Colorectal cancer in inflammatory bowel disease: The risk, pathogenesis, prevention and diagnosis (original) (raw)
Topic Highlight Open Access
Copyright ©2014 Baishideng Publishing Group Inc. All rights reserved.
World J Gastroenterol. Aug 7, 2014; 20(29): 9872-9881
Published online Aug 7, 2014. doi: 10.3748/wjg.v20.i29.9872
Colorectal cancer in inflammatory bowel disease: The risk, pathogenesis, prevention and diagnosis
Eun Ran Kim, Dong Kyung Chang, Division of Gastroenterology, Department of Internal Medicine, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul 135-710, South Korea
ORCID number: $[AuthorORCIDs]
Author contributions: All the authors contributed equally to this manuscript.
Correspondence to: Dong Kyung Chang, MD, PhD, Division of Gastroenterology, Department of Internal Medicine, Samsung Medical Center, Sungkyunkwan University School of Medicine, 50, Irwon-dong, Gangnam-gu, Seoul 135-710, South Korea. dkchang@skku.edu
Telephone: +82-2-34103409 Fax: +82-2-34106983
Received: November 13, 2013
Revised: March 4, 2014
Accepted: April 21, 2014
Published online: August 7, 2014
Processing time: 266 Days and 22.7 Hours
Abstract
Patients with inflammatory bowel disease (IBD) are at increased risk for developing colorectal cancer (CRC), although the overall incidence of IBD-associated CRC has been diminishing in recent decades in western countries. As demonstrated in previous studies, the risk of CRC in IBD increases with longer duration, extent of colitis, a familial history of CRC, coexistent primary sclerosing cholangitis, and the degree of inflammation. The pathogenesis of CRC in IBD is poorly understood. Similar to sporadic CRC, IBD-associated CRC is a consequence of sequential episodes of genomic alteration. Multiple inter-related pathways, including immune response by mucosal inflammatory mediators, oxidative stress, and intestinal microbiota, are also involved the pathogenesis of IBD-associated CRC. Continuing colonic inflammation appears to be a factor in the development of CRC; therefore, anti-inflammatory agents such as 5-aminosalicylate compounds and immune modulators have been considered as potential chemopreventive agents. Colonoscopic surveillance is widely accepted as being effective in reducing the risk of IBD-associated CRC, although no clear evidence has confirmed that surveillance colonoscopy prolongs survival in patients with extensive colitis. The traditional recommendation has been quadrantic random biopsies throughout the entire colon; however, several guidelines now have endorsed chromoendoscopy with a target biopsy because of increasing diagnostic yields and reduced workloads for endoscopists and pathologists. New technologies such as narrow band imaging, confocal endomicroscopy, and autofluorescence imaging have not yet been confirmed as surveillance strategies in IBD.
Core tip: An updated comprehensive review on the risk, pathogenesis, prevention and diagnosis of colorectal cancer in inflammatory bowel disease.
- Citation: Kim ER, Chang DK. Colorectal cancer in inflammatory bowel disease: The risk, pathogenesis, prevention and diagnosis. World J Gastroenterol 2014; 20(29): 9872-9881
- URL: https://www.wjgnet.com/1007-9327/full/v20/i29/9872.htm
- DOI: https://dx.doi.org/10.3748/wjg.v20.i29.9872
INTRODUCTION
Inflammatory bowel disease (IBD) is widely accepted as one of the important risk factors leading to colorectal cancer (CRC). IBD ranks as the third highest risk condition for CRC, behind only familial adenomatous polyposis (FAP) and hereditary nonpolyposis colorectal cancer syndrome (HNPCC)[1]. In fact, CRC accounts for one sixth of ulcerative colitis-related deaths[2].
Recently, a large nationwide Japanese study reported poorer survival for patients with ulcerative colitis (UC)-associated CRC than with sporadic CRC in the advanced stage. In addition, UC-associated CRC tended to be of higher histologic grade, such as mucinous or signet ring cell type, and had a greater multiplicity in number when compared to sporadic CRC[3]. This finding is similar to results reported in the large-scale nationwide population-based Danish study[4], where the overall mortality rate ratio for UC-associated CRC patients compared with sporadic CRC patients was 1.24 (95%CI: 1.02-1.51) in the first year and 1.17 (95%CI: 1.01-1.36) after 5 years of follow-up. Therefore, identifying the at-risk patients and carrying out appropriate preventions such as chemoprevention or surveillance for these patients is of great importance in managing the CRC risk in IBD.
This review discusses the established risk factors, pathogenesis, and prevention and diagnosis of colon carcinogenesis in patients with IBD.
RISK FACTORS
The most important and well-recognized risk factors identified for IBD-associated CRC are disease duration and extent. The meta-analysis by Eaden _et al_[5], which included 116 studies from a wide array of centers and geographic sites, was one of several milestones in establishing the incidence and cumulative risk of UC-associated CRC. This global meta-analysis reported that the incidence, which measures risk over time, was 0.3% per year, or 3 cases of CRC per 1000 patient-years of follow up. Based on long term follow up in a subset of studies included in the meta-analysis, the cumulative risk of CRC in patients with left-sided disease or pancolitis was estimated at 1.6% at 10 years, 8.3% at 20 years, and 18.4% at 30 years after the development of UC. However, the more recently published population studies demonstrated contradictory results (Table 1). A Danish population-based cohort study reported that the cumulative probability of CRC was 0.4% at 10 years, 1.1% at 20 years, and 2.1% at 30 years after the development of UC[6]. However, these results must be interpreted with caution because they reported high rates of proctocolectomy. Apart from this, several large studies from Hungary, the United Kingdom, and Sweden reported the cumulative risk of CRC in UC as 0.6%-1.5% at 10 years, 2.5%-5.4% at 20 years, and 7.5%-10.8% at 30 years; these studies suggest a low incidence rate of CRC. Recently, a nationwide Korean study involving 7061 patients with UC reported an estimated cumulative risk of ulcerative colitis-associated CRC of 0.7% at 10 years, 7.9% at 20 years, and 33.2% at 30 years[7]. These results were comparable to Eaden’s meta-analysis[5].
