Transcriptome profiling of the cold response and signaling pathways in Lilium lancifolium (original) (raw)
- Research article
- Open access
- Published: 17 March 2014
BMC Genomics volume 15, Article number: 203 (2014)Cite this article
- 14k Accesses
- 74 Citations
- 1 Altmetric
- Metrics details
Abstract
Background
Lilium lancifolium, a very important cold-resistant wild flower for lily cold resistance breeding, is widely distributed in southwestern and northeastern China. To gain a better understanding of the cold signaling pathway and the molecular metabolic reactions involved in the cold response, we performed a genome-wide transcriptional analysis using RNA-Seq.
Results
Approximately 104,703 million clean 90- bp paired-end reads were obtained from three libraries (CK 0 h, Cold-treated 2 h and 16 h at 4°C); 18,736 unigenes showed similarity to known proteins in the Swiss-Prot protein database, and 15,898, 13,705 and 1849 unigenes aligned to existing sequences in the KEGG and COG databases (comprising 25 COG categories) and formed 12 SOM clusters, respectively. Based on qRT-PCR results, we studied three signal regulation pathways —the Ca2+ and ABA independent/dependent pathways —that conduct cold signals to signal transduction genes such as LlICE and LlCDPK and transcription factor genes such as LlDREB1/CBF, LlAP2/EREBP, LlNAC1, LlR2R3-MYB and LlBZIP, which were expressed highly in bulb. LlFAD3, Llβ-amylase, LlP5CS and LlCLS responded to cold and enhanced adaptation processes that involve changes in the expression of transcripts related to cellular osmoprotectants and carbohydrate metabolism during cold stress.
Conclusions
Our study of differentially expressed genes involved in cold-related metabolic pathways and transcription factors facilitated the discovery of cold-resistance genes and the cold signal transcriptional networks, and identified potential key components in the regulation of the cold response in L lancifolium, which will be most beneficial for further research and in-depth exploration of cold-resistance breeding candidate genes in lily.
Background
Plants have a remarkable ability to cope with highly variable environmental abiotic stresses, including cold, drought, heat, salinity and nutrient deficiencies. Nevertheless, these stresses together represent the primary cause of plant injuries and losses worldwide, reducing the ornamental value and production of most major plants and crops by more than 50% [1]. As a wild cold-resistant plant, Lilium lancifolium is mainly distributed in the North Temperate Zone, where the winter temperate can fall as low as -35°C, but it can survive exposure, acclimate to low or freezing temperatures and continuously germinate in the next spring. In addition, studies have shown its capacity for resisting heat, drought and changing soil salinity. Nevertheless, the decline of L. lancifolium is gradually becoming more serious in recent years with the deterioration of its ecological environment. Therefore, further protection and a better understanding of the gene expression profile of L. lancifolium under cold stress is imperative, and it could be an ideal model to study cold tolerance mechanisms and signaling regulation for improving the quality of cold resistance in other plants using molecular biological techniques.
Cold responses have been observed in many plants, which initiate reactions of the freezing tolerance of plants after low temperature stress, including _Arabidopsis_[2], _Oryza sativa_[3], _Triticum aestivum_[4], and _Ammopiptanthus mongolicus_[5]. The initiation of most stress treatments correlates with a cytosolic calcium release, in some cases with stimulus-specific patterns of oscillation [6]. In addition, stimulus-specific changes in gene expression are often observed alongside a set of shared stress responses. For example, in a survey of 1, 300 Arabidopsis genes, the majority of cold and drought stress regulated genes were observed in the shared stress response [7]. Together, these observations support the hypothesis that a common set of signal transduction pathways are triggered during many stress responses. Many of the biochemical, molecular and physiological changes that occur during the cold response are considered to be important in the induction of freezing tolerance. During this process, plants alter the expression of certain genes as well as the biosynthesis of amino acids and soluble sugars [8]. At the gene expression level, DNA microarray analysis studies have revealed that exposure of Arabidopsis plants to a low temperature of 4°C resulted in up- or down-regulation of hundreds to thousands of cold-regulated (COR) genes, and that many of the cold inducible genes are linked with the accumulation of osmolytes, cryoprotectants, antioxidant detoxification enzymes, chaperones, transporters, dehydrins, late embryogenesis abundant (LEA) proteins and enzymes involved in lipid, carbohydrate and secondary metabolites, and in abscisic acid (ABA) and jasmonic acid (JA) biosynthesis [9].
Traditional cloning and genetic transformation methods are expensive and time consuming. In recent years, the development of novel high-throughput sequencing technologies, such as Solexa/Illumina RNA-Seq (RNA sequencing) and digital gene expression (DGE) has provided an opportunity to explore cold resistance and signaling-associated genes in different species by de novo assembly or mapping, and also facilitated rapid identification and analysis of the vast majority of transcriptomes [10]. Transcriptome sequencing is an efficient means to generate functional genomic data for non-model organisms or those with genome characteristics prohibitive to whole-genome sequencing [11]. Illumina/Solexa has been successfully applied to the transcriptome sequencing of many plant species, including _Populus euphratica_[12], _Aegilops variabilis_[13], _Aechmea fasciata_[14], _Brassica napus_[15], _Zea mays_[16], _Arachis hypogaea_[17], and _Picrorhiza kurrooa_[18]. For instance, transcriptome profiling of grapefruit flavedo following exposure to low temperature and conditioning treatments has increased our understanding of the principal molecular components involved in chilling tolerance and susceptibility [19]. Using Illumina/Solexa, we found that the study of differentially expressed genes involved in cold-related metabolic pathways and transcription factors could facilitate the discovery of cold-resistance genes for the desert shrub _Ammopiptanthus mongolicus_[20]. The transcriptomic analysis of Aechmea fasciata treated with ethylene has been revealed part of the ethylene signaling pathway and flowering process [14]. Genome-wide analysis approaches have been used to elucidate gene expression in response to drought stress in _Populus simoniia_[21].
However, few studies have been carried out to date on the cold-related metabolic pathways and transcription factors in L. lancifolium. Such studies would bridge the physiological and anatomical changes during cold acclimation with molecular data. Here, we present three cDNA libraries (two cold-treated and a control) of living L. lancifolium leaves which were subjected to short-term cold (4°C) stress treatment and describe the short-term cold response (0 to 16 h) of L. lancifolium using the next-generation Illumina/Solexa sequencing technology, and also compared the long-term cold acclimation (1 to 20d) of L. lancifolium and Oriental hybrids using anatomical and physiological analyses and biochemistry experiments. We had two specific objectives. First, to identify genes that change expressions in a stress-specific fashion and reveal the transcription factors that change in the key transcriptional, finding development networks and signal pathways in the response to cold stress. Second, to identify the expression of various genes that are co-regulated during the biological processes, such as intercellular osmoprotectant biosynthesis and biodegradation of carbohydrates of shared stress responses. This global view illustrates the “fluid” nature of the transcriptome and the challenge we face in understanding the complexity of any given stress response. In particular, the analyses on differentially expressed genes under cold stress furthers our understanding of the cold response mechanism of L. lancifolium, and these cold-related genes should also contribute to providing a method of developing cold-tolerant plants through genetic manipulation.
