Architecture and biogenesis of plus-strand RNA virus replication factories (original) (raw)
Topic Highlight Open Access
Copyright ©2013 Baishideng. All rights reserved.
World J Virol. May 12, 2013; 2(2): 32-48
Published online May 12, 2013. doi: 10.5501/wjv.v2.i2.32
Architecture and biogenesis of plus-strand RNA virus replication factories
David Paul, Ralf Bartenschlager, Department of Infectious Diseases, Molecular Virology, University of Heidelberg, 69120 Heidelberg, Germany
ORCID number: $[AuthorORCIDs]
Author contributions: Paul D and Bartenschlager R contributed equally to write this paper.
Supported by The DFG, SFB638, TP A5 and SFB/TRR83, TP 13
Correspondence to: Dr. Ralf Bartenschlager, Professor, Department of Infectious Diseases, Molecular Virology, University of Heidelberg, Im Neuenheimer Feld 345, 69120 Heidelberg, Germany. ralf_bartenschlager@med.uni-heidelberg.de
Telephone: +49-6221-564225 Fax: +49-6221-564570
Received: December 5, 2012
Revised: January 18, 2013
Accepted: January 23, 2013
Published online: May 12, 2013
Processing time: 155 Days and 3.1 Hours
Abstract
Plus-strand RNA virus replication occurs in tight association with cytoplasmic host cell membranes. Both, viral and cellular factors cooperatively generate distinct organelle-like structures, designated viral replication factories. This compartmentalization allows coordination of the different steps of the viral replication cycle, highly efficient genome replication and protection of the viral RNA from cellular defense mechanisms. Electron tomography studies conducted during the last couple of years revealed the three dimensional structure of numerous plus-strand RNA virus replication compartments and highlight morphological analogies between different virus families. Based on the morphology of virus-induced membrane rearrangements, we propose two separate subclasses: the invaginated vesicle/spherule type and the double membrane vesicle type. This review discusses common themes and distinct differences in the architecture of plus-strand RNA virus-induced membrane alterations and summarizes recent progress that has been made in understanding the complex interplay between viral and co-opted cellular factors in biogenesis and maintenance of plus-strand RNA virus replication factories.
- Citation: Paul D, Bartenschlager R. Architecture and biogenesis of plus-strand RNA virus replication factories. World J Virol 2013; 2(2): 32-48
- URL: https://www.wjgnet.com/2220-3249/full/v2/i2/32.htm
- DOI: https://dx.doi.org/10.5501/wjv.v2.i2.32
INTRODUCTION
As obligate intracellular parasites, all viruses depend on the host cell biosynthetic machinery in order to replicate their genome and generate progeny virus particles. A common feature among many different viruses is the induction of specialized membranous compartments, often forming organelle-like structures, within the cytoplasm of an infected cell. The unique features of those structures that facilitate virus propagation are best expressed by the commonly accepted term “viral replication factories”. This review focuses on animal plus-strand RNA virus induced replication compartments. However, the concept of viral replication factories also applies to plant plus-strand RNA viruses as well as to other animal RNA viruses or some large DNA viruses such as vaccinia virus. These virus groups are covered by other reviews and will not be discussed here[1-3]. By analogy to an industrial manufacturing unit, virus induced replication factories serve multiple purposes.
To increase the local concentration of all necessary factors thus ensuring high efficacy
Local enrichment of the viral replication factors and co-opted cellular proteins, together with all required metabolites, is the basis for highly efficient genome replication. This is achieved by reducing the three-dimensional (3-D) diffusion in the cytosol to two-dimensional diffusion on the surface of a membrane, thus increasing local reaction efficacy presumably by several orders of magnitude[4]. In fact, many replication enzymes encoded by plus-strand RNA viruses are membrane anchored as exemplified by the hepatitis C virus (HCV)[5]. In addition, also substrates such as ribonucleotides and metabolites providing energy for biosynthetic processes are enriched in replication compartments. Along these lines a recent study convincingly demonstrated locally elevated ATP levels at HCV replication sites[6].
To spatially coordinate different processes of the replication cycle such as RNA translation, replication and assembly
Compartmentalization allows the spatial separation and regulation of RNA translation, replication and packaging of the viral genome, thereby preventing an interference between these processes. Hence, ribosomes responsible for RNA translation and RNA-binding assembly factors are excluded from replication sites, thus avoiding interference with the replication machinery. Indeed, replication factories of all plus-strand RNA viruses are built up of ribosome-free membranes[7-11], and for all Flaviviridae (excess) amounts of core/capsid protein, binding RNA with high affinity, are frequently targeted to other cellular organelles such as lipid droplets (LDs) in the case of HCV and dengue virus (DENV)[12-14], or to the nucleus as observed for DENV[15,16] Japanese encephalitis virus[17] and West Nile virus (WNV)[18].
To create a protected environment shielding viral RNA and eventually also proteins from a hostile degradative environment
The generation of specialized membranous replication compartments protects viral replicase complexes and genomic RNA from degradation by cellular proteases or nucleases, respectively and “hides” the viral RNA genome from cytoplasmic sensors of the innate immune response. The RigI-like receptors efficiently recognize 5’ triphosphorylated RNAs as well as double-stranded RNA (dsRNA) in a length-dependent manner[19,20], leading to mitochondrial antiviral signaling-mediated induction of interferons and nuclear factor κB-mediated inflammation[21]. Minimizing the exposure of stimuli to the innate immune surveillance, by the induction of innate sensor-protected organelle-like replication factories, is therefore an important evolutionary conserved feature of plus-strand RNA virus infection.
In the following we will summarize recent insights into the 3-D ultrastructure of plus-strand RNA virus-induced membrane rearrangements and discuss possible mechanisms of their biogenesis. Furthermore, viral subversion of host cell membrane biology, by interference with signaling pathways and recruitment of host cell factors contributing to biogenesis and maintenance of viral replication factories are highlighted.
MORPHOLOGY OF PLUS-STRAND RNA VIRUS REPLICATION FACTORIES
In the last few years, electron tomography has been instrumental to decipher the 3-D architecture of viral replication factories (for technical review see[22,23]). This accounts for evolutionary diverse plus-strand RNA viruses such as flock-house virus (FHV)[24], rubella virus (RUBV)[8], the two enteroviruses coxsackievirus B3 (CVB3)[25] and poliovirus (PV)[26], severe acute respiratory syndrome coronavirus (SARS-CoV)[11], equine arterivirus (EAV)[27], the two flaviviruses DENV[10] and WNV[9] and HCV[28]. Despite many differences in host range, virion morphology, genome organization, or donor membrane usage (Table 1), these analyses revealed that plus-strand RNA viruses appear to induce one of two different membrane alterations: the invaginated vesicle (InV) or spherule type and the double membrane vesicle (DMV) type. These morphologies that will be used in this review to group plus-strand RNA viruses might reflect the use of different host cell pathways and factors exploited by these viruses to establish the membranous replication compartment.
Table 1 Overview of plus-strand RNA viruses and induced replication factories.
