistance with low arterial blood pressure. The understanding of the pathophysiology of splanchnic vasodilatation and hyperdynamic circulatory syndrome is essential for the prevention and treatment of the complications of portal hypertension.
Footnotes
P- Reviewers: Xie F, Kanda T, Singh V, Toso C S- Editor: Cui XM L- Editor: A E- Editor: Wang CH
References
| 6. | Jiménez W, Rodés J. Impaired responsiveness to endogenous vasoconstrictors and endothelium-derived vasoactive factors in cirrhosis. Gastroenterology. 1994;107:1201-1203. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 17. | Gitlin N, Grahame GR, Kreel L, Williams HS, Sherlock S. Splenic blood flow and resistance in patients with cirrhosis before and after portacaval anastomoses. Gastroenterology. 1970;59:208-213. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 19. | Vorobioff J, Bredfeldt JE, Groszmann RJ. Hyperdynamic circulation in portal-hypertensive rat model: a primary factor for maintenance of chronic portal hypertension. Am J Physiol. 1983;244:G52-G57. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 20. | Lautt WW, Legare DJ. Passive autoregulation of portal venous pressure: distensible hepatic resistance. Am J Physiol. 1992;263:G702-G708. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 26. | Benoit JN, Womack WA, Hernandez L, Granger DN. „Forward“ and „backward“ flow mechanisms of portal hypertension. Relative contributions in the rat model of portal vein stenosis. Gastroenterology. 1985;89:1092-1096. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 27. | Vallance P, Moncada S. Hyperdynamic circulation in cirrhosis: a role for nitric oxide? Lancet. 1991;337:776-778. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 37. | Whittle BJ, Moncada S. Nitric oxide: the elusive mediator of the hyperdynamic circulation of cirrhosis? Hepatology. 1992;16:1089-1092. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 49. | Bruix J, Bosch J, Kravetz D, Mastai R, Rodés J. Effects of prostaglandin inhibition on systemic and hepatic hemodynamics in patients with cirrhosis of the liver. Gastroenterology. 1985;88:430-435. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 53. | Clemens MG. Does altered regulation of ecNOS in sinusoidal endothelial cells determine increased intrahepatic resistance leading to portal hypertension? Hepatology. 1998;27:1745-1747. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 61. | Heller J, Schepke M, Gehnen N, Molderings GJ, Müller A, Erhard J, Spengler U, Sauerbruch T. Altered adrenergic responsiveness of endothelium-denuded hepatic arteries and portal veins in patients with cirrhosis. Gastroenterology. 1999;116:387-393. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 62. | Barter F, Tanner AR. Autonomic neuropathy in an alcoholic population. Postgrad Med J. 1987;63:1033-1036. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 65. | Gusberg RJ, Peterec SM, Sumpio BE, Meier GH. Splenomegaly and variceal bleeding--hemodynamic basis and treatment implications. Hepatogastroenterology. 1994;41:573-577. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 67. | Okuda K, Kono K, Ohnishi K, Kimura K, Omata M, Koen H, Nakajima Y, Musha H, Hirashima T, Takashi M. Clinical study of eighty-six cases of idiopathic portal hypertension and comparison with cirrhosis with splenomegaly. Gastroenterology. 1984;86:600-610. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 68. | Murata K, Shiraki K, Takase K, Nakano T, Tameda Y. Long term follow-up for patients with liver cirrhosis after partial splenic embolization. Hepatogastroenterology. 1996;43:1212-1217. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 71. | Williams R, Condon RE, Williams HS, Blendis LM, Kreel L. Splenic blood flow in cirrhosis and portal hypertension. Clin Sci. 1968;34:441-452. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 82. | Sacerdoti D, Bolognesi M, Merkel C, Angeli P, Gatta A. Renal vasoconstriction in cirrhosis evaluated by duplex Doppler ultrasonography. Hepatology. 1993;17:219-224. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 87. | Maroto A, Ginès P, Arroyo V, Ginès A, Saló J, Clària J, Jiménez W, Bru C, Rivera F, Rodés J. Brachial and femoral artery blood flow in cirrhosis: relationship to kidney dysfunction. Hepatology. 1993;17:788-793. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 88. | Hadengue A, Gadano A, Moreau R, Giostra E, Durand F, Valla D, Erlinger S, Lebrec D. Beneficial effects of the 2-day administration of terlipressin in patients with cirrhosis and hepatorenal syndrome. J Hepatol. 1998;29:565-570. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 94. | Bosch J, Arroyo V, Betriu A, Mas A, Carrilho F, Rivera F, Navarro-Lopez F, Rodes J. Hepatic hemodynamics and the renin-angiotensin-aldosterone system in cirrhosis. Gastroenterology. 1980;78:92-99. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 96. | Schrier RW, Arroyo V, Bernardi M, Epstein M, Henriksen JH, Rodés J. Peripheral arterial vasodilation hypothesis: a proposal for the initiation of renal sodium and water retention in cirrhosis. Hepatology. 1988;8:1151-1157. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 97. | Okumura H, Aramaki T, Katsuta Y, Satomura K, Akaike M, Sekiyama T, Terada H, Ohsuga M, Komeichi H, Tsutsui H. Reduction in hepatic venous pressure gradient as a consequence of volume contraction due to chronic administration of spironolactone in patients with cirrhosis and no ascites. Am J Gastroenterol. 1991;86:46-52. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 114. | Narahara Y, Kanazawa H, Taki Y, Kimura Y, Atsukawa M, Katakura T, Kidokoro H, Harimoto H, Fukuda T, Matsushita Y. Effects of terlipressin on systemic, hepatic and renal hemodynamics in patients with cirrhosis. J Gastroenterol Hepatol. 2009;24:1791-1797. [PubMed] [DOI] [Cited in This Article: ] [Cited by in Crossref: 68] [Cited by in F6Publishing: 57] [Article Influence: 3.8] [Reference Citation Analysis (0)] |
|---|