Results
High-throughput transcriptome sequencing and read assembly
To identify the number of genes involved in the transcriptome, a cDNA sample was prepared from an equal mixture of total RNA isolated from leaves for three libraries (cold-treated 2 h, 16 h and control 0 h samples), which were sequenced using the Illumina HiSeq™ 2000. We obtained approximately 74 million raw reads for the two cold-treated samples (CT2h and CT16h) and 41 million for the control sample (CK0h). We discarded low-quality reads, which contained adapters and unknown or low-quality bases, according to our bioinformatics analysis. After stringent quality checks and data cleaning, we obtained 115,421,520 raw reads containing a total of 11.6 Gb nucleotides. The average read size, Q20 percentage (sequencing error rate < 1%), and GC (guanine + cytosine) percentage were 90 bp, 98.1%, and 43.7%, respectively. Based on the high quality reads, 46,516 contigs were assembled with an average length of 793 bp. With paired-end joining and gap-filling, the contigs were further assembled into 39,154 scaffolds with an average length of 951 bp, including 11478 scaffolds larger than 1000 bp. After local assembly with the unmapped ends to fill in the small gaps within the scaffolds, the de novo assembly yielded 37,843 unigenes with an average length of 971 bp (Tables 1 and 2). To demonstrate the quality of sequencing data, ten unigenes were randomly selected and ten pairs of primers were designed for qRT-PCR, and then the products were confirmed by biological Sanger sequencing.
Table 1 Overview of the sequencing and assembly
Table 2 Summary for the Lilium lancifolium transcriptome
To validate and annotate the of assembled unigenes, using E-value < 1e-5, they were blast searched against the UniProt (date: 2013.04) and Swiss-Prot protein database (date: 2013.05) (http://www.expasy.ch/sprot) which has the largest and most detailed protein annotation database, including 24,889,084 proteins. The results indicated that among the 37,843 unigenes, 18,736 (49.5% of the total) had significant similarity to known proteins in the Swiss-Prot database. The lack of L. lancifolium genome and EST information meant that 19.107 (50.5% of the total) unigenes had no Swiss-Prot annotation (Table 2).
Gene annotation and functional classification
To further evaluate the completeness of our transcriptome library and the effectiveness of our annotation process, we randomly searched the annotated sequences for genes with COG (Clusters of orthologous groups) classifications. Of 18,736 Swiss-Prot hits, 13,705 sequences had a COG classification. Among the 25 COG categories, the cluster for 'Signal transduction mechanisms’ (8332, 17.54%) represented the largest group, followed by 'Cytoskeleton’ (7078, 14.89%) and 'General function prediction only’ (4239, 8.92%). The 'defense mechanisms’ (190, 0.39%) and 'extracellular structures’ (147, 0.30%) categories, represented the smallest groups (Figure 1). Compared with the control sample, the most-abundant cluster in the cold treated samples was 'Signal transduction mechanisms', indicating that STM genes play a vital regulation role in the L. lancifolium cold senescence and stress responses.
Figure 1
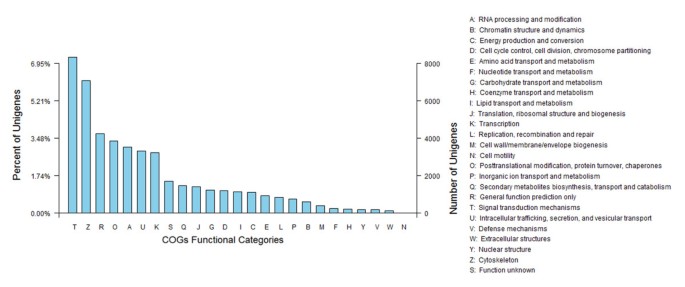
Clusters of orthologous groups (COG) classifications in Lilium lancifolium . These 13705 sequences have a COG classification within the 25 categories.
The Gene ontology (GO) assignments system was also used to classify the functions of the predicted L. lancifolium genes. Based on sequence homology, the 18,736 sequences were categorized into 53 functional groups (Figure 2). The unigenes were then assigned into three main categories: biological process, cellular component, and molecular function, with 7901 (42.17%), 2338 (12.47%), and 4459 (23.79%) unigenes, respectively, assigned to each. In each of the three main GO classifications, the 'metabolic process', 'cellular process', and 'catalytic activity’ terms, were dominant. We also noticed a high percentage of genes from the 'cellular process', 'cell part', and 'binding’ categories, but few from 'cell junction', 'symplast', 'extracellular region part’ and 'locomotion’ (Figure 2). The GO analysis indicated that a great number of identified genes were associated with various biological processes and molecular functions under low temperature. About 5331 sequences were annotated as belonging to the 'metabolic process’ category, which may allow for the identification of novel genes involved in secondary metabolism pathways in cold acclimation. The 'cellular component part’ suggested that the membrane and cell junction could function in the later cold resistance process (Figure 2).
Figure 2
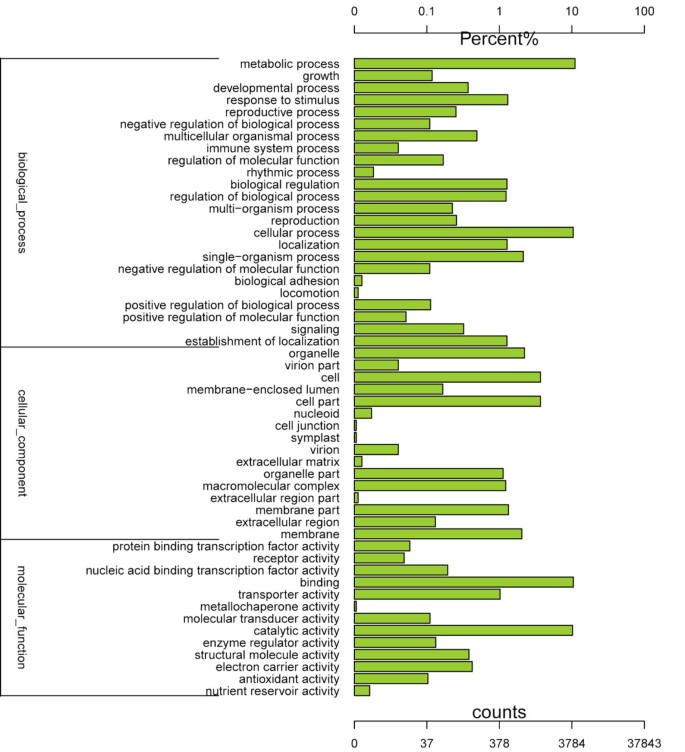
Histogram presentation of Gene Ontology classifications. The results are summarized in three main categories: biological process, cellular component, and molecular function. The y-axis on the right side indicates the percent of genes in a category, and the y-axis on the left side means the number of genes.
The KEGG (Kyoto Encyclopedia of Genes and Genomes) is a database resource for understanding the high-level functions and utilities of biological systems, such as the cell, the organism and the ecosystem, from molecular-level information, especially large-scale molecular datasets generated by genome sequencing and other high-throughput experimental technologies (see the release notes for new and updated features). The pathway database records networks of molecular interactions in cells, and variants specific to particular organisms. Based on a comparison against the KEGG database using BLASTx with an E-value cutoff of < 10-5, of the 37,843 unigenes, 15,898 (42%) had significant matches in the database and were assigned to 119 KEGG pathways (Table 3). The most represented pathways were 'metabolism pathways’ (1784 members, 11.23%), 'biosynthesis of secondary metabolites’ (946 members, 5.95%), and 'Microbial metabolism in diverse environments’ (400 members, 2.51%) (Table 3). These annotations provide a valuable resource for investigating the processes, functions, and pathways involved in the short-term cold response and long-term cold acclimation.