| Invaginated vesicle/spherule type | |||||||
|---|---|---|---|---|---|---|---|
| Superfamily/order | Alphavirus like | Alphavirus like | Alphavirus like | Alphavirus like | |||
| Family | Togaviridae | Togaviridae | Togaviridae | Bromoviridae | Nodaviridae | Flaviviridae | Flaviviridae |
| Genus | Alphavirus | Alphavirus | Rubivirus | Bromovirus | Alphanodavirus | Flavivirus | Flavivirus |
| Species | SFV | SINV | RUBV | BMV | FHV | DENV | WNV |
| Genome | ss(+) RNA | ss(+) RNA | ss(+) RNA | tripartite ss(+) RNA | bipartite ss(+) RNA | ss(+) RNA | ss(+) RNA |
| Gene order | NS-S | NS-S | NS-S | NS-S | NS-S | S-NS | S-NS |
| Genome size (bases) | 13000 | 12000 | 10000 | 8300 | 4500 | 10000 | 10000 |
| Virion size (nm) | 70 | 70 | 70 | 27 | 30 | 50 | 50 |
| Envelope | Yes | Yes | Yes | No | No | Yes | Yes |
| Host | Mosquitoes, humans | Mosquitoes, humans | Humans | Plants, yeast | Insects | Mosquitoes, humans | Mosquitoes, mammals |
| Disease | Encephalitis | Sinbis fever | German measels | Plant disease | Unknown | Dengue fever | Mostly asymtomatic |
| Type of replication factories | Spherules at PM and CPVs | Spherules at PM and CPVs | Invaginations at CPVs | Spherules at the ER | Spherules at mitochondria | Invaginated vesicles at the ER | Invaginated vesicles at the ER |
| Diameter of invaginations (nm) | 50 | 50 | 80-500 | 50-70 | 50 | 90 | 50-150 |
| Primary membrane source | PM, endosomes | PM, endosomes | Endosomes | ER | Outer mitochondrial membrane | ER | ER |
| Proposed sites of replication | Inside spherules | Inside spherules | Inside invaginated vesicles | Inside spherules | Inside spherules | Inside invaginated vesicles | Inside invaginated vesicles |
| Double membrane vesicle type | |||||||
| Superfamily/order | Picornavirales | Picornavirales | Nidovirales | Nidovirales | Nidovirales | ||
| Family | Flaviviridae | Picornaviridae | Picornaviridae | Coronaviridae | Coronaviridae | Arteriviridae | |
| Genus | Hepacivirus | Enterovirus | Enterovirus | Coronavirus | Coronavirus | Arterivirus | |
| Species | HCV | PV | CVB3 | SARS CoV | MHV | EAV | |
| Genome | ss(+) RNA | ss(+) RNA | ss(+) RNA | ss(+) RNA | ss(+) RNA | ss(+) RNA | |
| Gene order | S-NS | S-NS | S-NS | S-NS | S-NS | S-NS | |
| Genome size (bases) | 9600 | 8000 | 8000 | 30000 | 30000 | 13000 | |
| Virion size (nm) | 50 | 30 | 30 | 80-160 | 80-160 | 40-60 | |
| Envelope | Yes | No | No | Yes | Yes | Yes | |
| Host | Humans | Humans | Humans | Humans | Mice | Horses, donkeys | |
| Disease | Hepatitis | Poliomyelitis | HFM disease | SARS | HMM illness | Haemorragic fever | |
| Type of replication factories | DMVs | SMTs, DMVs | SMTs, DMVs | DMVs, VPs | DMVs | DMVs | |
| Length/diameter of replication factories | 150 nm | 100-200 nm, 100-300 nm | 650 nm, 150 nm | 150 nm, 1-5 μm | 200-350 nm | 90 nm | |
| Primary membrane source | ER | Golgi, ER | Golgi, ER | ER | ER | ER | |
| Proposed sites of replication | Possible role of DMVs but largely unknown | On SMT/DMV outer membrane | On SMT/DMV outer membrane | Inside DMVs | Inside DMVs | Inside DMVs |
Architecture of replication factories corresponding to the InV/spherule type
Viral replication factories of the InV/spherule type are induced by alphaviruses such as Semliki Forrest virus (SFV)[29,30] and Sindbis virus[31], by FHV[24], RUBV[8], DENV[10] and WNV[9]. Although no 3-D reconstruction of alphavirus replication factories has been published yet, pioneering classical electron microscopy (EM) studies from Grimley _et al_[29] describing SFV replication sites at modified membranous structures, date back to the 1960s. Alphavirus infection induces so called cytoplasmic vacuoles (CPVs) (600-2000 nm in size), containing small invaginations called “spherules” with an average diameter of approximately 50 nm[29,32-34]. Surprisingly, at early time points after alphavirus infection, spherules are frequently found at the plasma membrane[30,31]. These spherules are subsequently internalized and become part of the endo-lysosomal membrane system, giving rise to CPVs. The single membrane invagination of spherules is continuous with its donor membrane and an approximately 8 nm small opening connects its interior with the cytoplasm[7]. Viral replicase proteins nsp1 to nsp4 as well as newly synthesized viral RNA localize to spherules[29,30]. Interestingly, the spherules themselves are devoid of ribosomes and viral capsid protein, which are frequently found juxtaposed to the spherule openings[7]. The first 3-D reconstruction of a plus-strand RNA virus replication factory was published by Kopek _et al_[24]. Electron tomography of FHV-infected cells revealed InVs on the outer mitochondrial membrane (OMM) (Figure 1A). Similar to alphavirus spherules, InVs found in FHV-infected cells are approximately 50 nm in diameter[35]. The single membrane building up the spherule is continuous with the OMM and a membranous neck with an interior diameter of approximately 10 nm connects the spherule lumen to the cytoplasm[24]. The sole viral replicase factor protein A as well as nascent viral RNA localize to spherules. By using high resolution electron tomography in combination with biochemical analyses, Kopek _et al_[24] determined that each spherule contains approximately 100 copies of protein A, covering the complete interior surface of the spherule and one or two copies of minus-strand RNA, the replication intermediate. RUBV induces replication factories with similar architecture. Replicase proteins p150 and p90, as well as nascent viral RNA localize to remodeled endosomes/lysosomes, termed cytopathic vacuoles[36-39]. Fontana _et al_[8] conducted electron tomography studies of RUBV replicon cells and observed spherule-like membrane invaginations. In addition, the authors detected bigger vacuolar invaginations and rigid straight membrane sheets, which are continuous with the outer membrane of the CPV and whose interior is connected to the cytoplasm (Figure 1B). Mitochondria localized in close proximity to CPVs and were probably engaged in supplying energy for viral replication. Importantly, CPV membranes were free of ribosomes, but frequently surrounded by rough ER sheets and Golgi stacks, thus facilitating translation and encapsidation of viral RNA, respectively[8,40]. In contrast to the replication compartments of these plus-strand RNA viruses, replication factories of Flaviviridae, exemplified by WNV and DENV, are most likely derived from the ER. Apart from the rather undefined convoluted membranes (CMs) found in DENV-infected cells, our laboratory revealed by means of electron tomography vesicular invaginations of approximately 90 nm diameter in the rough ER, which are connected by approximately 10 nm diameter necked channel openings to the cytoplasm[10] (Figure 1C). Likewise, Gillespie _et al_[9] observed in WNV replicon-containing cells, InVs in the rough ER, which were connected via neck-like membranous pores to the cytoplasmic space. For both viruses, replicase proteins and dsRNA, assumed to represent a replication intermediate, localized to InVs[10,41], whereas ribosomes were excluded from the InVs, but localized in close proximity on ER membranes[9,10]. Interestingly, DENV virion budding was observed frequently close to or directly opposite of pore-like vesicle openings[10], highlighting the spatial orchestration of replication and assembly steps in the DENV replication cycle.