Table 1 Summary of cumulative risk of colorectal cancer in patients with inflammatory bowel disease in recent population-based studies and meta-analysis.
| Study | Country (n) | Population (n) | Observed period | Incidence of colorectal cancer |
|---|---|---|---|---|
| Eaden _et al_[5] | United States (4), United Kingdom (7), Scandinavia (3), other (5) | UC (meta-analysis of 19 studies) | 1925-1999 | Cumulative incidence |
| 1.6% by 10 yr of disease | ||||
| 8.3% by 20 yr of disease | ||||
| 18.4% by 30 yr of disease | ||||
| Winther _et al_[6] | Copenhagen County, Denmark | 1161 UC | 1962-1987 | Cumulative probability |
| 0.4% by 10 yr of disease | ||||
| 1.1% by 20 yr of disease | ||||
| 2.1% by 30 yr of disease | ||||
| Lakatos _et al_[14] | Veszprem, Hungary | 723 UC | 1974-2004 | Cumulative risk |
| 0.6% by 10 yr of disease | ||||
| 5.4% by 20 yr of disease | ||||
| 7.5% by 30 yr of disease | ||||
| Rutter _et al_[84] | London, United Kingdom | 600 long standing extensive UC | 1971-2000 | Cumulative incidence |
| 0% by 10 yr of disease | ||||
| 2.5% by 20 yr of disease | ||||
| 7.6% by 30 yr of disease | ||||
| 10.8% by 40 yr of disease | ||||
| Söderlund _et al_[8] | Sweden | 7607 UC | 1954-1989 | Cumulative risk |
| 1.5% by 10 yr of disease | ||||
| 3.8% by 20 yr of disease | ||||
| 7.6% by 30 yr of disease | ||||
| Canavan _et al_[9] | United States (3), Denmark (3), United Kingdom (2), Sweden (2), Canada (1), Israel (1) | 11545 CD (meta-analysis of 12 studies) | 1972-2004 | Cumulative risk |
| 2.9% by 10 yr of disease | ||||
| 5.6% by 20 yr of disease | ||||
| 8.3% by 30 yr of disease | ||||
| Kim _et al_[7] | South Korea | 7061 UC | 1970-2005 | Cumulative risk |
| 0.7% by 10 yr of disease | ||||
| 7.9% by 20 yr of disease | ||||
| 33.2% by 30 yr of disease |
This discrepancy between Western and Asian studies is thought to arise for several reasons. The incidence of ulcerative colitis seems to have reached a plateau in Western countries; thus, colitic cancer development may also be at a steady state or else decreasing[8]. However, in Asian countries, ulcerative colitis is still increasing; therefore, the occurrence of colitis cancer is also anticipated to increase. A meta-analysis of 12 published articles by Canavan _et al_[9] reported that the cumulative risk for patients with Crohn’s disease (CD) was 2.9% at 10 years, 5.6% at 20 years, and 8.3% at 30 years after the CD diagnosis. This study showed that cumulative risk of developing CRC, once diagnosed with CD, is comparable to the risk associated with UC[9].
The extent of colitis is an important risk factor, along with duration, for the development of CRC. The meta-analysis by Eaden _et al_[5] reported that an overall prevalence of 3.7% for CRC among patients with UC in all 116 studies, but when restricted to the 35 studies that stratified their analyses by extent of UC, the prevalence of CRC among patients with extensive involvement rose to 5.4%[5,10]. A recent Danish population-based cohort study involving 1515 patients diagnosed with UC also reported that the risk of CRC in patients diagnosed with UC was highest among patients with extensive colitis (standardized incidence ratio, 1.85; 95%CI: 0.60-4.32). Several studies have shown a general consensus indicating little or no increased risk of CRC in patients with proctitis or proctosigmoiditis, whereas the risk is intermediate in those with left-sided colitis and highest with pan-colitis[5,7,8,11-14].
Primary sclerosing cholangitis (PSC) is a chronic cholestatic liver disease that is strongly associated with IBD. An increased risk of CRC has been observed in patients with IBD complicated by PSC. A meta-analysis by Molodecky _et al_[15] reported that the pooled proportion of IBD in PSC cases was 68% (58%-77%). Among IBD subtypes, PSC was more common in UC than CD (80% vs 10% of cases)[16]. The association with IBD was stronger with more extensive colonic involvement: in patients with pancolitis, the prevalence of PSC was about 6%, in contrast to 1% in those with only distal colitis[17]. A meta-analysis of 11 studies involving 16844 patients with UC by Soetikno _et al_[18] reported that, overall, 21% of the patients with both UC and PSC developed colorectal neoplasms, compared with 4% of patients without PSC. The OR of developing dysplasia or cancer in patients with PSC was thus 4.8 (95%CI: 3.6-6.4), and this increased risk was present even when the risk of CRC alone was considered (OR = 4.3; 95%CI: 2.8-6.5).