Table 3 Categorization of Lilium lancifolium unigenes to KEGG biochemical pathways
Changes in gene expression profiles among the different cold stress stages
To investigate the annotated transcriptome assembly and gain statistical confirmation of the differences in gene expression that served as a reference for RNA-Seq profiling of stage-specific expression, we conducted a small RNA-Seq experiment using tangential cryosections of CK0h, CT2h and CT16h of L. lancifolium and mapped the resulting reads to our reference transcriptome. To minimize false positives and negatives, we concluded that a statistical analysis was reliable when applied to genes with an RPKM value ≥ 2 in at least one of the three stages. It should be noted that this statistical significance was based on expected sampling distributions. To determine which of the 37,843 genes were differentially expressed among the three stages, we filtered with an FDR ≤ 0.001 and |log2 (ratio)| ≥ 2; the expression of 2755 DEGs was found to be significantly changed during the three stages. On the one hand, some gene were down-regulated from the 0 h to 2 h, but up-regulated obviously at 16 h. On the other hand, some genes showed increased quantitative expression at 2 h, but decreased transcript abundance at the 16 h stage. To identify genes showing a significant change in expression during different cold stress stages, the differentially expressed tags between the three samples were identified using an algorithm developed from the heat-map. Some genes were immediately expressed at the initial stage of cold stress, while others were up-regulated subsequently indicating that transcription factors induced the expression of cold-related genes during the regulation of cold signaling (Figure 3A).
Figure 3
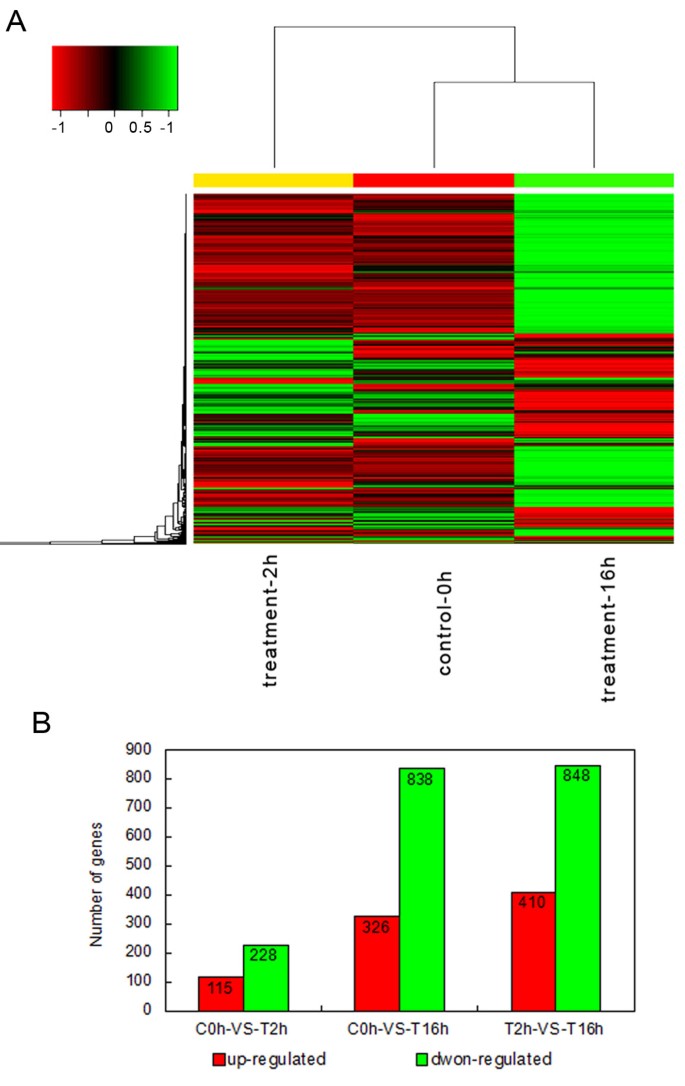
The expression of the gene changes among the different cold stress stages. A. The heat-map of the total differentially expressed genes (DEGs). Columns and rows in the heat maps represent samples and genes, respectively. Sample names are displayed below the heat maps. CK Results of controls, CT2h and 16h results of cold treatments. Color scale indicates fold changes of gene expression. A fold change of ≥1 is shown in green (increased transcript abundance), a fold change of ≤ -1 is shown in red (decreased transcript abundance), and no change is indicated in black. The results show that 1,028 transcripts were differentially expressed between the control and cold treatments 2 h and 16 h. B. Changes in gene expression profile among the different cold stress stages. The number of up-regulated and down-regulated genes between C0h-VS-T2h, C0h-VS-T16h and T2h-VS-T16h are summarized. Between the C0h and T2h Lilium lancifolium libraries, there are 115 genes up-regulated and 228 genes down-regulated, while there are 326 up-regulated genes and 828 down-regulated genes between the C0h and T16h Lilium lancifolium libraries, and 410 up-regulated genes 848 down-regulated genes between the C2h and T16h Lilium lancifolium libraries.
In addition, we compared the Ck0h and CT2h libraries, and 343 variable genes were found a total of 115 up-regulated and 228 down-regulated genes were detected between the two L. lancifolium libraries. There were also 326 up-regulated and 828 down-regulated genes between the C0h and T16h libraries, 410 up-regulated and 848 down-regulated genes between the C2h and T16h libraries (Figure 3B). This suggests that the differentiation of expressed genes between C2h and T16h is larger than that between C0h and T16h, while the difference between and the C0h and T2h is the smallest of the three. That means, in L. lancifolium, transcript abundance changed dramatically at these key switches among the cold stress stages of 2 h to 16 h which the cold response genes could be induced and expressed largely, but we should not ignore the genes expression during the short-term of 0 h to 2 h cold treatment, because many important cold-stress response genes were up- and down-regulated in this period, they would earliest determine the plant to play instantaneous reflection and response to the cold stress. These findings suggested forecast that our analysis was capable of identifying cold stress response genes and therefore suitable for further investigation of the biological functions of these genes.
SOM cluster analysis of gene expression
To facilitate cluster analysis of gene expression, the expression profiles of the differentially expressed genes were determined by SOM cluster analysis based on the k-means method using Pearson’s correlation distance. The total differential genes were divided into 12 groups based on their expression modulation with analysis of GO and KEGG pathway enrichment, representing the number of profiles as indicated by figure of merit analysis. Clusters were obtained by the k-means method using the gene expression profiles of the 1849 modulated genes. The most abundant group were Clusters 8 and 1, with 323 and 276 genes whose expression showed a positive slope during the T2h to T16h stage. The second most abundant group was Cluster 4, which contained 295 genes whose expression showed a negative slope from C0h to T2h. The functional category distribution frequency was then calculated for each cluster to identify differences in the distribution of genes among the three cold stress stages (Figure 4).