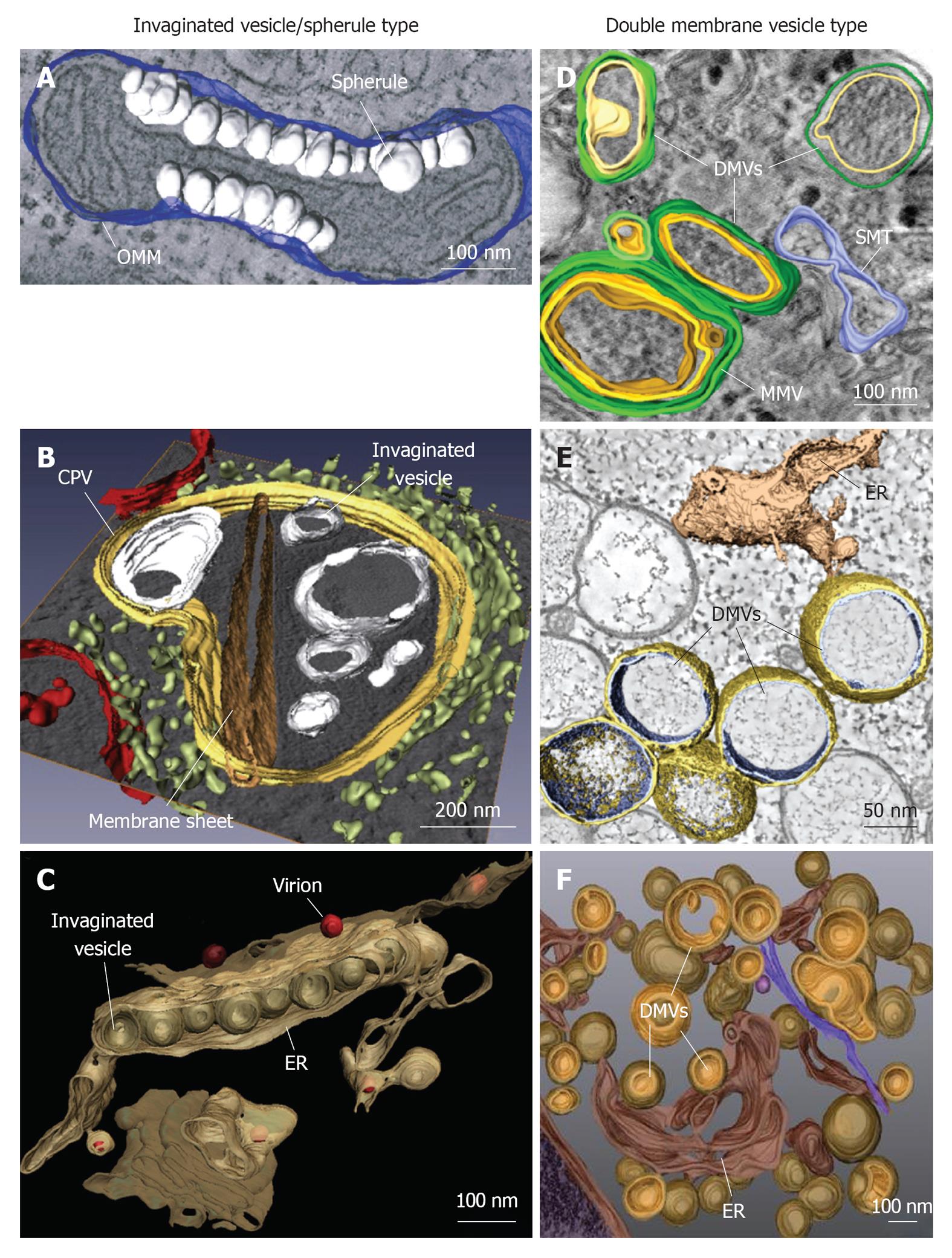
Figure 1 Classification and morphologies of plus-strand RNA virus-induced membrane alterations as revealed by electron tomography and three-dimensional reconstruction. Morphological similarities of membrane structures belonging to the invaginated vesicle (InV)/spherule type (A-C) or the DMV type (D-F). A: Flock-house virus (FHV); B: Rubella virus (RUBV); C: Dengue virus (DENV); D: Poliovirus (PV); E: Severe acute respiratory syndrome coronavirus (SARS-CoV); F: Hepatitis C virus (HCV). A: FHV-induced spherules (white) in the outer mitochondrial membrane (OMM; blue); B: InVs (white) and rigid membrane sheets (dark brown) in a modified endosome (the cytopathic vacuole, CPV) (yellow) in cells replicating RUBV. The rough endoplasmic reticulum (ER) is shown in light-green and mitochondria in red; C: Spherule-like invaginations (InVs) in the endoplasmic reticulum (ER) (brown) observed in DENV-infected cells. Newly formed progeny virions are shown in red; D: PV infection induces single membrane tubules (SMT) (blue), double membrane vesicles (DMVs) and multi membrane vesicles (MMVs). The DMV outer membrane is shown in green, the second membrane in yellow and a third membrane in case of MMVs in orange. These membranes are primarily derived from the Golgi; E: An interconnected network of ER-derived DMVs is found in SARS-CoV infected cells. The ER is colored in light-brown, interconnected outer membranes of DMVs in yellow and DMV inner membranes in blue; F: HCV-induced DMVs protruding from the ER (dark-brown). Outer membranes of DMVs are shown in light-brown and inner membranes in orange. Images are modified with permission from ©2007 Kopek _et al_[24] Plos Biol (A); ©2010 Elsevier Inc.[8] (B); ©2009 Elsevier Inc.[10] (C); ©2012 American Society for Microbiology[26] (D); ©2008 Knoops _et al_[11] Plos Biol (E); ©2012 Romero-Brey _et al_[28] Plos Pathog (F).
Architecture of DMV-like replication factories
DMV-like replication factories are formed by the enteroviruses CVB3[25] and PV[26], SARS-CoV[11], EAV[27] and HCV[28]. For PV, probably the best studied plus-strand RNA virus, structural alterations of cellular membranes were observed already more than 50 years ago. They were first described as membranous vesicles of heterogeneous size and termed clear vacuoles and U-bodies[42]. Recent electron tomography studies of PV-infected cells by Belov _et al_[26] revealed apparently empty and heterogeneous single membrane-branching tubular structures (100-200 nm diameter) that were detected at very early time points after infection. As infection progresses, these tubular structures developed into DMVs (100-300 nm in diameter), filled with presumably cytoplasmic material (Figure 1D). The very early membrane alterations were derived from the Golgi[26], but PV-induced vesicles were earlier shown to “bud” from the ER in a COPII-dependent manner and to contain lysosomal markers[43,44]. Replicase proteins 2C and 3A as well as nascent viral RNA localize to the outside of both single and double membrane structures[26,44]. A similar 3-D architecture of virus-induced membrane alterations was recently described for CVB3, another member of the Picornaviridae family. By using electron tomography, Limpens _et al_[25] observed interconnected single membrane tubular structures (approximately 650 nm in length and approximately 80 nm in diameter) at the onset of infection. These structures transformed into DMVs (approximately 160 nm in diameter) and multimembranous structures by an enwrapping process. Immunofluorescence analyses showed co-localization of CVB3 replicase proteins 3A and 3D with dsRNA[25] during the log phase of viral replication, when predominantly single membrane tubules were present in infected cells. However, the exact localization of viral proteins and RNA with respect to the rearranged membranes remains to be determined. In contrast to picornavirus-infected cells, Knoops _et al_[11] revealed that early after infection SARS-CoV replication factories appear as presumably ER-derived CMs and interconnected DMVs (250 nm in diameter) (Figure 1E), which subsequently merge into vesicle packets (VPs) (1-5 μm in size), most likely by fusion of the DMV outer membranes. Electron tomography analysis showed that SARS-CoV-induced DMVs do not exist as isolated structures, but their outer membrane is continuous to other DMVs, CMs, or the rER, thus explaining the transition from DMVs to VPs[11]. Viral replicase proteins nsp3, 5 and 8 were sporadically found in DMVs, whereas the majority localized to CMs. In contrast, dsRNA a marker for viral replication intermediates, was predominantly found inside DMVs or inside the vesicles of VPs[11]. The DMV interior was devoid of ribosomes, which were found on the outer membranes of DMVs or rER sheets in close proximity to DMVs. Virus budding was detected at the outer membrane of VPs, arguing for spatial organization of the different steps of the SARS-CoV replication cycle[11]. A comparable architecture of virus-induced membrane rearrangements was observed for EAV, a SARS-CoV related virus that also belongs to the order Nidovirales. By means of electron tomography, Knoops _et al_[27] unraveled a reticulovesicular network of interconnected DMVs in EAV-infected cells. However, in contrast to SARS-CoV induced DMVs, those observed for EAV were smaller in diameter (approximately 90 nm), but also exhibited continuous outer DMV membranes. Replicase proteins nsp3 and 9 associated with DMVs and surrounding membranes and as in case of SARS-CoV, dsRNA was prominently found inside DMVs[27]. In addition to the reticulovesicular DMV network, membrane tubules with approximately 43 nm diameter were associated with the EAV capsid protein N and frequently found in close proximity to DMVs[27], thus providing another example for the spatial coordination of replication and assembly steps. Although belonging to the Flaviviridae that includes DENV and WNV, HCV-induced viral replication factories are very different from the InV/spherule type, but more comparable to those of unrelated Picornaviridae or Coronaviridae. Electron tomography of HCV-infected cells conducted in our laboratory, revealed ER-derived DMVs as the predominant structures, which partially develop into MMVs at later stages of infection[28] (Figure 1F). HCV-induced DMVs have an average diameter of approximately 150 nm. Around 50% of DMVs were linked to the ER via the outer membrane giving rise to a neck-like structure, but later on DMVs seem to detach from their donor membrane. Only a small subset (approximately 10%) exhibited a pore-like opening to the cytoplasm, whereas the vast majority showed “sealed” inner and outer membranes.
By using correlative-light-EM, DMVs were shown to contain the viral replicase protein non-structural protein (NS)5A, which co-localizes with dsRNA as shown by fluorescence microscopy[28]. However, direct detection of the HCV replication site at the ultrastructural level was not successful. DMVs localized in close proximity to the rER and LDs, an important organelle for HCV assembly[13], arguing for a compartmentalization of viral RNA translation, replication and assembly steps[28].