Familial history of sporadic CRC increases the risk of CRC by at least two-fold when compared to patients with IBD without familial history for CRC[19,20]. A population-based cohort study of patients with IBD reported that a family history of CRC was associated with a more than 2-fold risk of IBD-associated CRC (adjusted RR = 2.5; 95%CI: 1.4-4.4); furthermore, patients with a 1st-degree relative diagnosed with CRC before 50 years of age also had a higher risk (RR = 9.2; 95%CI: 3.7-23)[20].
Chronic inflammation is believed to promote carcinogenesis[21]. Several lines of evidence indicate that chronic inflammation is a key risk factor for CRC in patients with IBD[22]. However, few studies have directly investigated whether the level of inflammation correlates with CRC risk in patients with IBD[21]. A retrospective case-control study[23] reported a significant correlation between the colonoscopic and histologic inflammation scores and the risk of colorectal neoplasia. However, multivariate analysis revealed that only the histologic inflammation score remained a significant risk factor. Another retrospective cohort study of 418 patients undergoing colonoscopic surveillance for UC showed similar results, where each unit increase in inflammation score conferred a 3.8-fold increase in the risk for high-grade dysplasia or CRC over time, based on use of a 4-point scale to classify histologic inflammation from a previous retrospective case control study[23,24]. On the other hand, Velayos _et al_[25] reported that the history of pseudopolyps increased the risk of CRC in UC by 2.5-fold (95%CI: 1.4-4.6), as a marker of more severe inflammation in the past.
PATHOGENESIS
The pathogenesis of CRC in IBD is poorly understood. Both genetic and environmental factors contribute to this pathogenesis, and the factors responsible for neoplastic changes in IBD can be summarized as follows: genetic instability; epigenetic alteration; immune response by mucosal inflammatory mediators; oxidative stress; and intestinal microbiota[21,26,27].
Many of the genetic alterations associated with development of sporadic CRC also play roles in colitis-associated CRC[27,28]. Colon carcinogenesis in IBD is thought to be similar to the adenoma-carcinoma sequence found in sporadic CRC. However, unlike sporadic CRC, which develops from dysplasia in 1 or 2 foci of the colon, cancer arising in colitis mucosa usually develops from multifocal dysplasia, indicating a “field change effect”[1,21]. Aneuploidy, a marker of genomic instability, is demonstrated at 20%-50% in dysplastic lesion and 50%-90% of cancers and is detected in long-standing UC[1,27,29,30]. Because aneuploidy is often more widespread than dysplasia in IBD, substantial genomic alterations must occur in colonic mucosa without disturbing morphology[1,21].
The two major types of genomic instability found in CRCs are chromosomal instability (CIN) and microsatellite instability (MSI)[31]. CIN and MSI in colitis-associated CRC appeared with the same frequency (85% CIN, 15% MSI) as seen in sporadic CRC, but they differed in the timing and frequency from the pattern seen with sporadic CRC (Figure 1)[10,21,27,32]. For example, loss of adenomatous polyposis coli (APC) function, considered to be a very common initiating event in sporadic colorectal carcinogenesis, is less frequent and usually occurs later in the colitis-associated dysplasia-carcinoma sequence[32-34]. Loss of P53 function is an important step in the progression of colitis- associated cancer. In contrast to sporadic CRC, P53 mutation in patients with IBD occurs early and is often detected in mucosa that is nondysplastic[26,27,35]. MSI, due to defective DNA mismatch repair, has been reported to occur at variable frequencies in IBD associated CRC[28,36-38]. The MSI-high (MSI-H) phenotype is observed in approximately 10% to 15% of sporadic CRC[36].
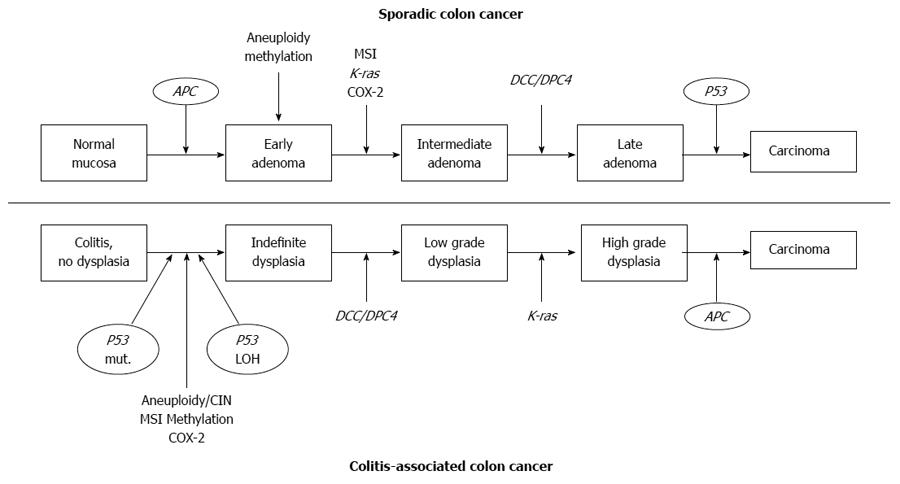
Figure 1 Molecular pathogenesis of sporadic colon cancer and colon cancer associated with inflammatory bowel disease. Many of the genetic alterations associated with development of sporadic colon cancer also play a role in colitis associated colon cancer. However, the frequency and sequence of these events differ between the cancers. Modified from Ref [21]. APC: Adenomatous polyposis coli; MSI: Microsatellite instability; KAS: Kirsten Rat Sarcoma Viral Oncogene Homolog; COX-2: Cyclooxygenase-2; DCC: Deleted in colorectal carcinoma; DPC4: Deleted in pancreatic carcinoma 4; CIN: Chromosomal instability.