Figure 4
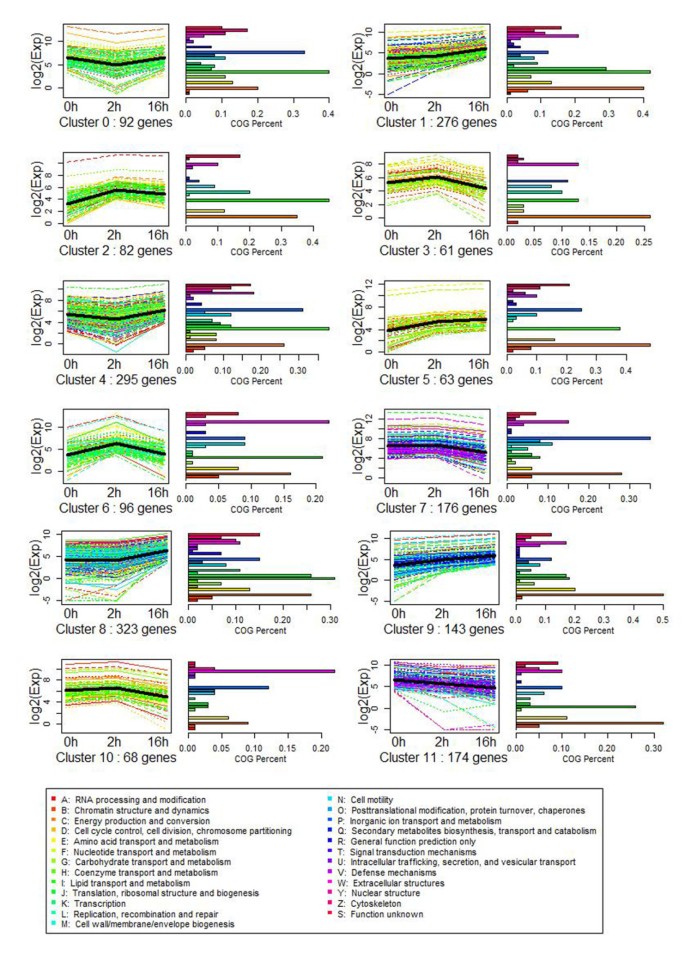
SOM cluster analysis of gene expression in the 12 different patterns. Clusters were obtained by the k-means method on the gene expression profiles of the 1849 modulated genes. The most abundant group is Cluster 8 and 1, with 323 and 276 genes whose expressions show positive slopes during stage of T2h to T16h embryogenesis. The second abundant group is Cluster 4, which contained 295 genes whose expression shows a negative slope from C0h to T2h.
Clusters 2, 3, 6, 7, and 10 comprised 483 genes up-regulation at CT2h but down-regulation in CT16h, including different pathways as the 'energy production and conversion', 'inorganic ion transport and metabolism', 'extracellular structures', 'lipid transport and metabolism’ and 'coenzyme transport and metabolism’genes. They indicate that cells accept the cold signal and instantly reflect using the ion transport and extracellular structure activities during the CT2h stage. In total, 802 genes up-regulated in clusters 1, 5, 8 and 9 in whole stages of cold response. They are specifically included in different pathways, such as 'carbohydrate transport and metabolism', 'amino acid transport and metabolism', 'translation, ribosomal structure and biogenesis’ 'energy production and conversion', and 'signal transduction and mechanism’ (Figure 4), suggesting that L. lancifolium initiates carbohydrate conversion and metabolism during CT2h to 16 h.
Response of important transcription factors to cold stress
Transcription factors (TF) play crucial roles not only as molecular switches for gene expression, but also as terminal points of signal transduction in the response to cold stress. At the 2 h and 16 h stages cold treatment for L. lancifolium, the genes whose transcript abundance exhibited highly dynamic changes (|log2(ratio)| ≥ 4, Figure 5) included genes for transcription factors (LlAP2/EREBP (KJ489026) transcription protein, _LlNA_C (KJ467622) domain transcription, LlERF2 transcription factor, LlBZIP transcription factor protein, LlMYBR (KJ467623) family domain class transcription factor), signal transport (ABC transporter, _LlCalcium_-transporting ATPase 4, LlZIP transporter, Zinc transporter, sugar transporter protein), stress kinases (_LlCalcium_-dependent protein kinase 1 (KJ467621), _LlCBL_-interacting protein kinase 25, Serine/threonine-protein kinase LlSAPK3, Putative LlsnRK/SAPK family protein kinase), low temperature induced-like proteins (DRE-binding protein LlDREB1/CBF (KJ467618), Cold-regulated LlCOR12 (KJ489025), Putative WRKY DNA-binding domain superfamily protein, Peptidyl-prolyl cis-trans isomerase, Copia LlLTR rider, Elicitor-inducible LlLRR, Arachidonic acid-induced LlDEA1), and stress-associated compound proteins (Ll30S ribosomal protein, LlClass III homeobox-leucine zipper protein, LlHeat shock cognate 70 kDa protein 2 (KJ467620), LlGPAT protein (KJ467617), Calcium-dependent calmodulin-independent protein (KJ467621); Figure 5, Additional file 1: Table S1).
Figure 5
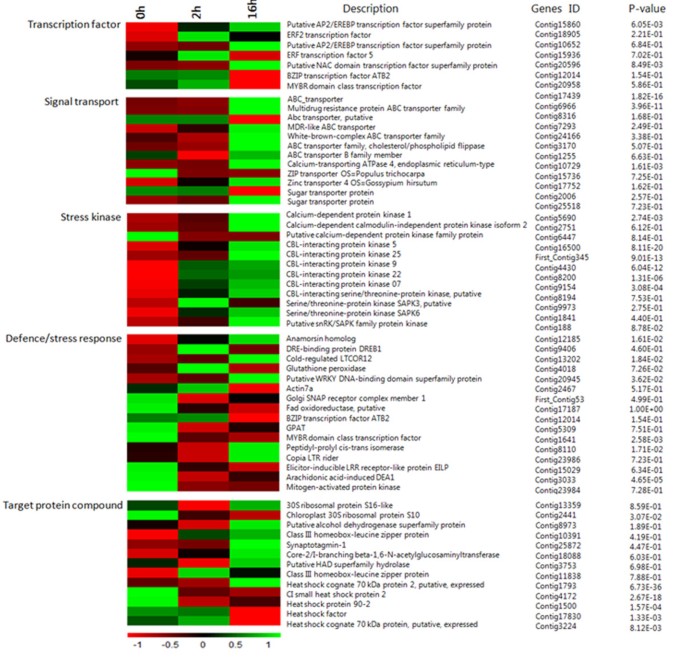
Heat-map of 65 differentially expressed genes involved in transcription factor, signal transport, stress kinase, defense/stress response, target protein compound in the cold response and acclimation of Lilium lancifolium . They were differentially expressed between the control 0 h, cold treatments 2 h and 16 h. The bar represents the scale of the expression levels for each gene (log10 RPKM (number of reads per kilobase per million clean reads)) in the cold response and acclimation as indicated by green/red rectangles. Green indicates up-regulation of genes and Red indicates down-regulation and no change is indicated in black. Complete information for each gene list can be found in Additional file 1: Table S1.
