Acute suppression of VLDL1 secretion rate by insulin is associated with hepatic fat content and insulin resistance (original) (raw)
Introduction
An increased plasma level of triacylglycerol (TG)-rich VLDL is not only a common characteristic of the dyslipidaemia associated with insulin resistance and type 2 diabetes but seems to be the central pathophysiological feature of the abnormal lipid profile [1]. Emerging evidence suggests that the fundamental defect is an overproduction of large VLDL particles (VLDL1), which initiates a sequence of lipoprotein changes, including higher levels of remnant particles and smaller LDL and lower levels of HDL particles [1]. Importantly, these atherogenic lipid abnormalities appear years before type 2 diabetes is diagnosed and may explain the ‘ticking clock’ hypothesis of cardiovascular disease [2]. Thus, pinpointing the mechanisms for the overproduction of VLDL1 particles in diabetic dyslipidaemia is of key importance.
The assembly of VLDL1 involves a step-wise loading of nascent VLDL with core lipids during biosynthesis [3, 4]. This results in a triacylglycerol-poor VLDL particle that is either secreted or further lipidated to form mature triacylglycerol-rich VLDL1 [5]. The importance of hepatic TG for the assembly and secretion of VLDL1 is supported by our recent observations in vivo demonstrating that the secretion of VLDL1 increases with increasing hepatic concentrations of lipids [6]. Interestingly, hepatic fat content is a major determinant of insulin resistance, leading to glucose overproduction, the hallmark of type 2 diabetes [7–9]. The coexistence of hepatic insulin resistance and steatosis even independently of diabetes and obesity is consistent with the tenet that hepatic steatosis precedes hepatic insulin resistance covering both glucose and lipid metabolism [10]. This is further supported by the reports that hepatic steatosis predicts diabetes [11]. Normally, insulin suppresses VLDL1 formation by controlling the substrate influxes for liver TG synthesis [12–15]. Consequently, an imbalance between insulin-sensitive and insulin-resistant action sites will be reflected in increased liver fat content and secondarily in increased VLDL1 secretion. In addition, emerging results indicate that insulin has a direct suppressive effect on the production of VLDL1 in the liver, independently of the availability of NEFAs [14, 15]. The molecular mechanism behind this effect of insulin remains to be elucidated, but tentative mechanisms have been reviewed [1, 15].
The objective of this study was to test the association between insulin resistance, liver fat and the acute suppression of VLDL1 by insulin.
Methods
Participants
We studied eight men with type 2 diabetes and 12 healthy male participants without diabetes, all of whom were of white European origin. The participants were recruited through advertisements in local newspapers and from a Finnish database, Health 2000 (a population-based cohort). All participants underwent a physical examination and laboratory tests to exclude hepatic diseases (other than hepatic steatosis) and renal, thyroid or haematological diseases. Participants with coronary heart disease, diabetic retinopathy or microalbuminuria, total cholesterol >7.0 mmol/l, TG >5.0 mmol/l or BMI >40 kg/m2 were excluded.
All the participants without diabetes had lipid values representative of the western population; one had impaired glucose tolerance, according to WHO criteria [16]. The diagnosis of type 2 diabetes was based on two separate glucose tolerance tests according to WHO criteria [16] or on the use of oral glucose-lowering medication. The age of onset of the type 2 diabetes was required to be at least 35 years. None of the participants with type 2 diabetes was taking lipid-lowering therapy or insulin. Three participants with type 2 diabetes and one participant without diabetes were taking acetylsalicylic acid. Two patients with type 2 diabetes were treated with diet alone, one with diet and metformin, two with diet and sulfonylurea, and four with a combination therapy of sulfonylurea, metformin and diet. One patient with type 2 diabetes was using antihypertensive therapy (felodipine). The medications were continued throughout the study. The mean duration of diabetes was 7.0 years (range 0–13 years). The study design was approved by the ethics committee of Helsinki University Central Hospital, and each participant gave written informed consent. All samples were collected in accordance with the Declaration of Helsinki. Nine of the participants with low liver fat and eight of the participants with high liver fat have been presented in previous papers [6, 17]. However, none of the present kinetic data have previously been reported in any form.
Euglycaemic–hyperinsulinaemic clamp studies
The patients were admitted at 0730 hours after an overnight fast. An indwelling cannula was inserted in an antecubital vein for infusions and a second cannula was inserted retrogradely into a heated hand vein to obtain arterialised venous blood for sampling. At 0 min a primed-continuous infusion of insulin (Actrapid Human; Novo Nordisk, Copenhagen, Denmark) was started (rate of continuous infusion 1 mU min−1 kg−1) (Fig. 1). Plasma glucose was maintained at its fasting level by a variable rate infusion of glucose (200 g/l) [18]. The infusion rate was determined empirically based on plasma glucose measurements, which were performed every 5–10 min from arterialised venous blood. Whole body insulin sensitivity (M value) was calculated from the mean values of the second hour of glucose infusion rate after correcting for changes in the glucose pool size [18] and was expressed per body weight (μmol min−1 kg−1). The insulin infusion continued for 8.5 h (510 min) and the participants remained fasting until 1700 hours.