TOPOLOGY OF PLUS-STRAND RNA VIRUS REPLICATION SITES
In order to identify viral replication sites, various methods have been applied to detect viral RNA at the ultrastructural level. Initial studies employed metabolic radiolabeling of nascent viral RNA with 3H-labeled uridine or adenosine and subsequent EM-based detection by autoradiography using film emulsions[29,33]. Others applied in situ hybridization methods by using nucleic acid probes and detection via colloidal gold particles[45], labeling of dsRNA replication intermediates by using a dsRNA-specific antibody[10,11,27,41], or antibody-based detection of bromouridine, incorporated into nascent viral RNA[24,26,30]. Importantly, for identification of viral replication sites the detection of metabolically labeled viral RNA is the only reliable method; hybridization techniques do not discriminate between replicating RNA or RNA used e.g., for translation or virion formation. The same applies to detection of viral RNA with the dsRNA-specific antibody that can bind to RNA structures present in genomes of plus-strand RNA viruses, thus excluding specificity for detection of active viral RNA replication. Indeed, by using the incorporation of ethynyl-uridine (EU) into actively replicating coronavirus RNA and subsequent detection via click-chemistry, Hagemeijer _et al_[46] observed a loss of overlap of the EU- signal with dsRNA labeling during the course of infection. For replication factories of the InV/spherule type it is commonly accepted that viral replicase complexes reside on the invaginated membrane and thus, RNA replication takes place in the spherule lumen[9,10,24,30,36,41,47] (Figure 2A). The neck-like connection to the cytoplasm allows import of all required metabolites (e.g., ribonucleotides) and export of newly synthesized RNA destined for translation or packaging into the capsid. By using bromouridine labeling of nascent viral RNA, this assumption has been confirmed for RUBV[36] alphaviruses[30], FHV[24] and brome mosaic virus (BMV)[47]. However, identification of flavivirus replication sites (DENV and WNV) up to now relies only on the immuno-detection of dsRNA[10,41]. DMV-type replication factories exhibit a topologically more complex situation, as both membranes of the DMV are often sealed and no connection to the cytosol is obvious[11,25-28]. Hence, for this group of plus-strand RNA viruses different topologies of replication sites are currently proposed (Figure 2B). In case of enteroviruses, replication is thought to occur on the cytoplasmic side of single and double-membrane structures[26,48]. However, the fact that during the log phase of viral replication predominantly single membrane tubules are found in PV-or CVB3-infected cells[25,26] suggests that active enteroviral replicase complexes primarily reside on these membrane structures. In this case one might speculate that RNA replication ceases after membrane enwrapping and DMV sealing, but this remains to be determined. In case of SARS-CoV and EAV, the bulk of viral RNA is found inside the lumen of DMVs as determined by dsRNA labeling[11,27]. Moreover, nascent EAV and mouse hepatitis virus (MHV) RNA is associated with DMV membranes as shown by bromouridine labeling[49,50] arguing that DMVs are sites of viral replication. However, if replication takes place inside the DMV lumen, it is unclear how import of metabolites and export of viral RNA occurs. Even with high resolution electron tomography, neck-like membrane discontinuities of approximately 10 nm, as observed for InV/spherule like replication factories, have not been found[11,27]. Thus, proteinaceous channels or transporters might be responsible for linking DMV interior to the cytosol. In fact, proteins such as the ER translocon complex or the mitochondrial transporter outer membrane and transporter inner membrane complexes mediating transport across one or two membranes, respectively (reviewed in[51,52]), have not been visualized by electron tomography so far. Thus, an up to now unidentified enzymatic function of viral proteins containing multiple transmembrane passages and/or recruitment of a cellular factor executing this transport function is in principle possible.
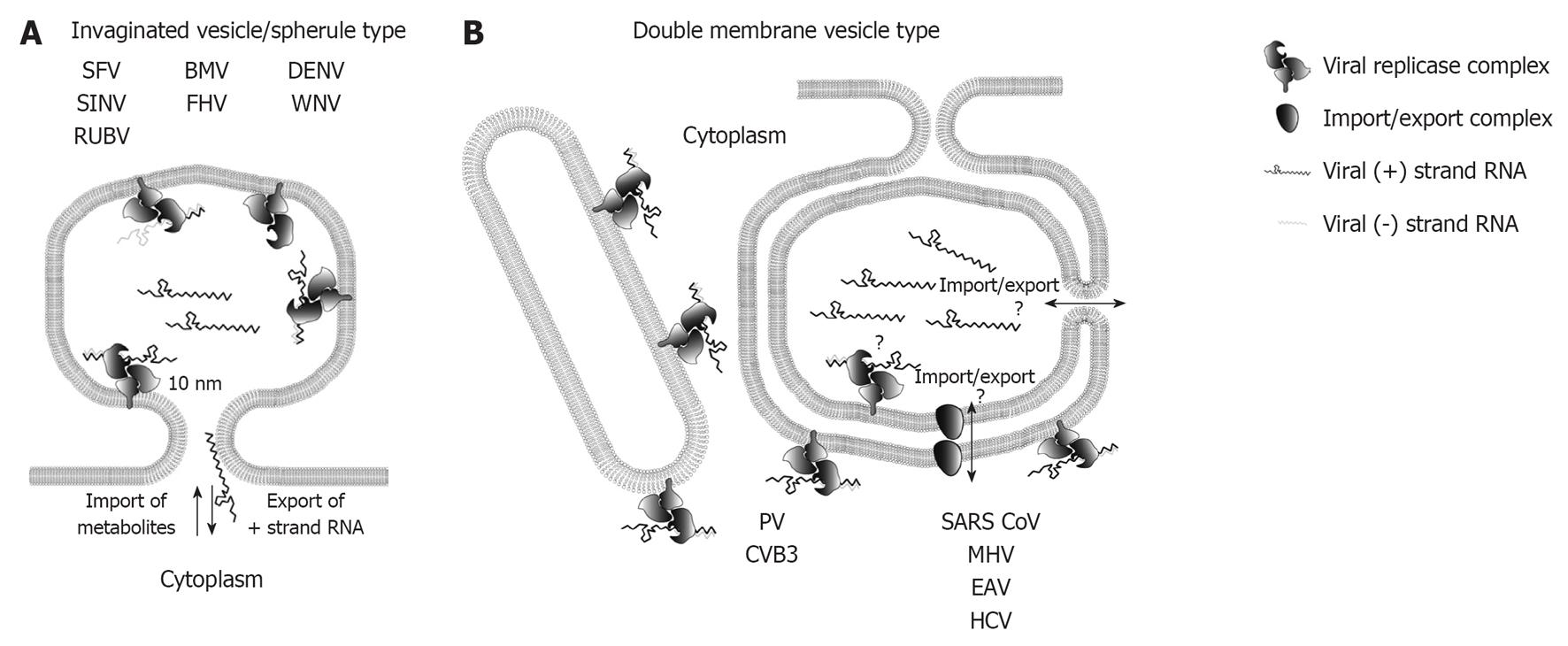
Figure 2 Topology of plus-strand RNA virus replication sites. A: RNA replication of plus-strand RNA viruses inducing invaginated vesicles/spherules is thought to occur in the spherule lumen. Examples of viruses inducing this type of remodeled membranes are denoted. The neck like opening to the cytoplasm allows export of progeny RNA destined for translation or packaging, as well as import of metabolites required for RNA replication; B: Possible topologies of RNA replication site for double membrane vesicle (DMV)-type factories. Enterovirus replication is thought to take place on the outer membrane of single membrane tubules and DMVs, whereas the exact replication site of nidovirales and hepatitis C virus (HCV) is still unclear as indicated by the question marks. Replication in the DMV lumen would require import and export across two membranes, which could be mediated via a proteinacous transport complex, or via an opening to the cytosol of not yet fully closed DMVs (see main text for details). SFV: Semliki Forrest virus; SINV: Sindbis virus; RUBV: Rubella virus; BMV: Brome mosaic virus; FHV: Flock-house virus; DENV: Dengue virus; WNV: West Nile virus; PV: Poliovirus; CVB3: Coxsackievirus B3; SARS-CoV: Severe acute respiratory syndrome coronavirus; MHV: Mouse hepatitis virus; EAV: Equine arterivirus.
Another hypothesis is discussed for HCV-induced DMVs, which accumulate during the log phase of viral RNA replication[28]. Only a small fraction of DMVs has an opening to the cytoplasm, whereas the majority exhibits entirely closed membranes. It is speculated that replication might occur in the interior of DMVs as long as they are linked to the cytosol, but upon membrane sealing DMVs would contain dead end replication complexes that are no longer active. Apart from this possibility, the other models described above also apply to HCV, but further studies are required to define the exact site of RNA replication.
MECHANISMS OF MEMBRANE ALTERATIONS INDUCED BY VIRAL PROTEINS
Given the highly complex architecture of plus-strand RNA virus replication factories on one hand and the small genetic coding capacity of these viruses on the other hand, it is obvious that membrane alterations are induced by the concerted action of viral and cellular factors. In this section we will discuss the intrinsic membrane-active properties of viral proteins; host cell factors contributing to formation of replication factories are summarized in section 6 of this article. In principle, membrane bending can be achieved by (1) local alterations of membrane lipid composition; or (2) asymmetric interaction of proteins with membranes (Figure 3). The latter includes scaffolding of the membrane by peripheral proteins, insertion of asymmetric proteins or protein complexes into the membrane, insertion of amphipathic helices into one leaflet of the lipid bilayer or interactions of the membrane with the cytoskeleton (reviewed in[53-55]) (Figure 3C-H). Negative membrane curvature is predominant for the InV/spherule like replication factories (Figure 3A), with spherules bending the membrane away from the cytoplasm, whereas DMV-like replication factories exhibit positive membrane curvature, i.e., the outer DMV membrane is bent towards the cytosol (Figure 3B). However, regions of positive membrane curvature are also present in neck like openings of spherules (Figure 3A) and the inner membranes of DMVs exhibit negative membrane curvature (Figure 3B), respectively, highlighting the complexity of membrane bending events in the induction of plus-strand RNA virus replication factories.