Therefore, the changes in the expression patterns of distinct transcripts suggest a requirement for different developmental events when L. lancifolium is under cold stress. For example, seven transcription factors preferentially expressed and 12 signal transport transcripts accumulated to a higher level at 2 h than 16 h, which indicated that transcription factors reacted more actively to cold in the initial short-term response rather than the long-term cold acclimation (Figure 5, Additional file 1: Table S1). Transcription factors (TF) act as switches and terminal points of signal transduction in a stress-specific fashion in the response to cold stress. In addition, 12 stress kinase protein genes and 13 stress-associated compound protein genes showed the highest accumulation in the cold-treatment 16 h stages which was expected, as expected for cold resistance. Interestingly, the 16 genes related to low temperature induced-like proteins showed different trends. Three of them showed higher expression in CT2h and then decreased in CT16h, such as DRE-binding protein LlDREB1, _LlCBL_-interacting protein kinase, Glutathione peroxidase and LlBZIP transcription factor ATB2, and five of them reached the highest expression value at 16 h, including Cold-regulated LlCOR12, LlWRKY DNA-binding domain superfamily protein, Copia LlLTR rider and Peptidyl-prolyl cis-trans isomerase; the rest of them were down-regulated from 0 h to 16 h (Figure 5, Additional file 1: Table S1). All of these results may contribute to identifying the signaling development networks in response in the cold reponse of L. lancifolium.
Verification of the gene expression profiles by qRT-PCR
To further verify the expression profiles of the genes in our Illumina sequencing analyses, we selected 10 DEGs for qRT-PCR using samples of leaves, stems, roots and bulbs originally used for RNA-Seq, all of which are known to be related to cold stress, including the genes encoding LlAP2 (putative AP2/EREBP transcription factor, KJ489026), LlLEA (late embryogenesis abundant protein, KJ489024), LlCIS (peptidyl-prolyl cis-trans isomerase, KJ467624), LlDREB1 (dehydration-responsive element binding, KJ467618), LlHOT (heat shock protein, KJ467620), LlCOR12 (cold-regulated LTCOR12, KJ489025), LlMYBR (MYBR domain class transcription factor, KJ467623), LlNAC1 (NAC domain protein, KJ467622), and LlCDPK (calcium-dependent protein kinase, KJ467621), LlGPAT (KJ467617). The Ct values of the LlTIP1 rRNA in all samples ranged from 24.0 to 26.0. All 10 transcripts showed the same expression pattern as the in silico differential analysis results from high-throughput sequencing. We have provided GenBank accession numbers for our gene nucleotide sequences in Table 4 and Additional file 2: Txt S3.
Table 4 Primers used in real-time quantitative PCR of Lilium lancifolium (RT-qPCR)
These genes were selected for their key roles in regulating stress signal transcription, cold responses, and cold acclimation. The results presented in Figure 6A and 6B showed that the expression levels of six genes were higher during CT2h to CT16h than in the other stages, including LlCIS, LlMYBR, LlGPAT, LlCOR12, LlDREB1 and LlCDPK, indicating their signal transduction and transcriptions reaction after receiving the cold signal. Four genes, encoding LlAP2, LlLEA, LlNAC1 and LlHOT, were more highly expressed between 24 h and 48 h than in the initial stages, and showed higher expression values than the former six genes (Figure 6A B), demonstrating that these genes may react slowly after the transcription factors and are related to cold acclimation. Also, the expression levels of the LlLEA, LlNAC1, LlCDPK and LlDREB1 genes were highest in bulb at 2 h and 16 h than in the other tissues, including roots and stems and reached a higher value in the stem CT2h sample. The LlCDPK genes were remarkably highly expressed in roots at 16 h (Figure 6C, D). We predicted that the bulb has played an important role in for the L. lancifolium to resistance and adaptation to cold stress. These results indicated that there was a close correlation between the expression changes (fold difference) measured by RNA-Seq and those by qRT-PCR (Table 4).
Figure 6
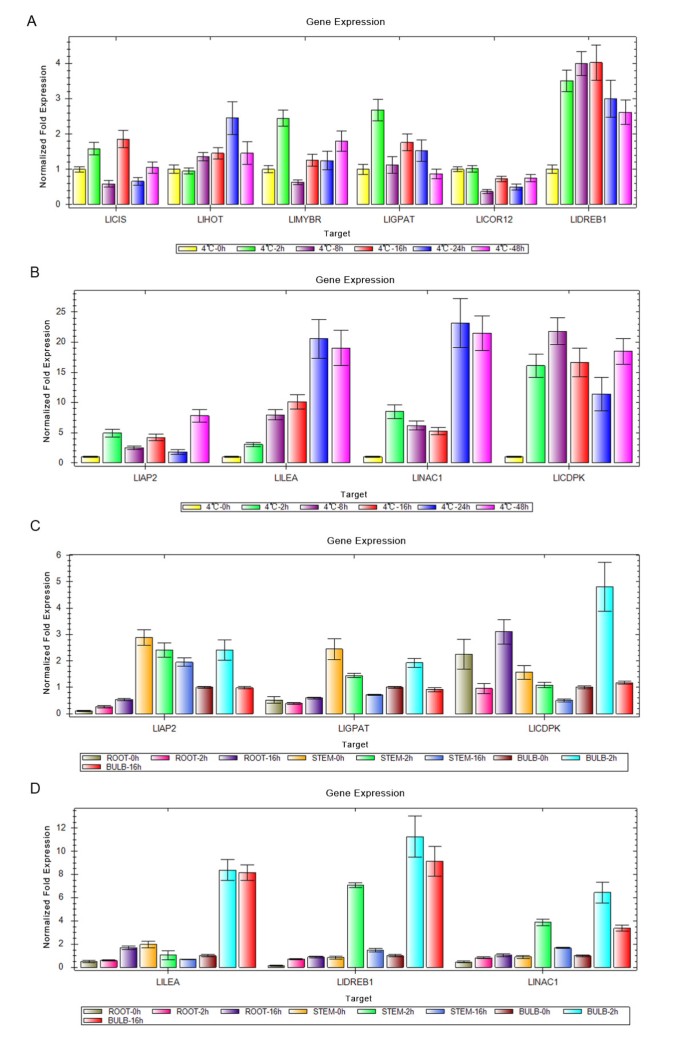
The expression profiles of 10 transcripts in Lilium lancifolium by the quantitative reverse transcription polymerase chain reaction (qRT-PCR). The Figures A and B indicated the expression of ten genes from their leaves; the Figures C and D implied the expression of six genes from their stems, roots and stems. The y-axes show normalized fold expression levels determined by qRT-PCR.
Soluble protein, starch, soluble sugar and Malondialdehyde concentration
To elucidate the mechanism underlying the cold response, it is important to determine how plants alter gene expression in response to this biological process. The levels of two measured soluble proteins increased sharply and rapidly following cold exposure of L. lancifolium and Oriental hybrid leaves, but the soluble protein levels of L. lancifolium declined more sharply and faster than those of the Oriental hybrids from 2 d to 14 d of cold exposure, and the totals of the two varieties showed significant differences that reached a maximum at 24 h (Figure 7A, Additional file 1: Table S2). On an area basis, starch tests showed that L. lancifolium leaves had increasing starch content between 1 h to 48 h of cold treatment, which rapidly fell during subsequent cold stress; although unlike L. lancifolium, the cold development slowly and incrementally increased the starch content of the Oriental hybrids leaves (Figure 7B, Additional file 1: Table S2). Soluble sugar levels also decreased, with linear regression significant on a CT basis, and exhibited a greater decrease in L. lancifolium than in the Oriental hybrids when measured at 4°C (Figure 7C, Additional file 1: Table S2); however, the data suggested a peak at 16–24 h. Total nonstructural carbohydrates (TNCs) responded non-linearly, because of the influence of starch and glucose. Both in L. lancifolium and Oriental hybrid leaves_,_ cold development increased MDA levels relative to the cold response from 1 h to 16 h, whereas, MAD content in L. lancifolium leaves declined significantly with increasing cold exposure duration at 4°C in contrast to the increase in the Oriental hybrids during the same stage (Figure 7D, Additional file 1: Table S2). The L. lancifolium leaves responded more strongly than Oriental hybrid leaves during the cold treatment.