The CpG island hypermethylation is a key epigenetic mechanism of gene silencing of tumor suppressor genes, including certain DNA repair genes where it acts through hypermethylation of their promoters[28,32]. Promoter hypermethylation of the mismatch repair gene hMLH1 is strongly associated with MSI. Among neoplastic samples from patients with colitis, hMLH1 hypermethylation was observed in 6 of 13 (46%) patients with high levels of MSI, 1 of 6 (16%) with low levels of MSI, and 4 of 27 (15%) without MSI, so it might cause MSI or contribute to its development[28,32,38]. Likewise, p16INK4a, a cell cycle inhibitor that is also implicated in sporadic colon cancer, is commonly hypermethylated in UC tissues, even in the absence of dysplasia[28,39].
Other key elements in the pathogenesis of CRC in IBD are related to chronic inflammation, such as the induction cyclooxygenase (COX)-2, inflammatory cytokines, and chemokines. Some evidence indicates that NSAIDs decrease the risk of CRC in IBD patients by 40%-50%[26,40]. NSAIDs exert their effects through their action on COX enzymes. Among three isoforms of COX enzyme, COX-2 is induced by inflammation and triggered by inflammatory stimuli such as IL-1, IFN-γ, and TNF-α[26]. Previous studies have shown that COX-2 expression is elevated in nearly 50% of adenomas and 85% of adenocarcinomas[41,42]. Agoff _et al_[43] reported that COX-2 messenger RNA and COX-2 protein overexpression occurred early in UC-associated neoplasia. TNF-α is released by activated macrophages and T cells; it binds to the receptor TNF-receptor (TNF-R) and has been reported to promote inflammation and colitis-associated cancer[21]. Several studies have assessed the relationship between the polymorphism of TNF-α-308 G>A and susceptibility to IBD and CRC[26]. Recently, a meta-analysis study suggested that the polymorphism of TNF-α-308 G>A participates in modifying the susceptibility to UC and CD in Europeans and Asians[44]. However, the increased risk of CRC in IBD patients should be clarified as the combined effects of polymorphisms in TNF-α and other cytokines and the interaction with environmental factors[44].
Oxidative stress also contributes to pathogenesis of the colon carcinogenesis by attacking proteins and nucleic acids, resulting in denaturation and a variety of alterations including base modifications, double-base lesions, and strand breaks[21,45,46]. Oxidative stress develops in response to inflammatory reactions in particular because the inflammatory cells, activated neutrophils, and macrophages produce large amounts of reactive oxygen and nitrogen species (RONs)[21,26,28]. Inflamed tissues from patients with active UC or CD demonstrate increased expression of nitric oxide synthase (NOS) and RONs[47-49]. RONs can interact with key genes involved in carcinogenic pathways such as P53 and DNA mismatch repair genes[28]. A previous study showed higher levels of P53 mutation frequencies of both G:C to A:T transitions at the CpG site of codon 248 and C:G to T:A transitions at codon 247 in the inflamed tissue than in the noninflamed tissue of the colon, as well as in colon tissue from normal adult controls[47]. Another study reported that noncytotoxic levels of hydrogen peroxide dramatically reduced the activities of the mismatch repair system[45].
Although intestinal microbiota have considered as contributors to development of colorectal neoplasia in IBD, the mechanism of intestinal microbiota-induced carcinogenesis remains largely unclear[26,27,50]. Several different rodent models of IBD have suggested that commensal or specific bacteria contribute to the development of colitis-associated cancer. For example, the IL-10 knock-out mice showed IBD (primarily colon and rectum) induced by Enterococcus faecalis, as well as rectal dysplasia and adenocarcinoma[51]. Under specific pathogen free conditions, all IL-10 gene knock-out mice also developed colitis after 3 mo of age, and CRC observed in 25% and 60% of the mice after 3 and 6 mo, respectively[52]. Modification of enteric flora in IL-10 knockout mice by probiotic lactobacilli was associated with a reduced prevalence of colon cancer and mucosal inflammatory activity[53].
CHEMOPREVENTION
Chemoprevention has been shown to reduce the risk of adenoma and CRC in the general population without IBD[54]. Pinczowski _et al_[55] first reported a chemopreventive effect of sulfasalazine in IBD, and several agents with chemopreventive potential have since been identified, including 5-aminosalicylate (5-ASA) compounds, immunomodulators, ursodeoxycholic acid (UDCA), and folic acid[56]. However, prospective randomized controlled trials on chemoprevention are lacking in patients with IBD because of insurmountable obstacles such as ethical problems; consequently, data on chemoprevention in patients with IBD are not clearly definitive and refer to either retrospective case-control or cohort studies[27,56].
Chronic inflammation has been accepted as one of the causative factors of IBD-associated CRC. The 5-ASA compounds are used as maintenance therapy in patients with UC because they have been shown to reduce inflammation in patients with UC. A meta-analysis of nine studies (three cohort and six case-control studies) that included 1932 UC patients reported that use of 5-ASA in UC was associated with a reduced risk of CRC, and the benefit occurred with regular use and use of at least 1.2 g of mesalamine equivalents per day[57]. However, subsequent to this meta-analysis, more studies reporting negative results have emerged[58-60]. A population based case control study showed that among patients with IBD (364 CRC cases, 1172 controls), exposure to 5-ASA therapy of any dose or duration during the 12 mo before CRC diagnosis was not associated with a reduced risk of CRC (OR = 0.97; 95%CI: 0.77-1.23)[58]. Recently, Bernstein _et al_[59] used the Manitoba IBD epidemiology database to show that 5-ASA was not chemoprophylactic in IBD for CRC in either a population-based case control or a cohort analysis. Although the chemopreventive role of 5-ASA compounds has biological plausibility, the chemopreventive effects of 5-ASA in IBD remain conflicting.