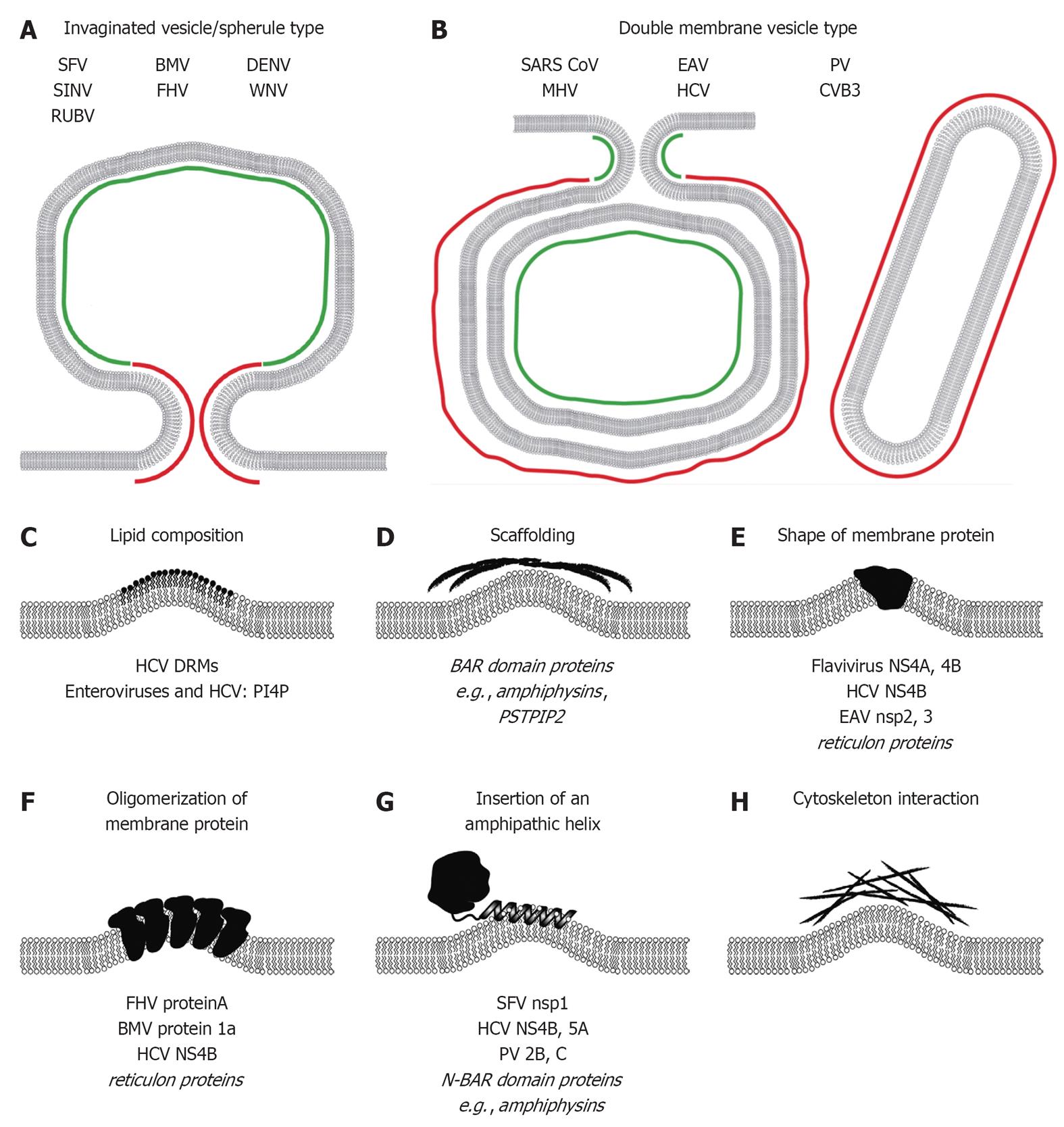
Figure 3 Mechanisms responsible for induction of membrane curvature likely contributing to formation of plus-strand RNA virus replication factories. Positive and negative membrane curvature for invaginated vesicle/spherule type (A) and double membrane vesicle type (B) replication factories is indicated in red and green, respectively. Mechanisms of membrane modifications/bending are schematically depicted in (C-H). Examples of viral and cellular (in italics) proteins are given at the bottom of each panel (see main text for details). SFV: Semliki Forrest virus; SINV: Sindbis virus; RUBV: Rubella virus; BMV: Brome mosaic virus; FHV: Flock-house virus; DENV: Dengue virus; WNV: West Nile virus; SARS-CoV: Severe acute respiratory syndrome coronavirus; MHV: Mouse hepatitis virus; EAV: Equine arterivirus; HCV: Hepatitis C virus; PV: Poliovirus; CVB3: Coxsackievirus B3; DRM: Detergent-resistant-membrane; BAR: Bin-Amphiphysin-Rvs; PSTPIP2: Proline-serine-threonine phosphatase interacting protein 2; NS4A: Non-structural protein 4A; NS4B: Non-structural protein 4B; N-BAR: N-terminal amphipathic helix containing BAR; PI4P: Phosphatidylinositol-4-phosphate.
Several viral proteins were identified that play crucial roles in the induction of membrane rearrangements. For instance, the membrane-associated protein nsp1 of alphaviruses interacts with anionic phospholipids via an amphipathic helix (AH) (Figure 3G)[56] and is additionally tethered to membranes via a palmitic acid residue in the carboxy-terminal region of the protein[57]. AH-mediated nsp1 membrane association proved crucial for SFV replication[58], whereas palmitoylation mutants could be rescued by second site compensatory muations _in vitro_[59] but are attenuated _in vivo_[60]. Nonetheless, only expression of the P123 protein precursor induced spherule-like structures as seen in SFV-infected cells[61], demonstrating the need of nsp1 to recruit and interact with other viral and/or cellular proteins to induce spherule-like membrane rearrangements. The FHV replicase factor protein A exhibits binding affinity to specific anionic phospholipids, namely phosphatidic acid, phosphatidylglycerol and cardiolipin[62], which are enriched in mitochondrial membranes. Protein A is inserted into the OMM by an N-terminal mitochondria-targeting transmembrane sequence[63]. Self-interaction of protein A is required for FHV replication and mediated by multiple domains (Figure 3F)[64]. Nevertheless, protein A is not sufficient to induce spherule like membrane invaginations in the OMM, which requires in addition a replication-competent template RNA and protein A’s polymerase activity[65]. Interestingly, these requirements do not apply to the FHV-related plant virus BMV, for which expression of protein 1a is sufficient to induce spherule-like membrane invaginations[47]. All flaviviral proteins are associated directly or indirectly with membranes either by amphipathic helices or transmembrane domains or both (Figure 3E-G). Fully processed DENV and WNV NS4A is sufficient to induce rearrangements of the ER membrane[66,67], although membrane structures induced by sole NS4A expression are morphologically distinct from those induced in infected cells. Studies of the membrane topology of DENV NS4A revealed an ER luminal helix in the central region of the protein, lying in plane in the luminal membrane leaflet[66]. This topology is compatible with the negative curvature of InVs, arguing that NS4A increases the surface area of the ER membrane on the luminal side. However, the role of NS4A self-interaction and a possible interplay with other viral (e.g., NS4B) and cellular factors, in membrane remodeling remain to be determined. Also for HCV that in contrast to DENV and WNV induces DMV-type replication factories, membrane remodeling activity has been attributed to viral proteins, most prominently to NS4B. It has been proposed that expression of this protein is sufficient to induce the membranous HCV replication compartment (the “membranous web”)[68]. NS4B is a highly hydrophobic protein that contains at least four transmembrane passages[69]. It is palmitoylated at its C-terminus[70] and in addition contains multiple AHs in its N-and C-terminal domains[71-73] (Figure 3E-G). The membrane-associated C-terminal domain of NS4B or the AH within it have been reported to alter membrane integrity (Figure 3G)[74,75], corroborating membrane remodeling activity of NS4B. In addition, NS4B self-interacts in a homo- and heterotypic fashion[76], which is required for HCV RNA replication[76,77] (Figure 3F). Interestingly, mutations in NS4B that inhibit HCV RNA replication also alter NS4B self-interaction and morphology of HCV-induced DMVs[77], implying a functional relationship between DMV morphology and HCV RNA replication. Inspite of the membrane remodeling activity of NS4B, we recently found that only an NS3-5A polyprotein fragment is sufficient to induce DMV structures with morphological similarity to those observed in HCV-infected cells[28]. In fact, MMVs and a small number of DMVs were observed in cells over-expressing just NS5A, which is a dimeric and eventually oligomeric replicase factor with an N-terminal AH[78-82] (Figure 3G). Based on these observations we assume that a concerted action of HCV replicase factors is required for the biogenesis of the membranous web. Similar observations have been made for Nidovirales. For instance, coronavirus nsp3, 4 and 6 contain multiple membrane-spanning domains[83-85]. These proteins appear to play a central role in membrane remodeling (Figure 3E), because mutations e.g., in nsp4 exhibit impaired viral replication and altered DMV morphology[86]. Nsp3 of the related EAV also contains multiple transmembrane segments and is another key player in the induction of DMVs. Expression of nsp2-3 suffices to induce DMVs[87] and their morphology is dramatically altered by nsp3 mutations affecting an ER luminal loop[88].