Figure 7
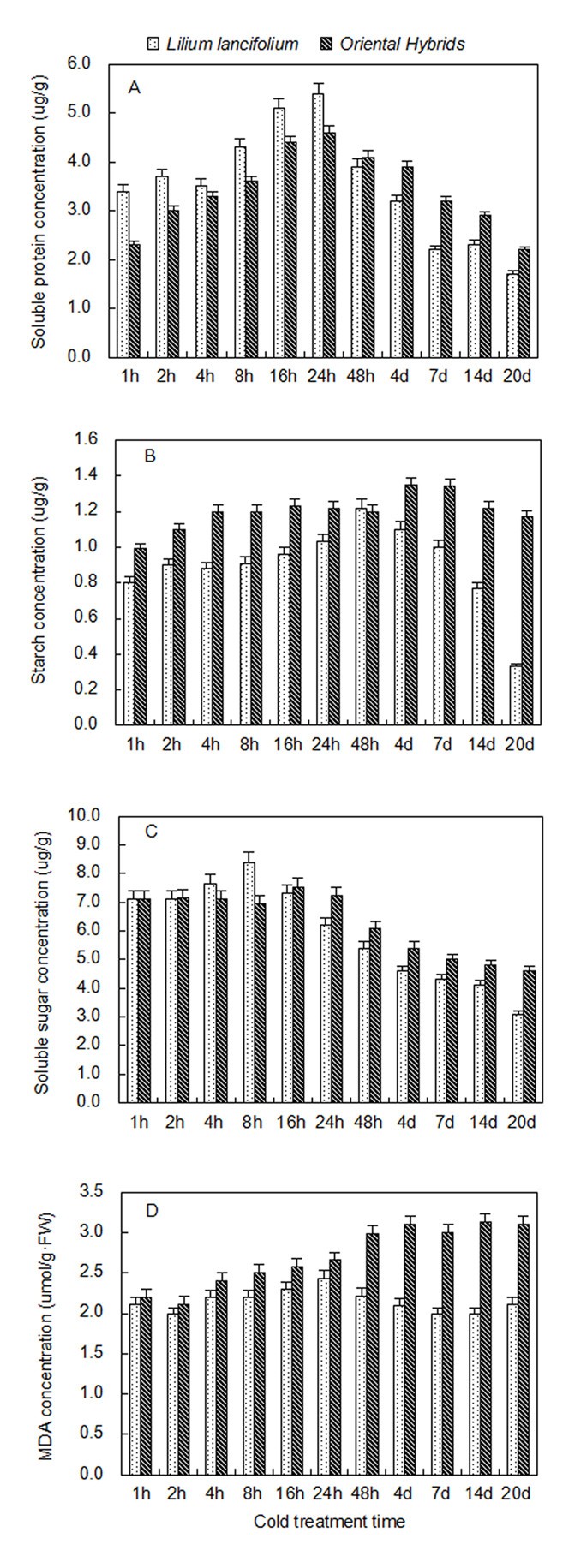
Soluble sugar, starch, soluble protein and MDA concentration per unit 4°C cold treatment (CT) of Lilium lancifolium and Oriental hybrids ’ leaves. A. Two cultivars’ soluble protein content in different stages under 4°C cold treatments B. Two cultivars’ starch content in different stages under 4°C cold treatments C. Two cultivars’ soluble sugar content in different stages under 4°C cold treatments D. Two cultivars’ MDA content in different stages under 4°C cold treatments Black squares: Lilium lancifolium; grey squares: Oriental hybrids’fructose; Bars refer to standard errors.
Membrane systems and cellular osmoprotectant
Membrane systems, which are known to be the primary site of freezing injury in plants, suffer multiple forms of damage caused by freeze-induced cellular dehydration [22]. During cold acclimation, plants experience improved cold tolerance with increased cellular metabolic activity, and increased concentrations of unsaturated fatty acids and phospholipids [23]. Correspondingly, we identified a total of 1153 genes (4.46%) involved in 'Lipid transport and metabolism’ and 3333 genes (7.01%) involved in 'Intracellular trafficking, secretion, and vesicular transport’ according to the COG classification. Furthermore, according to the metabolic pathway enrichment analysis, the 'biosynthesis of unsaturated fatty acids’ (ko01040), 'fatty acid elongation’ (ko00062) and 'regulation of actin cytoskeleton’ (ko04810) pathways were all involved in lipid metabolism. Increases in the biosynthesis of unsaturated fatty acids improve cold defense and prevent damage caused by low temperatures [24]. Thirty genes in this process showed significant regulation of their transcripts after cold stress. For example, two chloroplast omega-3 fatty acid desaturase genes (Contig10730_All, Contig17187_All) were up-regulated by 0.38 to 1.15-fold. The FAD3 gene in L. lancifolium encodes a chloroplast membrane-associated omega-3 fatty acid desaturase, which contributes to freezing tolerance by altering the lipid composition [25]. In our present study, the analysis of the structural characteristics of L. lancifolium leaf cells showed that leaf thickness doubles compared with room-temperature controls during cold treatment. This thickening is a result of palisade cell length elongation and an enlargement of the intercellular spaces caused by a more loosely packed spongy parenchyma matrix [26]. The intercellular spacing and spongy parenchyma packing were homoplastically altered following the cold treatment, they were close together at 16 h, but the palisade cell length and spacing increased prior to 48 h, and was most altered after 7d of treatments (Figure 8).
Figure 8
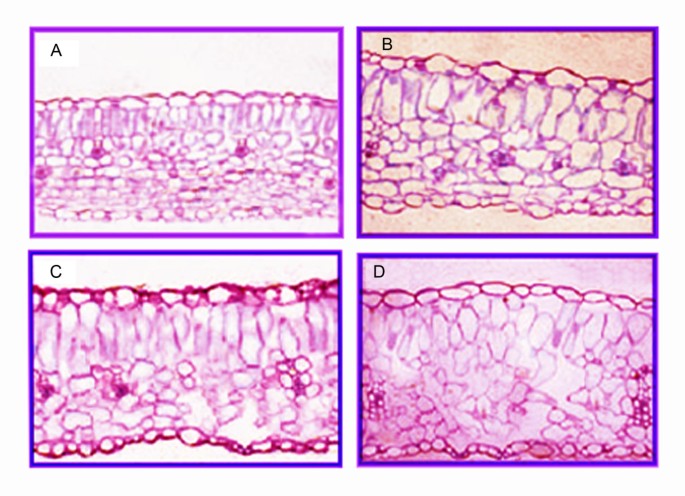
Lilium lancifolium leaf structures in different stages under 4°C cold treatments. A. Controlled 0 h(grow up in room-temperature) leaf structure; B. cold treatment 16 h leaf structure; C. cold treatment 48 h leaf structure; D. cold treatment 7 days leaf structure.