Fig. 1
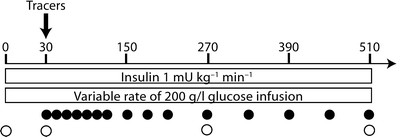
The insulin infusion (1 mU kg−1 min−1) was initiated at 0 min and was continued for 510 min. To keep the plasma glucose levels constant, a 20% glucose infusion was given at a variable rate, determined by plasma glucose measurements taken every 5–10 min. The plasma concentrations of ApoB and TG in the VLDL1 and VLDL2 subfractions were determined at 0, 30, 270 and 510 min (open circles). At 30 min, the stable isotopes [2H3]leucine and [2H5]glycerol were given as a bolus injection, and enrichment in ApoB and TG in VLDL1 and VLDL2 was determined at 15 time-points (closed circles)
Determination of fat compartments and liver fat and biochemical analyses
Subcutaneous and intra-abdominal fat volumes and liver fat content were measured using magnetic resonance imaging and magnetic proton spectroscopy as previously described [6]. Biochemical analyses of TG, NEFAs, cholesterol, insulin and glycerol were performed as previously described [19] and alanine aminotransferase was determined according to European Committee for Clinical Laboratory Standards. HOMA-IR was estimated from insulin (pmol/l)× plasma glucose (mmol/l)/405.
Isolation of lipoproteins, and ApoB and TG turnover protocol
Isolation and analysis of VLDL1 and VLDL2 was performed as described [19]. Particle composition and ApoB mass of the VLDL1 and VLDL2 fractions were determined at 0, 30, 270 and 510 min after the start of the insulin infusion to determine pool sizes (Fig. 1). To estimate turnover of ApoB and TG, a bolus injection of [2H3]leucine and [2H5]glycerol was given at 30 min and blood samples were drawn as previously described [19]. A theoretical problem with using labelled plasma glycerol as tracer for VLDL-TG is that during the insulin infusion, the release of unlabelled glycerol from adipose tissue is reduced, resulting in an increased enrichment of the glycerol derived from plasma. On the other hand, recruitment of hepatic lipid storage as a source of VLDL-TG synthesis together with increased production of glycerol from glucose will probably compensate for this by increasing the unlabelled portion of glycerol. To test if this theoretical problem influenced our data, we compared the plasma glycerol enrichment in insulin infusion participants (n = 4) and control participants (n = 8). The results did not show any significant difference in glycerol enrichment (data not shown). However, the numbers in this study was too low to prove no difference.
Kinetics analysis
Modelling of VLDL1 and VLDL2 kinetics of ApoB and TG was performed as described in detail by Adiels et al. [19] with modifications to allow for changes in the secretion rates of VLDL1 and VLDL2 as well as changes in the transfer and catabolic rates. Allowing the VLDL1 secretion rate to decrease could not by itself account for the decrease in VLDL1 pool size in all participants; thus it was necessary to allow the VLDL1 fractional catabolic rate (FCR) to increase in order to achieve the desired change in pool size. However, decreasing the VLDL1 secretion rate and/or increasing the transfer to VLDL2 resulted in a decrease in the VLDL2 pools. Therefore, it was necessary to allow an increased VLDL2 secretion rate. A detailed description of the model can be found in the Electronic supplementary material.
Non-steady-state variables
VLDL1 and VLDL2 secretion rates are expressed as percentage of total VLDL (i.e. VLDL1 + VLDL2) secretion rate at baseline (t = 0). The FA index is the suppression of NEFA at 60 min of the insulin injection, expressed as the percentage change from baseline.
Statistical analysis
Correlations were evaluated by linear regression. For non-normally distributed variables, values were log transformed before correlation analysis. Differences were considered significant at p < 0.05. Differences groups were evaluated with unpaired, two-sided t tests. Statistical analysis was performed with SPSS and Microsoft Excel.
Results
Participant characteristics
The clinical characteristics of the participants are given in Table 1.
Table 1 Participant characteristics
Plasma glucose, insulin, NEFA and glycerol
Plasma glucose levels were almost constant during the study and after an initial rise; the insulin levels were almost constant from 150 to 510 min. The plasma concentration of NEFA was, on average, suppressed by 63% (FA index) and the plasma glycerol pool was suppressed by 33% after 60 min.
Relationships between VLDL1 and VLDL2 pool sizes and metabolic measures
On average, VLDL1-TG and VLDL1-ApoB pool sizes were decreased by 20 and 30%, respectively (p < 0.05) at 510 min. The VLDL2 pools showed a non-significant change. The VLDL1-TG and ApoB pools correlated with liver fat, M value, fasting serum glucose, FA index (Table 2), fasting serum insulin (TG r = 0.51, p < 0.05; ApoB r = 0.64, p < 0.01), homeostasis model assessment of insulin resistance (HOMA-IR; TG r = 0.55, p < 0.05; ApoB r = 0.66, p < 0.001) and NEFA at 60 min (data not shown). The suppression of the VLDL1 pools correlated with liver fat, M value, FA index (Table 2) and NEFA at 60 min (data not shown). There was also a correlation between VLDL1-TG with fasting serum insulin (r = 0.51, p < 0.05) and HOMA-IR (r = 0.56, p < 0.05). No variables correlated with the change in the VLDL2 pool sizes.
Table 2 Correlations (r) of metabolic measures and VLDL1-TG and -ApoB pool sizes, secretion rates and secretion
Relationship between VLDL1 and VLDL2 secretion rates and metabolic measures
The baseline (t = 0 min) VLDL1 secretion rates of both TG and ApoB correlated with liver fat; this correlation was even stronger during the insulin infusion (Table 2). VLDL1 secretion rates did not correlate with the baseline NEFA concentration but to NEFA at 60 min and to the FA index. Furthermore, the VLDL1 secretion rates correlated negatively with the M value.