In case of enteroviruses, proteins 2B, 2C or 3A are membrane-associated via transmembrane passages or AHs[89-91] and the expression of 2BC either alone[92] or in concert with 3A[93] induces structures similar to those in infected cells. Interestingly, expression of only 2C induces more dramatic membrane remodeling including tubular membrane swirls inside a highly dilated ER[92], implying a regulatory role of 2B in 2C membrane remodeling. Taken together these data suggest that membrane-associated proteins of plus-strand RNA viruses have intrinsic membrane remodeling properties. However, in most cases more than one viral factor, eventually in concert with recruited cellular proteins are involved in generation of viral replication factories. Further studies using e.g., recombinant viral proteins and model membranes are required to elucidate the intrinsic membrane remodeling properties of these viral proteins.
POSSIBLE ROLE OF AUTOPHAGY IN THE FORMATION OF DMV LIKE REPLICATION FACTORIES
Autophagy is an evolutionary conserved catabolic mechanism for degradation of long-lived organelles and cytoplasmic material and is crucial for cell homeostasis (reviewed in[94]). Virus-host co-evolution has shaped multiple mechanisms involving autophagy that either promote or restrict viral replication (reviewed in[95]). Due to morphological similarities between DMV-like replication factories and the also double-membrane nature of autophagosomes, it has been suggested that autophagy plays a role in biogenesis of viral replication compartments of this type as induced by enteroviruses[44,96,97], coronaviruses[98,99] and HCV[100,101]. Indeed, lipidation of microtubule - associated protein 1 light chain 3 (LC3), a key event in the induction of autophagy, was observed after enterovirus infection[96,97,102] or over-expression of 2BC[102]. Interestingly, knock-down of central autophagy components or pharmacological inhibition of the pathway decreased viral replication only slightly, whereas generation of progeny virus was clearly reduced arguing that autophagy contributes primarily to virus assembly and release[103]. In case of PV, electron tomography studies of infected cells revealed primarily single membrane structures during the log phase of viral replication, which subsequently developed into DMVs by collapsing and/or enwrapping events[26]. These data argue against a central role of autophagy per se in PV replication, although some factors of this pathway might contribute to heterogeneity of vesicular structures at later stages of infection. Similar observations have been made for the Coronaviridae. MHV infection induces lipidation of LC3 that co-localizes with nsp2/3 in infected cells[99]. Moreover, the non-lipidated form of LC3 is prominently found on isolated membrane fractions from MHV-infected cells, together with components (e.g., EDEM1) of the ER-associated degradation pathway[99]. However, functional studies based on knockouts of central autophagy factors led to contradictory results. In one study, ATG5 knockout decreased virus propagation dramatically[98], whereas others found that MHV replication is independent of ATG5[104] or ATG7[99]. Hence, a central role of the complete autophagy pathway in generation of coronavirus DMVs seems unlikely, although some single factors such as LC3 might add to it. Conflicting observations concerning a role of autophagy in virus propagation have also been made in case of HCV. One study claims that autophagy is implicated in translation of incoming viral RNA, but is dispensable for RNA replication[105]. Other studies suggest that autophagosomes are sites of active replication[101] or promote assembly and release of progeny virus[106]. In addition, a recent study showed an autophagy-mediated down-regulation of innate immune response[107]. In this case, knock-down of autophagy components reduces HCV RNA replication due to a stronger innate immune response. The reasons for these discrepant results are not known, but might be due, at least in part, to the use of different Huh7 cell clones, which are known to differ in their capacity to mount innate antiviral defenses. Although LC3 lipidation is observed upon HCV infection[105] and the protein is associated with HCV membrane fractions[100], EM-based studies of membrane remodeling events argues for a role of autophagy in formation of MMVs eventually as part of a cellular stress response induced by massive membrane alterations[28]. Taken together, the role of autophagy in biogenesis of DMV like replication factories remains rather elusive. Single components of the conventional cellular autophagy are possibly involved, whereas a direct contribution of the complete pathway in generation of DMV replication compartments is rather unlikely. Finally, autophagy might be an epiphenomenom, being activated as a cellular stress response to tremendous amounts of virus induced cytoplasmic membrane alterations, engaged in cell homeostasis and survival during viral infection.
PLUS-STRAND RNA VIRUS SUBVERSION OF CELLULAR MEMBRANE BIOLOGY
Since plus-strand RNA viruses induce massive remodeling of cytoplasmic membranes, but most often have very limited genetic coding capacity it is not surprising that these viruses utilize membrane-active host cell factors and exploit cellular pathways involved in membrane homeostasis.
Viral utilization of co-opted membrane-active proteins of the host cell
Enteroviruses and the plant virus BMV co-opt cellular reticulon proteins, which are required for membrane remodeling and viral RNA replication[108,109]. The evolutionary conserved reticulon protein family is characterized by a common reticulon homology domain (RHD) involved in shaping the ER by inducing and stabilizing highly curved ER tubules[110,111]. Morphogenic properties can be attributed to elongated hydrophobic, partially membrane-spanning hairpin structures within the RHD[112], which in concert with its oligomerization properties[113] increases the surface on the cytoplasmic membrane leaflet, thereby inducing positive curvature (Figure 3E and F). Reticulon 3 has been shown to directly interact with the enterovirus 2C protein[108] and is thus likely engaged in induction and/or stabilization of positive membrane curvature of enterovirus replication factories. BMV replicase protein 1a directly binds to and recruits reticulon proteins to spherule-like membrane invaginations and might stabilize positive membrane curvature in neck-like openings to the cytoplasm or facilitate expansion of the spherule volume by partially neutralizing overall negative membrane curvature[109]. It remains to be determined whether reticulon proteins are co-opted also by other plus-strand RNA viruses, especially those deriving their replication compartments from the ER. Another example of cellular membrane-shaping proteins are amphiphysins that are involved in formation of alphavirus replication factories[114]. These Bin-Amphiphysin-Rvs (BAR) domain containing proteins play pivotal roles in endocytosis and intracellular membrane trafficking (reviewed in[115]). Structural analyses revealed dimerization of the BAR domain mediated by helical coiled-coil interactions[116], giving rise to a concave banana-shaped structure. This complex has a positively charged inner surface, which interacts with negatively charged membrane phospholipids (reviewed in[117]). Thus, BAR domain-containing proteins sense and stabilize membrane curvature by scaffolding mechanisms, to which in case of N-terminal amphipathic helix containing BAR proteins membrane insertion of N-terminal amphipathic helices contributes (Figure 3D and G). Alphavirus nsp3 binds to SH3 domains in amphiphysin via a proline-rich sequence and mediates the recruitment to viral replication factories. Knock-down experiments proved an important role of amphiphysins in alphavirus replication[114]. Although their contribution to membrane remodeling during alphavirus infection remains to be discovered, one could envisage a similar mechanism as proposed for BMV and reticulon proteins. Proline-serine-threonine phosphatase interacting protein 2 (PSTPIP2) also belongs to the BAR protein superfamily (Figure 3D) and was recently shown to be involved in membrane alterations induced by HCV[118]. PSTPIP2 binds to NS4B and NS5A and thereby is recruited to the membranous replication compartment[118]. In fact, knockdown experiments showed the crucial role of PSTPIP2 in HCV membrane remodeling and RNA replication[118]. Interestingly, upon over-expression PSTPIP2 induces cytoplasmic tubular membranes[118], highlighting its ability to induce positive membrane curvature. Hence, PSTPIP2 is probably engaged in inducing and/or stabilizing positive membrane curvature of HCV replication factories. Another mechanism by which viruses can rearrange intracellular membranes has become evident for enteroviruses. PV appears to hijack components of the cellular secretory pathway, explaining why PV replication is sensitive to brefeldin A (BFA) treatment[119]. Viral proteins 3A and 3CD recruit the ADP ribosylation factor (Arf)-GEFs (GTP exchange factors) Golgi-specific BFA-resistance guanine nucleotide-exchange factor 1 (GBF1) and BFA-inhibited guanine nucleotide-exchange protein 1/2 (BIG1/2) to replication sites, leading to elevated levels of activated Arf-GTP in infected cells[120]. Arf proteins are central regulators of membrane dynamics and vesicle budding in the secretory pathway. Upon activation by Arf-GEFs, the GTP-bound form exerts its function on target membranes by recruitment of effector proteins such as lipid modifying enzymes and coat complexes, thus facilitating vesicle budding (reviewed in[121,122]). GBF1-mediated Arf activation is crucial for PV replication, although typical membrane alterations can be observed in BFA-treated cells that over-express viral proteins[123]. Nevertheless, PV replication factories of BFA-treated cells were shown to be inactive, implying a functional difference of remodeled membranes independent from morphological abnormalities[123]. Hence, PV subversion of cellular Arf-GEFs is important for maintenance of functional replication factories, rather than primary membrane remodeling. In summary, plus-strand RNA viruses recruit and utilize membrane-active host cell proteins in order to generate and/or maintain replication factories. Donor membrane usage defines engaged cellular factors, as enteroviruses employ Arf-GEFs from the Golgi, alphaviruses subvert amphiphysins localized at the plasma membrane and enteroviruses and BMV utilize ER-resident reticulon proteins (Figure 4).