Proline, one of the most effective organic osmolytes, participates in responses to various environmental stresses [27]. Moreover, according to the metabolic pathway enrichment analysis, 'proline metabolism’ (ko00330) involved 69 genes that regulate and relieve the osmotic stress caused by cold-induced dehydration. Two genes (Contig7616, Contig7617) for delta1-pyrroline-5-carboxylate synthetase (P5CS) were significantly up-regulated by 0.88 and 1.29-fold after cold stress, respectively. As a key enzyme in proline synthesis, DREB1 participates in the cold-stress response and shows high expression, which promotes the synthesis of proline for cold tolerance [28]. The accumulation of sucrose and other simple sugars also contributes to the stabilization of membranes, as these molecules protect membranes against freeze-induced damage in vitro. Five LEA (late embryogenesis abundant)-related genes (Contig24352_All, Contig19954 _All, Contig17189_ALL, Contig26956, Contig18777) were also differentially expressed with fold changes in their expression ranging from 0.23 to 1.30-fold. The LEA protein functions as an antioxidant, as well as a membrane and protein stabilizer, during cold stress [29]. Our studies have analogously suggested that the electrical conductivity of L. lancifolium leaves decreases at 2 h after but increases at 12 h, and then keeps a positive slope until 24 h cold treatment It is reasonable to infer that hydrophilic and LEA polypeptides stabilize membranes against freezing-induced injury in _L. lancifolium_[30]. Proline levels further enhanced the electrical conductivity of L. lancifolium leaves in different stages under 4°C cold treatment (Figure 9). The electrical conductivity dropped at 2 h, but kept gradually increasing with prolonged cold treatment. It can be inferred that the accumulation and effects of Proline may contribute to a remarkable control of cellular electrical conductivity, which reflects the destruction of cell wall and cytoplasm motivation, furthermore regulating outstanding cold tolerance for L. lancifolium (Figure 9).
Figure 9

Electrical conductivity expression of Lilium lancifolium leaves in different stages under 4°C cold treatments.
Discussion
Illumina paired-end sequencing, assembly, and functional annotation
Transcriptome analysis is important for elucidating the molecular constituents of cells and tissues and interpreting the functional elements of the genome [31]. In our study of the transcriptome of L. lancifolium (3n = 27), we sampled the pooled transcriptomes of leaves using Illumina paired-end sequencing technology to generate a large-scale EST database. Approximately 10.7 GB of data was generated and assembled into 37,843 unigenes. This large number of reads with paired-end information produced much longer unigenes (mean, 973 bp) than those in other lily studies. This increased coverage depth of the transcriptome facilitated de novo assembly, enhanced sequencing accuracy, and avoided possible contamination. Of the 37,843 L. lancifolium unigenes, 18,736 (49.30%) had homologs in the Swiss-Prot database. More importantly, we were able to assign a number of these unigenes to a wide range of GO categories and COG classifications (Figures 1 and 2), indicating that diverse transcripts are involved in the cold response and which are represented in the sequence data of this species, and also reflecting the complexity of differential low temperature signal transcription in L. lancifolium.
Furthermore, during cold responses and tolerance, plants receive low temperature signals and initiate a defense mechanism, including physical structure adaptations (changes in lipid composition and extracellular metabolic activity), increases in intercellular osmoprotectants (such as soluble sugars, proline and betaine), and up-regulated synthesis of anti-oxidants (superoxide dismutase, pathogen defense, catalase and ascorbic acid reductase), enabling restoration of the balance of biosynthesis and carbohydrate metabolism and enhancing survival in cold environments [32]. Interestingly, part of the expression patterns of a large number of genes during cold stress stimuli and transcription factors have been detected using transcriptome sequencing and microarray technologies. Most representative unigenes were annotated to specific pathways, such as metabolic pathways, biosynthesis of secondary metabolites, microbial metabolism in diverse environments, the mRNA surveillance pathway, ribosomes, pyrimidine metabolism, biosynthesis of amino acids, the cell cycle, carbon metabolism and plant hormone signal transduction using the KEGG databases (Table 3), leading us to conclude that most of the genes we identified are involved in the cold response and signaling regulation.
Carbohydrate metabolism
A principal factor in carbohydrate metabolism under stress conditions is regulation of the balance between biosynthesis and breakdown of proteins. Proteolysis plays a dynamic and vital role in the regulation of different metabolic processes, and in the cell’s response to environmental stimuli. It controls metabolic fluxes by regulating the levels of key rate-limiting enzymes, while also irreversibly irreversibly polypeptides into soluble sugars that may interfere with these pathways. Much of this directed protein turnover is performed by proteases that require ATP and the Clp protease is one of the best characterized to date [33]. Through our KEGG analysis, we discovered 186 regulated genes in the 'Starch and sucrose metabolism’ pathway and 208 regulated genes in the 'Protein processing in endoplasmic reticulum’ pathway (Table 3). Moreover, the protein of L. lancifolium increased with the rapid cold stimulus and then degraded into sugars with a faster reduction trend than the Oriental hybrids during the cold treatment. This demonstrated that L. lancifolium could adapt to 4°C cold treatment better than the Oriental hybrids, by accumulating protein and converting it into sugar; increasing the cell liquid concentration and reducing the freezing point, so as to prevent the frost damage (Figure 7A, Additional file 1: Table S2). This supports the fact that L. lancifolium can acclimate to a cold environment and elicit a series of physiological and biochemical responses to low temperature, such as the transformation and combination of soluble sugars (Figure 7C, Additional file 1: Table S2). The reason for the difference is that Oriental hybrids grow in slightly lower latitudes than L. lancifolium and their optimum growth temperature is 25°C. Interestingly, in the L. lancifolium transcriptome_,_ we found related proteolytic enzymes, including an ATP-dependent Clp protease proteolytic subunit, with a fold change of 0.94 (down-regulation) from 0 h to 2 h but a fold change of 1.28 (up-regulation) during 2 h to 16 h (Contig24056_All), a Clp-like energy-dependent protease, which showed a 1.56 fold change (Contig1979_All), and also Sucrose phosphate synthase and Glucose-6-phosphate 1-dehydrogenase, which were up-regulated with 1.67 and 1.12 fold changes, respectively (Contig7878_All, Contig3504_All). Overall, the data suggested that transfer of L. lancifolium leaf soluble protein is crucial for determining soluble synthesis, and induces better cold resistance than Oriental hybrid leaves, which cannot respond and acclimate to low temperature (Figure 7C, Additional file 1: Table S2).