The suppression of VLDL1 secretion (in % of total VLDL at baseline) correlated with liver fat, NEFA at 60 min and to the FA index (Table 2). At 270 min the suppression also correlated with the M value (Table 2). The VLDL2 secretion rates did not correlate with any metabolic measures either at baseline or during the insulin infusion. The FA index (NEFA suppression by insulin at 60 min) correlated inversely with liver fat (r = −0.64, p < 0.01; Fig. 2a) and positively with the M value (r = 0.56, p < 0.001; Fig. 2b).
Fig. 2
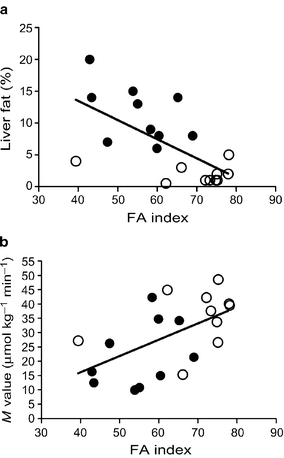
FA index vs liver fat (a) and M value (b) in low and high liver-fat groups. The FA index (NEFA suppression at 60 min) correlated negatively with liver fat (r = −0.72, p < 0.001) and positively with the M value (r = 0.67, p < 0.01). Closed circles, participants with high liver fat; open circles, participants with low liver fat
Furthermore, the FA index correlated with the intra-abdominal fat compartment (r = −0.53, p < 0.05), but not with the subcutaneous fat compartment (data not shown). Liver fat correlated also with plasma glucose (r = 0.66, p < 0.01), M value (r = −0.65, p < 0.01), HOMA-IR (r = 0.57, p < 0.01) and insulin (r = 0.49, p < 0.05).
Grouping of participants based on liver fat content
Both VLDL1 pools and production rates showed linear correlations in all participants regardless of the characteristics (Fig. 3a,c). Importantly, analysis of the correlation between VLDL1-ApoB and -TG productions separated the participants in two distinct groups: one group with no change in production rates and one with large suppression in the productions rates (Fig. 3d). Discriminant analysis showed that liver fat and FA index were better predictors of VLDL1 response, compared with insulin, diabetes per se, fasting plasma glucose or intra-abdominal fat volume (data not shown). Likewise, if diabetes or the median of M value was used as grouping variable, the difference in response in both VLDL1 pool sizes and secretion was smaller.
Fig. 3
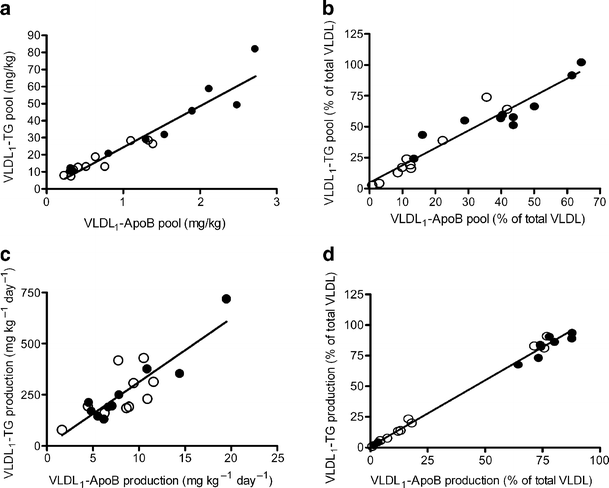
The VLDL1-ApoB and VLDL1- TG pools showed a strong correlation at the baseline (a) and throughout the study (510 min) (b). Similarly, the VLDL1-ApoB and VLDL1-TG secretion rates correlated at baseline (c) and throughout the study (510 min) (d). At the end of the study there was a clear separation of participants with high and low response to insulin infusion, and most of the participants with low liver fat were in the high-response group (d). Closed circles, participants with high liver fat; open circles, participants with low liver fat. To convert TG values from mg/kg to mmol/l multiply by 0.0282 and from mg kg−1 day−1 to μmol kg−1 day−1 multiply by 1.113; to convert ApoB values from mg/kg to pmol/l multiply by 48.8 and from mg kg−1 day−1 to pmol kg−1 day−1 multiply by 1.95
Based on the fact that liver fat content was the best prediction for VLDL1 response in the discriminant analyses, the participants were divided into two groups according to whether they had high (>5.5%) or low (<5.5%) liver fat based on the median in our population. This subdivision coincided with the cut-off from a recent study using magnetic resonance spectroscopy in a large population [20], where the 95th percentile of normal liver fat was below 5.56%, close to traditional diagnoses of hepatic steatosis (above 5%) [21, 22]. All eight participants with type 2 diabetes were in the high liver-fat group. The two groups showed significant differences in intra-abdominal fat, M value, weight, HDL concentration, LDL size and plasma glucose concentrations (Table 1).
Plasma glucose, insulin, NEFA and glycerol in subgroups
Plasma glucose levels were constant during the study (Fig. 4a). After an initial rise, the insulin levels remained almost constant, and there was no significant difference between the insulin levels in the two groups (Fig. 4b). The plasma concentration of NEFA was suppressed by 69% in the low liver-fat group but only by 57% in the high liver-fat group following 60 min of insulin infusion (p < 0.05 high liver fat vs low liver fat; Fig. 4c). The plasma glycerol pools were suppressed by 33% after 60 min in both the low and high liver-fat groups (Fig. 4d).