Immunomodulators also have anti-inflammatory properties and are used as maintenance therapy in IBD patients. Only limited evidence exists for a chemopreventive role for immunomodulators in IBD. A recent population-based study using a database linked to a nationwide pathology network from the Netherlands demonstrated a significant chemopreventive effect of thiopurine in patients with IBD (the adjusted hazard ratio: 0.10; 95%CI: 0.01-0.75). However, the majority of studies have not shown a chemopreventive effect for immunomodulators[25,58,61,62]. An 8-year follow up study reported no significant differences in rates of progression to advanced neoplasia or to any neoplasia between azathiopurine/6-MP user and never-user patients with UC[62]. Another retrospective cohort study also failed to demonstrate any preventive effect of azathiopurine/6-MP[24].
The chemopreventive role of UDCA therapy is well-supported from studies of patients with concomitant with IBD and PSC. The colonic concentration of additional bile acids has been implicated as a carcinogen, as this is cytotoxic to colonic epithelial cells and induces hyperproliferation[56]. Therefore, studies have suggested that treatment with UDCA may reduce the risk of CRC in IBD[63]. A post hoc analysis of a randomized, placebo-controlled trial of patients with UC and PSC suggested a significant chemoprotective effect for UDCA in these patients, with a 74% reduction seen in the risk for dysplasia or cancer in those assigned to the UDCA group[64]. Tung _et al_[65]. reported that UDCA use was strongly associated with a decreased prevalence of colonic dysplasia (OR: 0.18; 95%CI: 0.05-0.61). However, a retrospective analysis of data from a randomized controlled trial using a high dose UDCA (28-30 mg/kg body weight) showed that long-term use of high-dose UDCA was associated with an increased risk of colorectal neoplasia in patients with UC and PSC[66]. The chemoprotective effect of using UDCA for patients with UC but without PSC has not been explored.
Low folate intake has been associated with an increased risk for developing CRC and colon adenoma in sporadic CRC[27,56]. Folate deficiency is associated with alteration of the normal DNA methylation process, imbalance of steady-state levels of DNA precursors, and changes in chromosome and chromatin[67]. Patients with IBD are at risk of folate deficiency because sulfapyridine, a moiety of sulfasalazine used in IBD therapy, can lead to an impairment of folate absorption[68,69]. A meta-analysis showed that both sulfasalazine and folate supplementation have a protective effect in CRC development in patients with longstanding UC[70]. However, recent studies failed to find a chemopreventive effect of folic acid supplementation[56,60,71].
DIAGNOSIS AND SURVEILLANCE
One key element to decrease the risk of CRC in IBD may be the early diagnosis by colonoscopic surveillance and treatment of precancerous lesions[10]. However, randomized controlled trials have not been performed to verify that surveillance colonoscopy is effective. A previous Japanese study reported that close surveillance results in the detection of 48% of the cancers, 61% of which are early cancers[1,72]. A recent Cochrane analysis concluded that, for patients undergoing surveillance, cancers tend to be detected at an earlier stage and hence have a better prognosis, even if lead-time bias may contribute to the apparent benefit of surveillance[1,73,74]. Indirect evidence appears to support an effectiveness of surveillance in reducing the risk of death from IBD-associated CRC and that surveillance may be acceptably cost-effective, although no clear evidence was shown that surveillance colonoscopy prolonged survival in patients with extensive colitis[73,75].
Currently, several guidelines are available for recommending a specific surveillance program in IBD patients[76-79]. Almost all the guidelines agree on the following: (1) that a screening colonoscopy should be performed on patients during clinical remission of the disease in order to avoid confusion of inflammatory changes with dysplasia; (2) that surveillance colonoscopy should be started 8-10 years after the onset of symptoms for patients with left sided or extensive colitis; (3) that regular surveillance schedules need to be followed after the initial colonoscopy; and (4) that two to four random biopsy specimens should be taken with a jumbo forceps every 10 cm along the entire colon, with additional samples being taken in suspicious areas. Some distinctions are also made, such as the surveillance interval. These are summarized in Table 2.
Table 2 Summary of the screening and surveillance recommendation from international guidelines for patients with inflammatory bowel disease.
| ECCO 2008 | BSG 2010 | AGA 2010 | ACG 2010 | |
|---|---|---|---|---|
| 1st screening | 8-10 yr | 10 yr | Max 8 yr | 8-10 yr |
| Extensive: 2 yearly to 20 yr then annually | By risk: low 5 yr | 1-3 yr | ||
| Surveillance interval | Left sided: 2 yearly starting at 15 yr | Intermediate 3 yr | More often at high risk e.g., PSC | 1-2 yr |
| PSC: 1 yr | High 1 yr | |||
| Random biopsy | Recommended | Recommended | Recommended | Recommended |
| Chromoendoscopy | Superior to white light endoscopy | ≥ 33 if no chromo | ≥ 33 | ≥ 33 |
The success of surveillance colonoscopy will depend on improvement of the diagnostic yield of dysplasia or early colon cancer. As stated previously, many current guidelines recommend quadrantic random biopsies every 10 cm throughout the entire colon[63,80]. However, a random biopsy only samples 0.03% of the mucosal surface and has a detection rate of < 2 per 1000 biopsies[80,81]. A retrospective analysis estimated that 33 and 64 biopsy specimens are required to detect dysplasia with 90% and 95% probabilities, respectively[78,82]. This has served as the basis for the surveillance practice recommendation, and current guidelines for dysplasia surveillance recommend a minimum of 33 biopsies[10,78]. However, this biopsy protocol has failed to clearly demonstrate any significant gain in mortality rates and cost effectiveness[83-85]. Recent data have shown that most gastroenterologists do not follow the biopsy protocol. A previous study from the Netherlands showed that only 27% of gastroenterologists obeyed the recommended number of 33 random biopsies[86]. Another study showed that more than 50% of the gastroenterologists surveyed obtained fewer than 10 colonic mucosal biopsies per endoscopic surveillance examination[87,88].