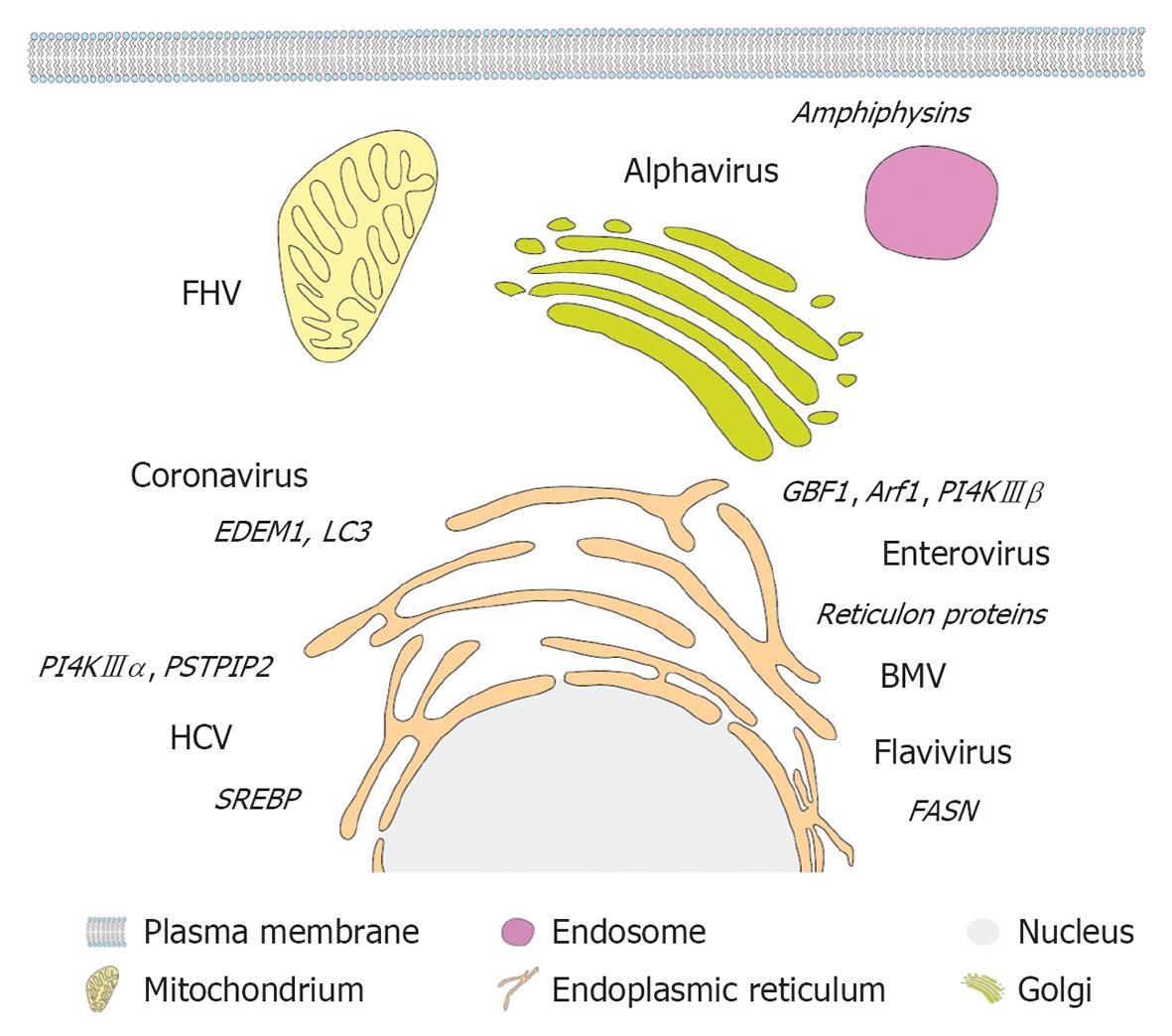
Figure 4 Primary membrane source and host cell factors subverted by different plus-strand RNA viruses. A eukaryotic cell and (endo)membrane organelles are depicted schematically as indicated on the bottom. Viruses are displayed next to their primary membrane source organelle and recruited host cell factors involved in lipogenesis and membrane remodeling are denoted in italics adjacent to each virus name. FHV: Flock-house virus; PSTPIP2: Proline-serine-threonine phosphatase interacting protein 2; HCV: Hepatitis C virus; SREBP: Sterol regulatory element-binding protein; PI4K: Phosphatidylinositol-4-kinase; GBF1: Golgi-specific BFA-resistance guanine nucleotide-exchange factor 1; BMV: Brome mosaic virus; FASN: Fatty acid synthase.
Viral exploitation of pathways regulating homeostasis of cellular membranes
Apart from remodeling existing intracellular membranes, virus infection often induces de novo lipid and membrane biosynthesis in order to increase membrane surface area, which is required for the formation of viral replication factories. Indeed, alteration of cellular lipid homeostasis and virus-induced lipogenesis has been reported for a broad range of plus-strand RNA viruses, including FHV[124], BMV[125], RUBV[126], DENV[127,128], WNV[129], enteroviruses[130] and HCV[131-133]. FHV mainly stimulates glycerophospholipid metabolism[124] and inhibition of phospholipid synthesis leads to destabilization of protein A and decreases viral replication[134]. In case of DENV, high-throughput analysis revealed multiple changes in the cellular lipidome of infected cells, including specific up or down regulation of main structural lipids species, highlighting the link to virus-induced membrane remodeling[128]. Additionally, DENV NS3 recruits fatty acid synthase (FASN), which catalyzes the rate limiting step in lipid biosynthesis at the sites of viral replication[127]. Thus, DENV appears to subvert FASN for de novo lipid synthesis in order to generate new membranes for the formation of viral replication factories. This feature is also observed for the closely related flavivirus WNV[129], stressing the impact of this particular virus host interaction to generate flavivirus replication factories.