Fig. 4
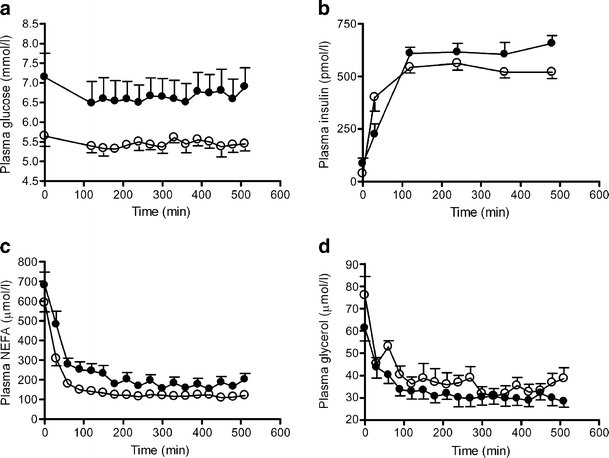
Mean plasma concentration of glucose (a), insulin (b), plasma NEFA (c) and glycerol (d) in low and high liver-fat groups. a Both baseline plasma glucose levels and plasma glucose levels during the study were lower in the low liver-fat group (p < 0.05 vs high liver-fat group). b Plasma insulin levels increased rapidly and were steady between 120 and 510 min. c Plasma NEFA levels were significantly suppressed in both groups after 60 min. The suppression was higher in the low liver-fat group (p < 0.05 vs high liver-fat group). d Plasma glycerol levels were significantly suppressed in both groups during the study. Filled circles, participants with high liver fat; open circles, participants with low liver fat. Means ± SEM, n = 10 in both groups
VLDL1 and VLDL2 pools in subgroups
Analysis of the VLDL1- and VLDL2-ApoB and VLDL1- and VLDL2-TG pools showed higher baseline concentrations in the high liver-fat group compared with the low liver-fat group (p < 0.05; Fig. 5a–d). In the low liver-fat group, VLDL1-ApoB and -TG pools decreased significantly during insulin infusion (all time-points, p < 0.01; Fig. 5a,b), but the VLDL2-ApoB and -TG pools did not change significantly (Fig. 5c,d). In the high liver-fat group, neither the VLDL1 nor the VLDL2 pools changed significantly during the insulin infusion (Fig. 5a–d).
Fig. 5
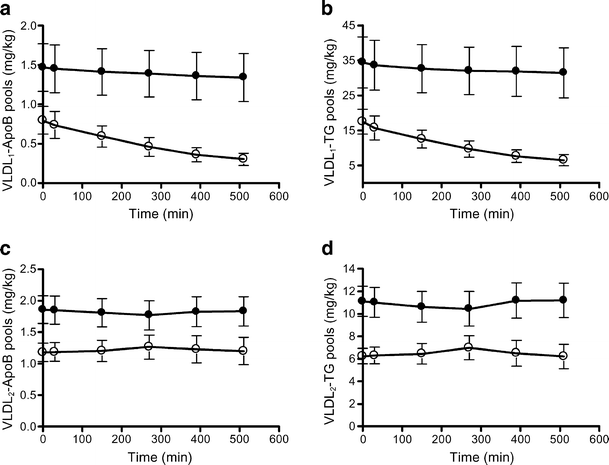
VLDL1-ApoB (a) and VLDL1-TG (b) and VLDL2 ApoB (c) and TG (d) pools in low liver-fat and high liver-fat groups. The VLDL pool sizes were significantly higher in the high liver-fat group than in the low liver-fat group (p < 0.05). In the low liver-fat group, the VLDL1 ApoB (a) and TG (b) pool sizes were significantly reduced at all time-points (p < 0.01 vs baseline). In the high liver-fat group, the VLDL1-ApoB (a) and -TG pool sizes (b) did not change significantly during the study. Filled circles, participants with high liver fat; open circles, participants with low liver fat. Means ± SEM, n = 10 in both groups. To convert TG values from mg/kg to mmol/l multiply by 0.0282; to convert ApoB values from mg/kg to pmol/l multiply by 48.8
VLDL1- and VLDL2-TG and VLDL1- and VLDL2-ApoB secretion rates in subgroups
In the low liver-fat group, a rapid and marked drop in VLDL1 secretion rate compared with baseline was observed at the start of insulin infusion (Fig. 6a,b). Both the VLDL1-ApoB and -TG secretion rates were decreased by 61% at 510 min (p < 0.01) (Fig. 6a,b). In contrast, neither VLDL1-ApoB nor -TG secretion rates were significantly changed over the study duration in the high liver-fat group (Fig. 6a,b).