The random biopsy protocol is now increasingly criticized, and increased focus is being placed on target biopsies supported by chromoendoscopy or other newer endoscopic techniques[10,63,89]. Chromoendoscopy involves the topical application of a dye onto colonic mucosa to improve detection of subtle colonic lesions, characterization, or diagnosis that are not visible with white light endoscopy[63,78,88]. Previous studies have demonstrated that the use of chromoendoscopy increases the detection rate of dysplasia by approximately 2 to 3 fold and gives a per lesion increase of 4 to 5 fold[83,90,91]. Comparable diagnostic yields have been obtained with both methylene blue and indigo carmine[90,92]. Although chromoendoscopy has a demerit of being more time consuming than white light endoscopy, recent guidelines have endorsed chromoendoscopy with target biopsies[76,77,80,88]. Moreover, recent back-to-back colonoscopy studies have showed that withdrawal times were similar, at 10 min for colonoscopy with random biopsy and 11 min for chromoendoscopy with target biopsy. In addition, a targeted biopsy protocol with pancolonic chromoendoscopy required fewer biopsies to detect dysplastic lesions[90].
Narrow band imaging (NBI) is another available technique that can provide clear imaging of the microvascular structure. However, NBI was not any more efficacious than conventional colonoscopy in detecting patients with neoplasia in tandem study and multicenter randomized studies[88,93-95]. Confocal laser endomicroscopy and fluorescence endoscopy are other promising techniques, but only limited data are available. Confocal laser endomicroscopy visualizes the histology of the mucosa and it has been proposed as an addition to chromoscopic detection of lesions to help target biopsy[80,88]. A recent study showed that endomicroscopy targeted biopsies increased the diagnostic yield of intraepithelial neoplasias by 2.5-fold when compared to chromoendoscopy guided biopsies alone[96]. Kiesslich _et al_[83] demonstrated that the combined use of chromoscopy and endomicroscopy detected 4.75-fold more neoplasias than could be detected with conventional colonoscopy and 50% fewer biopsy specimens were required.
Fluorescence endoscopy assesses intraepithelial neoplasia after topical or systemic sensitization with 5-aminolevulinic acid (5-ALA), which is converted intracellularly into the sensitizer protoporphyrin IX and accumulates selectively in neoplastic tissue[88,97]. In a previous study of 37 patients with UC, fluorescence endoscopy after 5-ALA sensitization showed excellent sensitivity, ranging from 87% to 100% after local sensitization, and negative predictive values of non-fluorescent mucosa for exclusion of dysplasia were also very high[97]. A recent back-to-back study that used autofluorescence imaging, where short-wavelength light provided the excitation of endogenous tissue fluorophores, showed lower neoplasia miss rates for autofluorescence imaging than for white light endoscopy[98].
CONCLUSION
Recent data about IBD-associated CRC have indicated several new trends. The overall incidence of CRC has been at a steady state or diminishing in western countries in recent decades. However, an anticipated increase in the occurrence of IBD-associated CRC has appeared in Asian countries as a consequence of the increasing incidence of IBD. Studies on the pathogenesis of IBD-associated CRC are now focused on genetic alteration. Interest is rising with regard to the role of several factors, such as oxidative stress, immune responses, and bacterial flora, in colon carcinogenesis in patients with IBD. Prospective randomized controlled trials on chemoprevention in patients with IBD are lacking and data on chemoprevention on patients with IBD are not clearly definitive; however, anti-inflammatory agents such as the 5-aminosalicylate compounds and immune modulators have been considered as potential chemopreventive agents. Several guidelines for surveillance strategy have also recently have endorsed the use of a target biopsy that incorporates chromoendoscopy or some new technologies instead of multiple random biopsies. However, the effectiveness of new technologies like narrow band imaging, confocal endomicroscopy, and autofluorescence imaging remain to be proven.