HCV induces lipogenesis via the sterol regulatory element-binding protein (SREBP) pathway[131]. SREBPs are major transcription factors for expression of genes required for lipid biosynthesis. SREBPs reside as inactive membrane-bound precursors in the ER, which upon stimulation traffic to the Golgi. There they are proteolytically activated by site 1 protease (S1P) and S2P and subsequently stimulate gene transcription (reviewed in[135]). Proteolytic cleavage of SREBP and transactivating phosphorylation has been observed in HCV-infected cells or upon over-expression of NS4B, leading to elevated levels of transcripts involved in lipogenesis such as FASN[131,136]. However, by using S1P-specific inhibitors SREBP-mediated lipogenesis was found to be dispensable for HCV replication but required for assembly and release of progeny virus[137]. In addition, the HCV replicase complex was shown to reside in detergent-resistant membranes[138]. These membrane micro domains, designated lipid rafts, are enriched for cholesterol, sphingolipids and certain proteins and they form nanoscale-ordered protein-lipid assemblies (reviewed in[139]). Sphingolipid synthesis is stimulated upon and required for HCV replication[140] and it was shown that the NS5B RdRP is activated by sphingomyelin in a genotype-specific manner[141]. A shared feature of plus-strand RNA viruses inducing DMV-like replication factories is their dependence on members of the phosphatidylinositol-4-kinase (PI4K) family and their product, phosphatidylinositol-4-phosphate (PI4P). Both enteroviruses and HCV rely on PI4P for functional replication factories, which is generated by recruitment of PI4KIIIβ in case of enteroviruses[130] and PI4KIIIα in case of HCV[142]. In non-infected cells PI4P localizes to the Golgi and the inner leaflet of the plasma membrane, where it fulfills important functions by providing “signatures” to distinct membrane compartments and by recruiting multiple factors involved in vesicle budding and lipid biosynthesis[143,144]. Subversion of PI4KIIIβ by enteroviruses is executed via 3A-GBF1 interaction, activating Arf that in turn recruits PI4KIIIβ to viral replication factories[130]. Locally elevated PI4P levels allow specific binding of the viral 3D RdRp to the membrane favoring viral replication. Knock-down as well as pharmacological inhibition of PI4KIIIβ dramatically decreases enteroviral replication[130].
In case of HCV, the replicase proteins NS5A and NS5B directly interact with PI4KIIIα, which is thereby recruited to HCV replication sites[142]. Importantly, in HCV-infected cells PI4P that is usually enriched at the Golgi and the plasma membrane, is prominently enriched at the ER-derived sites of HCV replication[142,145]. Numerous siRNA screens identified PI4KIIIα as a major host dependency factor for HCV replication[142,146-149], and pharmacological inhibition of PI4KIIIα activity efficiently blocks viral replication[145]. In the absence of PI4KIIIα, expression of HCV proteins NS3-5B still induces DMVs, but these vesicles are smaller in diameter, very homogeneous and tend to cluster[142]. This morphological change of the overall structure of the membranous web correlates with impaired replication although DMV morphology per se is only moderately affected. This phenomenon is reminiscent to what has been shown for BFA-treated cells over-expressing PV proteins[123]. These findings argue for a role of PI4P in recruiting viral factors such as PV 3D RdRp or so far unidentified cellular proteins, rather than acting as a structural lipid building up membranous replication factories. Importantly, the closely HCV-related flaviviruses DENV and WNV, which form replication factories of the InV/sperule type, do not depend on PI4KIIIα/β or PI4P[129,142], highlighting a functional relationship between PI4P and DMV-like replication factories. Therefore it would be very interesting to investigate the dependence of EAV and coronavirus replication on PI4P. We note that for the latter PI4KIIIβ was shown to be required for virus entry[150].
Taken together, the strong dependence of positive strand RNA viruses on cellular membrane-active proteins and on pathways implicated in cellular membrane homeostasis, renders those host cells factors very attractive targets for future antiviral drug development.
CONCLUSION
Although important discoveries on the 3D architecture of plus-strand RNA virus replication factories have been made, current knowledge is largely descriptive and important information about mechanisms is missing. For instance, the exact topology of RNA replication sites for DMV-type replication factories is elusive. Identification of these sites will require novel experimental techniques such as metabolic labeling of nascent viral RNA and its visualization by using microscopy methods with high resolution and specificity. Likewise, membrane remodeling events responsible for the biogenesis of replication factories are mostly unknown. They are probably mediated by a complex interplay of viral and cellular factors, but precise contributions of individual factors and their temporal and spatial coordination remain to be discovered. Studying the impact of single proteins or combinations thereof on model membranes in vitro or on membranes in cellulo by using correlative light-EM based methods as described recently for studies of membrane remodeling events during endocytosis[151] are possible ways to address this topic. Furthermore, determining the proteome and lipidome of purified viral replication factories will shed light on viral and host cell factors involved in biogenesis and activity of these membranous compartments. This approach has been used with great success for small intracellular vesicles such as COPI vesicles or neuronal transport vesicles[152,153] providing insight into individual membranous structures with unprecedented detail. Another emerging field is the specific in-membrane interaction of proteins with certain lipids, as recently shown for COPI machinery protein p24 and the sphingolipid SM 18 being implicated in regulation of COPI vesicle budding[154]. The tight membrane association of proteins of plus-strand RNA viruses suggests that such specific protein-lipid interactions also occur for this large virus group. Finally, time-resolved (imaging) techniques might shed light onto coupling of viral RNA translation and replication as recently shown for HCV[155] and furthermore onto transport processes of viral and cellular components inside and outside replication factories. These studies will be instrumental to integrate their functional role into the complete viral replication cycle.
ACKNOWLEDGMENTS
We want to express our gratitude to Ahlquist P, Belov G, Risco C, Romero-Brey I, Snijder E and Welsch S for providing EM reconstruction images and apologize to all colleagues whose work could not be cited due to space limitations.
Footnotes
P- Reviewers Chi-Ho C, Shih WL S- Editor Gou SX L- Editor A E- Editor Zheng XM
References
| 29. | Grimley PM, Berezesky IK, Friedman RM. Cytoplasmic structures associated with an arbovirus infection: loci of viral ribonucleic acid synthesis. J Virol. 1968;2:1326-1338. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 33. | Friedman RM, Levin JG, Grimley PM, Berezesky IK. Membrane-associated replication complex in arbovirus infection. J Virol. 1972;10:504-515. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 34. | Grimley PM, Levin JG, Berezesky IK, Friedman RM. Specific membranous structures associated with the replication of group A arboviruses. J Virol. 1972;10:492-503. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 36. | Kujala P, Ahola T, Ehsani N, Auvinen P, Vihinen H, Kääriäinen L. Intracellular distribution of rubella virus nonstructural protein P150. J Virol. 1999;73:7805-7811. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 39. | Fontana J, Tzeng WP, Calderita G, Fraile-Ramos A, Frey TK, Risco C. Novel replication complex architecture in rubella replicon-transfected cells. Cell Microbiol. 2007;9:875-890. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 41. | Westaway EG, Mackenzie JM, Kenney MT, Jones MK, Khromykh AA. Ultrastructure of Kunjin virus-infected cells: colocalization of NS1 and NS3 with double-stranded RNA, and of NS2B with NS3, in virus-induced membrane structures. J Virol. 1997;71:6650-6661. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 44. | Schlegel A, Giddings TH, Ladinsky MS, Kirkegaard K. Cellular origin and ultrastructure of membranes induced during poliovirus infection. J Virol. 1996;70:6576-6588. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 49. | Pedersen KW, van der Meer Y, Roos N, Snijder EJ. Open reading frame 1a-encoded subunits of the arterivirus replicase induce endoplasmic reticulum-derived double-membrane vesicles which carry the viral replication complex. J Virol. 1999;73:2016-2026. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 85. | Oostra M, Hagemeijer MC, van Gent M, Bekker CP, te Lintelo EG, Rottier PJ, de Haan CA. Topology and membrane anchoring of the coronavirus replication complex: not all hydrophobic domains of nsp3 and nsp6 are membrane spanning. J Virol. 2008;82:12392-12405. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 87. | Snijder EJ, van Tol H, Roos N, Pedersen KW. Non-structural proteins 2 and 3 interact to modify host cell membranes during the formation of the arterivirus replication complex. J Gen Virol. 2001;82:985-994. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 89. | van Kuppeveld FJ, Galama JM, Zoll J, van den Hurk PJ, Melchers WJ. Coxsackie B3 virus protein 2B contains cationic amphipathic helix that is required for viral RNA replication. J Virol. 1996;70:3876-3886. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 99. | Reggiori F, Monastyrska I, Verheije MH, Calì T, Ulasli M, Bianchi S, Bernasconi R, de Haan CA, Molinari M. Coronaviruses Hijack the LC3-I-positive EDEMosomes, ER-derived vesicles exporting short-lived ERAD regulators, for replication. Cell Host Microbe. 2010;7:500-508. [PubMed] [DOI] [Cited in This Article: ] [Cited by in Crossref: 270] [Cited by in F6Publishing: 320] [Article Influence: 22.9] [Reference Citation Analysis (0)] |
|---|
| 104. | Zhao Z, Thackray LB, Miller BC, Lynn TM, Becker MM, Ward E, Mizushima NN, Denison MR, Virgin HW. Coronavirus replication does not require the autophagy gene ATG5. Autophagy. 2007;3:581-585. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 119. | Maynell LA, Kirkegaard K, Klymkowsky MW. Inhibition of poliovirus RNA synthesis by brefeldin A. J Virol. 1992;66:1985-1994. [PubMed] [DOI] [Cited in This Article: ] |
|---|