Fig. 6
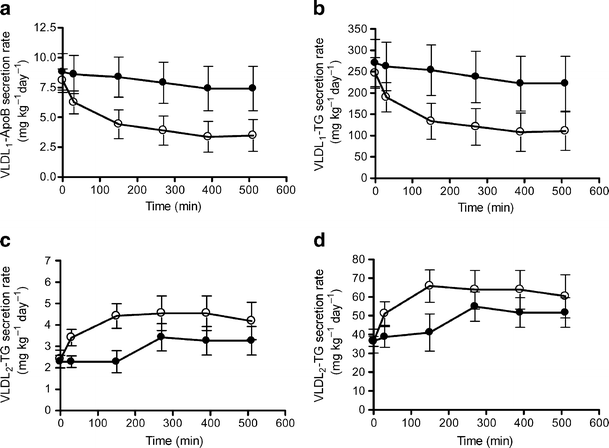
VLDL1-ApoB (a) and VLDL1-TG (b) and VLDL2-ApoB (c) and VLDL1-TG (d) secretion rates in low liver-fat and high liver-fat groups. Both the VLDL1-ApoB (a) and VLDL1-TG (b) secretion rates were significantly suppressed in the low liver-fat group (p < 0.01 vs baseline). VLDL2-ApoB (c) and VLDL2-TG (d) secretion rates were significantly increased in the low liver-fat group (p < 0.05 vs baseline). No significant changes were observed in the high liver-fat group. Closed circles, participants with high liver fat; open circles, participants with low liver fat. Means ± SEM, n = 10 in both groups. To convert TG values from mg kg−1 day−1 to ìmol kg−1 day−1 multiply by 1.113; to convert ApoB values from mg kg−1 day−1 to pmol kg−1 day−1 multiply by 1.95
Interestingly, the VLDL2-ApoB and -TG secretion rates displayed inverse kinetics compared with VLDL1-ApoB and -TG secretion rates and increased rapidly during insulin infusion (all time-points, p < 0.05) in the low liver-fat group (Fig. 6c,d). The VLDL2-ApoB and -TG secretion rates were increased by 73% at the end of insulin infusion (p < 0.05). Neither VLDL2-ApoB nor -TG secretion rates were significantly increased over the study duration in the high liver-fat group (Fig. 6c,d).
Total VLDL (i.e. VLDL1+VLDL2)-ApoB secretion rates decreased by 24% after insulin infusion (p < 0.05) in the low liver-fat group, but remained unchanged over the study in the high liver-fat group. Likewise, the total VLDL-TG secretion rates decreased by 39% (p < 0.01) in the low liver-fat group, but remained unchanged in the high liver-fat group.
VLDL1- and VLDL2-TG and VLDL1- and VLDL2-ApoB FCRs
The lipolytic rates, characterised by the FCR, of VLDL1 and VLDL2 were allowed to vary in the model. In the high liver-fat group, the FCRs of VLDL1 or VLDL2 did not change significantly during the study. However, in the low liver-fat group, the VLDL1 FCR of both ApoB and TG increased rapidly (by 22%, p < 0.05) at 30 min and the VLDL2 FCR increased by 75% (ApoB) and 83% (TG) from 270 to 510 min (p < 0.05).
Discussion
This study highlights the acute regulation of VLDL1 secretion rate by insulin and the impaired response in participants with high liver fat. Furthermore, it is the first non-steady-state kinetic study in which VLDL1- and VLDL2-ApoB and VLDL1- and VLDL2-TG have been simultaneously determined. The results showed a rapid and marked drop in VLDL1-ApoB and VLDL1-TG secretion in participants with low liver fat during the insulin infusion. In contrast, participants with high liver fat showed no significant changes in either VLDL1-ApoB or VLDL1-TG secretion rates over the study duration. This study confirms our recent results that liver fat determines baseline VLDL1 production [23], but extends this relationship to hold also under insulin infusion. Importantly the suppression itself correlated with the liver fat content.
Our results showed that the VLDL1 suppression following insulin infusion correlated with the suppression of NEFA, and that the ability of insulin to suppress the plasma NEFA was impaired in participants with high liver fat. The fact that the NEFA suppression was inversely related to intra-abdominal fat volume, but not to subcutaneous fat, supports the important role of visceral fat as the source of NEFA flux to the liver. The data pinpoint that adipose tissue insulin resistance together with endocrine overactivity contributes to hepatic steatosis. Overall, the influx of NEFA from adipose tissue to the liver normally accounts for 60–80% of the hepatic TG [24]. Insulin decreases TG biosynthesis in the liver by reducing this influx [12–14, 25, 26], possibly leading to a reduced synthesis of the intracellular lipid droplets required for the formation of VLDL1 from VLDL2. This is in agreement with the finding that acute lowering of NEFAs using the antilipolytic agent acipimox resulted in the secretion of smaller and denser VLDL2 particles [14], The increased hepatic biosynthesis in participants with insulin resistance may also stabilise ApoB and thereby reduce post-translational degradation of ApoB [3]. Indeed, total VLDL ApoB secretion was significantly reduced after insulin infusion for 8.5 h in the low liver-fat group, but remained unchanged over the study in the high liver-fat group.
Insulin also seems to have direct suppressive effects on the formation of VLDL1 in the liver, independently of the availability of NEFAs [14, 27]. First, insulin regulates the expression of key factors that regulate the initiation of VLDL assembly and the maturation of VLDL particles [28–31]. Furthermore, increased insulin levels promote formation of cytosolic lipid droplets [32, 33]. Activation and upregulation of genes that acutely shift TG and NEFA from VLDL formation and towards cytosolic storage may play important roles [32, 33].
A novel finding was the inverse response between VLDL1 and VLDL2 secretion in participants with low liver fat: VLDL1 secretion decreased acutely after insulin infusion whereas VLDL2 secretion increased. Results in vitro support a step-wise loading of nascent VLDL with core lipids during biosynthesis [3, 4]. If TG is not added to the nascent VLDL leading to the formation of VLDL1, the TG-poor VLDL particle will be secreted as a VLDL2 particle [3]. The finding that VLDL1 secretion decreased acutely after insulin infusion in participants with low liver fat, whereas VLDL2 secretion increased, supports in vivo a step-wise loading of VLDL particles during biosynthesis and suggests that insulin inhibits the bulk lipid addition that converts TG-poor VLDL into mature VLDL1. Furthermore, the shift from VLDL1 to VLDL2 explains why secretion of total VLDL-TG was reduced to a greater extent than the secretion of total VLDL-ApoB following insulin infusion.