Footnotes
P- Reviewer: Jiang CP, Mazzanti R, Wang ZH S- Editor: Gou SX L- Editor: A E- Editor: Ma S
References
| 2. | Jess T, Loftus EV, Velayos FS, Harmsen WS, Zinsmeister AR, Smyrk TC, Schleck CD, Tremaine WJ, Melton LJ, Munkholm P. Risk of intestinal cancer in inflammatory bowel disease: a population-based study from olmsted county, Minnesota. Gastroenterology. 2006;130:1039-1046. [PubMed] [DOI] [Cited in This Article: ] [Cited by in Crossref: 358] [Cited by in F6Publishing: 312] [Article Influence: 17.3] [Reference Citation Analysis (0)] |
|---|
| 5. | Eaden JA, Abrams KR, Mayberry JF. The risk of colorectal cancer in ulcerative colitis: a meta-analysis. Gut. 2001;48:526-535. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 6. | Winther KV, Jess T, Langholz E, Munkholm P, Binder V. Long-term risk of cancer in ulcerative colitis: a population-based cohort study from Copenhagen County. Clin Gastroenterol Hepatol. 2004;2:1088-1095. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 7. | Kim BJ, Yang SK, Kim JS, Jeen YT, Choi H, Han DS, Kim HJ, Kim WH, Kim JY, Chang DK. Trends of ulcerative colitis-associated colorectal cancer in Korea: A KASID study. J Gastroenterol Hepatol. 2009;24:667-671. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 8. | Söderlund S, Brandt L, Lapidus A, Karlén P, Broström O, Löfberg R, Ekbom A, Askling J. Decreasing time-trends of colorectal cancer in a large cohort of patients with inflammatory bowel disease. Gastroenterology. 2009;136:1561-157; quiz 1818-9. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 10. | Lakatos PL, Lakatos L. Risk for colorectal cancer in ulcerative colitis: changes, causes and management strategies. World J Gastroenterol. 2008;14:3937-3947. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 17. | Olsson R, Danielsson A, Järnerot G, Lindström E, Lööf L, Rolny P, Rydén BO, Tysk C, Wallerstedt S. Prevalence of primary sclerosing cholangitis in patients with ulcerative colitis. Gastroenterology. 1991;100:1319-1323. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 18. | Soetikno RM, Lin OS, Heidenreich PA, Young HS, Blackstone MO. Increased risk of colorectal neoplasia in patients with primary sclerosing cholangitis and ulcerative colitis: a meta-analysis. Gastrointest Endosc. 2002;56:48-54. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 19. | Nuako KW, Ahlquist DA, Mahoney DW, Schaid DJ, Siems DM, Lindor NM. Familial predisposition for colorectal cancer in chronic ulcerative colitis: a case-control study. Gastroenterology. 1998;115:1079-1083. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 20. | Askling J, Dickman PW, Karlén P, Broström O, Lapidus A, Löfberg R, Ekbom A. Family history as a risk factor for colorectal cancer in inflammatory bowel disease. Gastroenterology. 2001;120:1356-1362. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 23. | Rutter M, Saunders B, Wilkinson K, Rumbles S, Schofield G, Kamm M, Williams C, Price A, Talbot I, Forbes A. Severity of inflammation is a risk factor for colorectal neoplasia in ulcerative colitis. Gastroenterology. 2004;126:451-459. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 27. | Triantafillidis JK, Nasioulas G, Kosmidis PA. Colorectal cancer and inflammatory bowel disease: epidemiology, risk factors, mechanisms of carcinogenesis and prevention strategies. Anticancer Res. 2009;29:2727-2737. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 29. | Befrits R, Hammarberg C, Rubio C, Jaramillo E, Tribukait B. DNA aneuploidy and histologic dysplasia in long-standing ulcerative colitis. A 10-year follow-up study. Dis Colon Rectum. 1994;37:313-319; discussion 319-320. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 30. | Willenbucher RF, Zelman SJ, Ferrell LD, Moore DH, Waldman FM. Chromosomal alterations in ulcerative colitis-related neoplastic progression. Gastroenterology. 1997;113:791-801. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 31. | Hardy RG, Meltzer SJ, Jankowski JA. ABC of colorectal cancer. Molecular basis for risk factors. BMJ. 2000;321:886-889. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 32. | Itzkowitz S. Colon carcinogenesis in inflammatory bowel disease: applying molecular genetics to clinical practice. J Clin Gastroenterol. 2003;36:S70-S74; discussion S94-96. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 33. | Aust DE, Terdiman JP, Willenbucher RF, Chang CG, Molinaro-Clark A, Baretton GB, Loehrs U, Waldman FM. The APC/beta-catenin pathway in ulcerative colitis-related colorectal carcinomas: a mutational analysis. Cancer. 2002;94:1421-1427. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 34. | Tarmin L, Yin J, Harpaz N, Kozam M, Noordzij J, Antonio LB, Jiang HY, Chan O, Cymes K, Meltzer SJ. Adenomatous polyposis coli gene mutations in ulcerative colitis-associated dysplasias and cancers versus sporadic colon neoplasms. Cancer Res. 1995;55:2035-2038. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 37. | Schulmann K, Mori Y, Croog V, Yin J, Olaru A, Sterian A, Sato F, Wang S, Xu Y, Deacu E. Molecular phenotype of inflammatory bowel disease-associated neoplasms with microsatellite instability. Gastroenterology. 2005;129:74-85. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 38. | Fleisher AS, Esteller M, Harpaz N, Leytin A, Rashid A, Xu Y, Liang J, Stine OC, Yin J, Zou TT. Microsatellite instability in inflammatory bowel disease-associated neoplastic lesions is associated with hypermethylation and diminished expression of the DNA mismatch repair gene, hMLH1. Cancer Res. 2000;60:4864-4868. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 39. | Hsieh CJ, Klump B, Holzmann K, Borchard F, Gregor M, Porschen R. Hypermethylation of the p16INK4a promoter in colectomy specimens of patients with long-standing and extensive ulcerative colitis. Cancer Res. 1998;58:3942-3945. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 40. | Smalley WE, DuBois RN. Colorectal cancer and nonsteroidal anti-inflammatory drugs. Adv Pharmacol. 1997;39:1-20. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 41. | Elzagheid A, Emaetig F, Alkikhia L, Buhmeida A, Syrjänen K, El-Faitori O, Latto M, Collan Y, Pyrhönen S. High cyclooxygenase-2 expression is associated with advanced stages in colorectal cancer. Anticancer Res. 2013;33:3137-3143. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 46. | Klaunig JE, Xu Y, Isenberg JS, Bachowski S, Kolaja KL, Jiang J, Stevenson DE, Walborg EF. The role of oxidative stress in chemical carcinogenesis. Environ Health Perspect. 1998;106 Suppl 1:289-295. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 47. | Hussain SP, Amstad P, Raja K, Ambs S, Nagashima M, Bennett WP, Shields PG, Ham AJ, Swenberg JA, Marrogi AJ. Increased p53 mutation load in noncancerous colon tissue from ulcerative colitis: a cancer-prone chronic inflammatory disease. Cancer Res. 2000;60:3333-3337. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 48. | Kimura H, Hokari R, Miura S, Shigematsu T, Hirokawa M, Akiba Y, Kurose I, Higuchi H, Fujimori H, Tsuzuki Y. Increased expression of an inducible isoform of nitric oxide synthase and the formation of peroxynitrite in colonic mucosa of patients with active ulcerative colitis. Gut. 1998;42:180-187. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 49. | Rachmilewitz D, Stamler JS, Bachwich D, Karmeli F, Ackerman Z, Podolsky DK. Enhanced colonic nitric oxide generation and nitric oxide synthase activity in ulcerative colitis and Crohn’s disease. Gut. 1995;36:718-723. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 50. | Actis GC, Tarallo S, Rosina F. Cutting edge: chemoprevention of colorectal neoplasia in inflammatory bowel disease. Inflamm Allergy Drug Targets. 2013;12:1-7. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 53. | O’Mahony L, Feeney M, O’Halloran S, Murphy L, Kiely B, Fitzgibbon J, Lee G, O’Sullivan G, Shanahan F, Collins JK. Probiotic impact on microbial flora, inflammation and tumour development in IL-10 knockout mice. Aliment Pharmacol Ther. 2001;15:1219-1225. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 55. | Pinczowski D, Ekbom A, Baron J, Yuen J, Adami HO. Risk factors for colorectal cancer in patients with ulcerative colitis: a case-control study. Gastroenterology. 1994;107:117-120. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 61. | Lashner BA, Provencher KS, Seidner DL, Knesebeck A, Brzezinski A. The effect of folic acid supplementation on the risk for cancer or dysplasia in ulcerative colitis. Gastroenterology. 1997;112:29-32. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 62. | Matula S, Croog V, Itzkowitz S, Harpaz N, Bodian C, Hossain S, Ullman T. Chemoprevention of colorectal neoplasia in ulcerative colitis: the effect of 6-mercaptopurine. Clin Gastroenterol Hepatol. 2005;3:1015-1021. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 65. | Tung BY, Emond MJ, Haggitt RC, Bronner MP, Kimmey MB, Kowdley KV, Brentnall TA. Ursodiol use is associated with lower prevalence of colonic neoplasia in patients with ulcerative colitis and primary sclerosing cholangitis. Ann Intern Med. 2001;134:89-95. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 67. | Biasco G, Di Marco MC. Folate and prevention of colorectal cancer in ulcerative colitis. Eur J Cancer Prev. 2005;14:395-398. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 68. | Baggott JE, Morgan SL, Ha T, Vaughn WH, Hine RJ. Inhibition of folate-dependent enzymes by non-steroidal anti-inflammatory drugs. Biochem J. 1992;282:197-202. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 69. | Reisenauer AM, Halsted CH. Human jejunal brush border folate conjugase. Characteristics and inhibition by salicylazosulfapyridine. Biochim Biophys Acta. 1981;659:62-69. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 70. | Diculescu M, Ciocîrlan M, Ciocîrlan M, Piţigoi D, Becheanu G, Croitoru A, Spanache S. Folic acid and sulfasalazine for colorectal carcinoma chemoprevention in patients with ulcerative colitis: the old and new evidence. Rom J Gastroenterol. 2003;12:283-286. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 72. | Takashi H, Tmomyuki K, Yukihide K. Colorectal cancer in patients with inflammatory bowel disease in Japan. Stomach Intestine. 2002;37:887-893. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 82. | Rubin CE, Haggitt RC, Burmer GC, Brentnall TA, Stevens AC, Levine DS, Dean PJ, Kimmey M, Perera DR, Rabinovitch PS. DNA aneuploidy in colonic biopsies predicts future development of dysplasia in ulcerative colitis. Gastroenterology. 1992;103:1611-1620. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 86. | van Rijn AF, Fockens P, Siersema PD, Oldenburg B. Adherence to surveillance guidelines for dysplasia and colorectal carcinoma in ulcerative and Crohn’s colitis patients in the Netherlands. World J Gastroenterol. 2009;15:226-230. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 87. | Eaden JA, Ward BA, Mayberry JF. How gastroenterologists screen for colonic cancer in ulcerative colitis: an analysis of performance. Gastrointest Endosc. 2000;51:123-128. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 90. | Rutter MD, Saunders BP, Schofield G, Forbes A, Price AB, Talbot IC. Pancolonic indigo carmine dye spraying for the detection of dysplasia in ulcerative colitis. Gut. 2004;53:256-260. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 97. | Messmann H, Endlicher E, Freunek G, Rümmele P, Schölmerich J, Knüchel R. Fluorescence endoscopy for the detection of low and high grade dysplasia in ulcerative colitis using systemic or local 5-aminolaevulinic acid sensitisation. Gut. 2003;52:1003-1007. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 98. | van den Broek FJ, Fockens P, van Eeden S, Reitsma JB, Hardwick JC, Stokkers PC, Dekker E. Endoscopic tri-modal imaging for surveillance in ulcerative colitis: randomised comparison of high-resolution endoscopy and autofluorescence imaging for neoplasia detection; and evaluation of narrow-band imaging for classification of lesions. Gut. 2008;57:1083-1089. [PubMed] [DOI] [Cited in This Article: ] [Cited by in Crossref: 193] [Cited by in F6Publishing: 201] [Article Influence: 12.6] [Reference Citation Analysis (0)] |
|---|