Today the question whether hepatic insulin resistance is a consequence of increased liver fat or vice versa remains a matter of debate [34]. The key question is whether the effects of liver fat per se can be separated from the effects of diabetes/hepatic insulin resistance. The fact that the participants with high liver fat were predominantly people with diabetes is a weakness but we purposefully aimed to recruit participants with a wide range of liver fat. Recognising the fact that hepatic steatosis is highly prevalent in patients with type 2 diabetes we were unable to avoid the abundance of participants with type 2 diabetes in the high liver-fat group. Thus, we cannot exclude that impaired suppression of VLDL1 production rate may also reflect some direct effect of diabetes beyond liver fat accumulation. However, as seen in Fig. 3a,c, our data indicate that, independently of the characteristics of the participant, both pools and production rates fall on the same line and forms a continuum. Furthermore, a discriminant analysis showed that liver fat and NEFA suppression were better predictors of VLDL1 response to insulin (Fig. 3d) than diabetes per se, fasting plasma glucose, fasting insulin or intra-abdominal fat volume (data not shown). Moreover, liver fat and the NEFA suppression were the best correlates with the actual changes in the VLDL1 pools in the VLDL1 secretion rate itself at both baseline and during insulin infusion. Thus, we believe that liver fat and the ability of insulin to regulate NEFA flux are important determinants of synthesis and secretion of VLDL1 and VLDL2 particles.
Once hepatic insulin resistance has developed, it is likely that it will further contribute to fat accumulation in the liver, creating a vicious cycle. Indeed, recent evidence indicates that defective suppression of hepatic glucose production (i.e_._ the hallmark of hepatic insulin resistance) is closely correlated with liver fat accumulation in both healthy control participants and patients with type 2 diabetes [7, 10, 35]. Furthermore, emerging data link the metabolic pathways of carbohydrate and TG homeostasis by the action of nuclear receptors [36]. It is therefore hard to ascertain whether the observed correlation between high liver fat and reduced suppression of VLDL1 by insulin is the result of fatty liver or merely the consequence of hepatic insulin resistance, or alternatively by a combination of both. To specifically address this, it would be of interest to perform the simultaneous analysis of both hepatic glucose and lipid metabolism in patients with either disturbed hepatic glucose metabolism or disturbed lipid metabolism.
However, emerging data from both humans and animals support a causal role of intracellular hepatic accumulation of TG [36–39] and, probably more importantly, of certain intracellular fatty acid metabolites (fatty acyl CoAs, diacylglycerol, ceramides and glycosphingolipids) in the pathogenesis of hepatic insulin resistance [40–42]. Importantly preventing liver fat accumulation in animal models is associated with reversal of hepatic insulin sensitivity [43, 44]. Clinically, this relationship seems to be also reversible, as modest weight loss in humans is associated with a marked reduction of hepatic fat and reversal of hepatic insulin resistance [45, 46].
What are the consequences of the impaired suppression of VLDL1 secretion in participants with high liver fat? LDL particles can be converted to small dense LDL by interaction with triacylglycerol-rich lipoproteins, a reaction catalysed by hepatic lipase and cholesteryl-ester transfer protein, enzymes that are increased in type 2 diabetes [1]. In addition, increased levels of VLDL1 alter the composition of HDL, ultimately leading to an increased catabolism of these particles [47]. These findings are consistent with our observation of an inverse correlation of both LDL particle size and HDL concentration with VLDL1 production [6]. Thus, the two hallmarks of the highly atherogenic diabetic dyslipidaemia, low HDL levels and increased generation of small dense LDL, are both strongly linked to overproduction of VLDL1. The inability for the liver to regulate VLDL secretion in high liver-fat participants may also contribute to postprandial lipaemia, further contributing to the dyslipidaemia seen in high liver-fat participants.
In summary, our results show that insulin downregulates VLDL1 secretion and increases VLDL2 secretion in participants with low liver fat. In contrast, the acute suppression of VLDL1 secretion by insulin is impaired in participants with high liver fat, resulting in overproduction of VLDL1. Whether a reduction of liver fat is associated with decreased production of VLDL1 particles and a reversal of the atherogenic dyslipidaemia remains to be established, but this suggests a potential target for the treatment of diabetic dyslipidaemia.
References
- Taskinen MR (2003) Diabetic dyslipidaemia: from basic research to clinical practice. Diabetologia 46:733–749
Article PubMed Google Scholar - Zimmet P, Alberti KG, Shaw J (2001) Global and societal implications of the diabetes epidemic. Nature 414:782–787
CAS PubMed Google Scholar - Olofsson SO, Boren J (2005) Apolipoprotein B: a clinically important apolipoprotein which assembles atherogenic lipoproteins and promotes the development of atherosclerosis. J Intern Med 258:395–410
Article CAS PubMed Google Scholar - Alexander CA, Hamilton RL, Havel RJ (1976) Subcellular localization of B apoprotein of plasma lipoproteins in rat liver. J Cell Biol 69:241–263
Article CAS PubMed Google Scholar - Stillemark P, Boren J, Andersson M et al (2000) The assembly and secretion of apolipoprotein B-48-containing very low density lipoproteins in McA-RH7777 cells. J Biol Chem 275:10506–10513
Article CAS PubMed Google Scholar - Adiels M, Taskinen MR, Packard C et al (2006) Overproduction of large VLDL particles is driven by increased liver fat content in man. Diabetologia 49:755–765
Article CAS PubMed Google Scholar - Seppala-Lindroos A, Vehkavaara S, Hakkinen AM et al (2002) Fat accumulation in the liver is associated with defects in insulin suppression of glucose production and serum free fatty acids independent of obesity in normal men. J Clin Endocrinol Metab 87:3023–3028
Article CAS PubMed Google Scholar - Kelley DE, McKolanis TM, Hegazi RA, Kuller LH, Kalhan SC (2003) Fatty liver in type 2 diabetes mellitus: relation to regional adiposity, fatty acids, and insulin resistance. Am J Physiol Endocrinol Metab 285:E906–E916
Article CAS PubMed Google Scholar - Ryysy L, Hakkinen AM, Goto T et al (2000) Hepatic fat content and insulin action on free fatty acids and glucose metabolism rather than insulin absorption are associated with insulin requirements during insulin therapy in type 2 diabetic patients. Diabetes 49:749–758
Article CAS PubMed Google Scholar - Bugianesi E, Gastaldelli A, Vanni E et al (2005) Insulin resistance in non-diabetic patients with non-alcoholic fatty liver disease: sites and mechanisms. Diabetologia 48:634–642
Article CAS PubMed Google Scholar - Targher G, Bertolini L, Poli F et al (2005) Nonalcoholic fatty liver disease and risk of future cardiovascular events among type 2 diabetic patients. Diabetes 54:3541–3546
Article CAS PubMed Google Scholar - Malmstrom R, Packard CJ, Watson TD et al (1997) Metabolic basis of hypotriglyceridemic effects of insulin in normal men. Arterioscler Thromb Vasc Biol 17:1454–1464
Article CAS PubMed Google Scholar - Malmstrom R, Packard CJ, Caslake M et al (1999) Effect of heparin-stimulated plasma lipolytic activity on VLDL APO B subclass metabolism in normal subjects. Atherosclerosis 146:381–390
Article CAS PubMed Google Scholar - Malmstrom R, Packard CJ, Caslake M et al (1998) Effects of insulin and acipimox on VLDL1 and VLDL2 apolipoprotein B production in normal subjects [published erratum appears in Diabetes 1998, 47:1532]. Diabetes 47:779–787
Article CAS PubMed Google Scholar - Lewis GF, Carpentier A, Adeli K, Giacca A (2002) Disordered fat storage and mobilization in the pathogenesis of insulin resistance and type 2 diabetes. Endocr Rev 23:201–229
Article CAS PubMed Google Scholar - Report of a WHO consultation (1999) Definition, diagnosis and classification of diabetes mellitus and its complications. Part 1 Diagnosis and classification of diabetes mellitus. World Health Organization, Department of Noncommunicable Disease Surveillance, Geneva
- Adiels M, Boren J, Caslake MJ et al (2005) Overproduction of VLDL1 driven by hyperglycemia is a dominant feature of diabetic dyslipidemia. Arterioscler Thromb Vasc Biol 25:1697–1703
Article CAS PubMed Google Scholar - DeFronzo RA, Tobin JD, Andres R (1979) Glucose clamp technique: a method for quantifying insulin secretion and resistance. Am J Physiol 237:E214–E223
CAS PubMed Google Scholar - Adiels M, Packard C, Caslake MJ et al (2005) A new combined multicompartmental model for apolipoprotein B-100 and triglyceride metabolism in VLDL subfractions. J Lipid Res 46:58–67
Article CAS PubMed Google Scholar - Szczepaniak LS, Nurenberg P, Leonard D et al (2005) Magnetic resonance spectroscopy to measure hepatic triglyceride content: prevalence of hepatic steatosis in the general population. Am J Physiol Endocrinol Metab 288:E462–E468
Article CAS PubMed Google Scholar - Thomsen C, Becker U, Winkler K, Christoffersen P, Jensen M, Henriksen O (1994) Quantification of liver fat using magnetic resonance spectroscopy. Magn Reson Imaging 12:487–495
Article CAS PubMed Google Scholar - Hoyumpa AM Jr, Greene HL, Dunn GD, Schenker S (1975) Fatty liver: biochemical and clinical considerations. Am J Dig Dis 20:1142–1170
Article CAS PubMed Google Scholar - Adiels M, Taskinen MR, Packard C et al (2006) Overproduction of large VLDL particles is driven by increased liver fat content in man. Diabetologia 49:755–765
Article CAS PubMed Google Scholar - Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ (2005) Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest 115:1343–1351
Article CAS PubMed PubMed Central Google Scholar - Adeli K, Taghibiglou C, Van Iderstine SC, Lewis GF (2001) Mechanisms of hepatic very low-density lipoprotein overproduction in insulin resistance. Trends Cardiovasc Med 11:170–176
Article CAS PubMed Google Scholar - Lewis GF (1997) Fatty acid regulation of very low density lipoprotein production. Curr Opin Lipidol 8:146–153
Article CAS PubMed Google Scholar - Lewis GF, Uffelman KD, Szeto LW, Weller B, Steiner G (1995) Interaction between free fatty acids and insulin in the acute control of very low density lipoprotein production in humans. J Clin Invest 95:158–166
Article CAS PubMed PubMed Central Google Scholar - Carpentier A, Taghibiglou C, Leung N et al (2002) Ameliorated hepatic insulin resistance is associated with normalization of microsomal triglyceride transfer protein expression and reduction in very low density lipoprotein assembly and secretion in the fructose-fed hamster. J Biol Chem 277:28795–28802
Article CAS PubMed Google Scholar - Au WS, Kung HF, Lin MC (2003) Regulation of microsomal triglyceride transfer protein gene by insulin in HepG2 cells: roles of MAPKerk and MAPKp38. Diabetes 52:1073–1080
Article CAS PubMed Google Scholar - Phung TL, Roncone A, Jensen KL, Sparks CE, Sparks JD (1997) Phosphoinositide 3-kinase activity is necessary for insulin-dependent inhibition of apolipoprotein B secretion by rat hepatocytes and localizes to the endoplasmic reticulum. J Biol Chem 272:30693–30702
Article CAS PubMed Google Scholar - Brown AM, Gibbons GF (2001) Insulin inhibits the maturation phase of VLDL assembly via a phosphoinositide 3-kinase-mediated event. Arterioscler Thromb Vasc Biol 21:1656–1661
Article CAS PubMed Google Scholar - Andersson L, Bostrom P, Ericson J et al (2006) PLD1 and ERK2 regulate cytosolic lipid droplet formation. J Cell Sci 119:2246–2257
Article CAS PubMed Google Scholar - Magnusson B, Asp L, Bostrom P et al (2006) Adipocyte differentiation-related protein promotes fatty acid storage in cytosolic triglycerides and inhibits secretion of very low-density lipoproteins. Arterioscler Thromb Vasc Biol 26:1566–1571
Article CAS PubMed Google Scholar - Utzschneider KM, Kahn SE (2006) Review: the role of insulin resistance in nonalcoholic fatty liver disease. J Clin Endocrinol Metab 91:4753–4761
Article CAS PubMed Google Scholar - Bajaj M, Suraamornkul S, Piper P et al (2004) Decreased plasma adiponectin concentrations are closely related to hepatic fat content and hepatic insulin resistance in pioglitazone-treated type 2 diabetic patients. J Clin Endocrinol Metab 89:200–206
Article CAS PubMed Google Scholar - Gaemers IC, Groen AK (2006) New insights in the pathogenesis of non-alcoholic fatty liver disease. Curr Opin Lipidol 17:268–273
Article CAS PubMed Google Scholar - Westerbacka J, Lammi K, Hakkinen AM et al (2005) Dietary fat content modifies liver fat in overweight nondiabetic subjects. J Clin Endocrinol Metab 90:2804–2809
Article CAS PubMed Google Scholar - Kabir M, Catalano KJ, Ananthnarayan S et al (2005) Molecular evidence supporting the portal theory: a causative link between visceral adiposity and hepatic insulin resistance. Am J Physiol Endocrinol Metab 288:E454–E461
Article CAS PubMed Google Scholar - Samuel VT, Liu ZX, Qu X et al (2004) Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J Biol Chem 279:32345–32353
Article CAS PubMed Google Scholar - Boden G, She P, Mozzoli M et al (2005) Free fatty acids produce insulin resistance and activate the proinflammatory nuclear factor-{kappa}B pathway in rat liver. Diabetes 54:3458–3465
Article CAS PubMed Google Scholar - Hammond LE, Neschen S, Romanelli AJ et al (2005) Mitochondrial glycerol-3-phosphate acyltransferase-1 is essential in liver for the metabolism of excess acyl-CoAs. J Biol Chem 280:25629–25636
Article CAS PubMed Google Scholar - Shulman GI (2000) Cellular mechanisms of insulin resistance. J Clin Invest 106:171–176
Article CAS PubMed PubMed Central Google Scholar - Samuel VT, Liu ZX, Wang A et al (2007) Inhibition of protein kinase C epsilon prevents hepatic insulin resistance in nonalcoholic fatty liver disease. J Clin Invest 117:739–745
Article CAS PubMed PubMed Central Google Scholar - Savage DB, Choi CS, Samuel VT et al (2006) Reversal of diet-induced hepatic steatosis and hepatic insulin resistance by antisense oligonucleotide inhibitors of acetyl-CoA carboxylases 1 and 2. J Clin Invest 116:817–824
Article CAS PubMed PubMed Central Google Scholar - Tiikkainen M, Bergholm R, Vehkavaara S et al (2003) Effects of identical weight loss on body composition and features of insulin resistance in obese women with high and low liver fat content. Diabetes 52:701–707
Article CAS PubMed Google Scholar - Petersen KF, Dufour S, Befroy D, Lehrke M, Hendler RE, Shulman GI (2005) Reversal of nonalcoholic hepatic steatosis, hepatic insulin resistance, and hyperglycemia by moderate weight reduction in patients with type 2 diabetes. Diabetes 54:603–608
Article CAS PubMed Google Scholar - Rashid S, Watanabe T, Sakaue T, Lewis GF (2003) Mechanisms of HDL lowering in insulin resistant, hypertriglyceridemic states: the combined effect of HDL triglyceride enrichment and elevated hepatic lipase activity. Clin Biochem 36:421–429
Article CAS PubMed Google Scholar