The enzymes Alpha-amylase, β-amylase and Isoamylase degrade starch to soluble sugar, the further conversion of which leads to increased maltose, glucose, fructose and sucrose levels [34]. The effects of CT and cold duration on the expression of alpha-amylase, β-amylase, and isoamylase in L. lancifolium were shown by Illumina sequencing, with 2.25-fold up-regulation from 0 h to 2 h and 0.93-fold down-regulation from 2 h to 16 h, 0.73-fold up-regulation from 0 h to 2 h; and 1.78-fold up-regulation from 0 h to 2 h and 0.70-fold down-regulation, from 2 h to 16 h, respectively (Contig5436_all, First_Contig433_all, Contig13233_all). Thus, it is unsurprising that a subset of starch hydrolysis enzyme genes display a highly specific, largely up-regulation response between 0 h to 2 h of 4°C cold stress, because hydrolysis enzymes affect the degradation of carbohydrate during the early period of cold stress [35]. On an area basis, starch tests showed that the starch content of L. lancifolium leaves had increased between the 1 h to 48 h of cold treatment, and fell during subsequent cold stress as it was degraded into soluble sugar. The limited starch degradation of Oriental hybrid leaves indicated its poor ability to resist and adapt to cold exposure (Figure 7B, Additional file 1: Table S2). In L. lancifolium, we detected that the expression of 47 soluble sugar synthase DEGs out of 1326 different enzyme DEGs was significantly and markedly induced by 2 h and 16 h cold stress, including five up-regulated and 19 down-regulated glucose synthase genes, two up-regulated and four down-regulated fructose synthase genes and three up-regulated and 14 down-regulated sucrose synthase genes. Thus, exposure to 4°C cold treatment clearly favored starch degradation, which would result in increased accumulation of soluble sugars. In addition to the effects of chilling on β-amylase transcript levels, we found that CT increased the expression levels of a specific Sucrose-phosphate synthase7 (SPS7) transcript (First_Contig33) by 0.73-fold. Because SPS1 catalyzes sucrose synthesis, it is possible that increased accumulation of sucrose in L. lancifolium may contribute to cold tolerance (Figure 7B, C, Additional file 1: Table S2).
MDA (malondialdehyde) reflects the ability of plants to resist and acclimate to abiotic stress [36]. In L. lancifolium under 4°C cold treatment, MDA showed no obvious change during the early cold period but declined during the 48 h to 20 d period, which demonstrated the better cold resistance characteristics of in L. lancifolium compared with the Oriental hybrid (Figure 7D, Additional file 1: Table S2). To some extent, it is possible that MDA is related with proline, and the up-regulation of the delta1-pyrroline-5-carboxylate synthetase (P5CS, Contig7616_all, 1.29 fold) would enhance proline concentration to strengthen the plant’s osmotic adjustment ability, resulting in the mitigation of cellulose peroxidation.
Signaling pathways
In endocrine signaling, hormones act on distant target cells in paracrine signaling, a molecule released from a cell acts on nearby targets and in autocrine signaling, a cell produces a signaling molecule to which it also responds [36]. The binding of most signaling molecules to their receptors initiates a series of intracellular reactions that regulate virtually all aspects of cell behavior, including metabolism, movement, proliferation, survival, and differentiation. Understanding the molecular components of these pathways and how they are regulated has thus become a major area of research in contemporary cell biology [37]. Two major pathways of intracellular signaling are based on the use of second messengers derived from the membrane phospholipid phosphatidylinositol 4,5-bisphosphate (PIP). PIP1 and PIP2 are minor components of the plasma membrane, localized to the inner layer of the phospholipid bilayer. A variety of hormones and growth factors stimulate the hydrolysis of PIP by a phospholipase C-a reaction that produces two distinct second messengers: diacylglycerol and inositol 1,4,5-trisphosphate(IP3), which can stimulate distinct downstream signaling pathways (protein kinase C and Ca2+ mobilization, respectively), so that PIP1 and PIP2 hydrolysis triggers a two-armed cascade of intracellular signaling. In the L. lancifolium trancriptome, six genes in the PIP process showed significant transcript regulation after cold stress (Figure 10). For example, two Plasma membrane intrinsic protein genes (Contig1854_All, Contig1406_All) were up-regulated by 0.58 and 1.36-fold. Thus, the LlPIP 1 and LlPIP 2 genes in L. lancifolium encode plasma membrane intrinsic proteins, which contribute to triggering the protein kinase C and stimulating Ca2+ signaling pathways in response to low temperature [38].
Figure 10
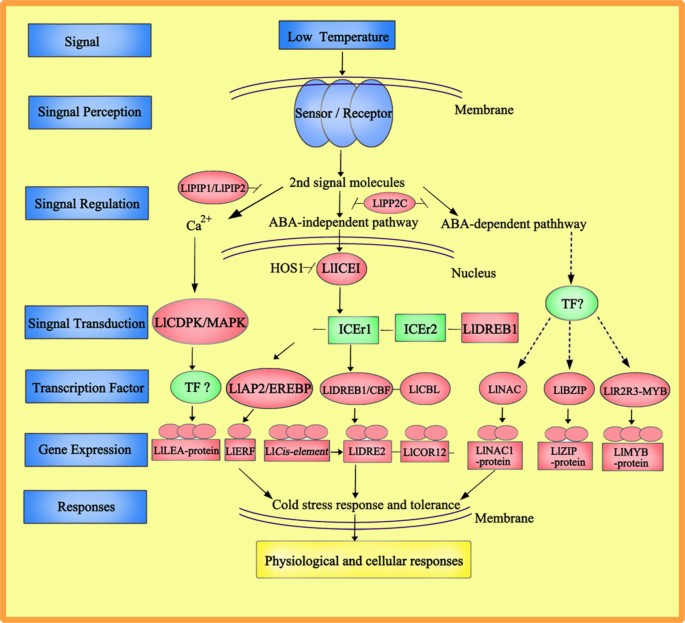
Models describing the signaling pathways involved in the acquisition of cold tolerance. The red box showed the indentified genes in Lilium lancifolium transcriptome; and the blue box demonstrated the unknown transcription factors and pathways in Lilium lancifolium.
Here, in L. lancifolium, protein kinase C including the receptor protein kinase LlCLAVATA1, a protein kinase Ck2 regulatory subunit and 2Serine/threonine-protein kinase cdk9 (Contig18123_All, Contig8528, Contig14527), showed up-regulated expression from 0.33- to 1.01-fold during the cold treatment, bridging the process of signal transduction. Another two contrasting themes are apparent in the expression of the signaling pathways initiated by Ca2+ and cAMP. These two second messengers may operate towards a similar goal. The activation of phosphorylase kinase through either PKA or Ca2+ is an example of such convergence. Ca2+ and cAMP-mediated pathways may also be coordinated through Ca-calmodulin dependent isoforms of adenylyl cyclase [36]. As an important second messenger, Ca2+ plays a vital role in the plant cold-stress response. The concentration of Ca2+ increases rapidly during cold stress, followed by a number of signals mediated by combinations of protein phosphorylation cascades [39]. As a large subfamily of plant kinases, Calcium dependent protein kinases (CDPKs) are implicated as important sensors of Ca2+ flux in plants in response to a variety of biotic and abiotic stress stimuli [40]. We identified two genes (Contig22048_All, Contig2751_All) related to CDPK, with fold changes ranging from 0.89- to 3.28-fold in their expression after cold stress (Figure 9); they have been demonstrated to activate a stress and ABA-inducible promoter. These results demonstrate the connection of particular CDPKs to specific signaling pathways in vivo and the usefulness of engineering CDPKs to enhance abiotic stress tolerance in L. lancifolium (Figure 10).
In many plant cells, Abscisic acid (ABA) also plays a crucial role in the cold tolerance of plants. The type 2C protein phosphatases (PP2C), which negatively regulate ABA responses, play a key role in ABA signal transduction [8, 39]. In this study, two DEGs (Contig24025_All, Contig19208_All) related to PP2C were identified that showed significant down-regulation, with fold changes ranging from 0.95 to 0.35, in their expression after cold stress. The cold response has been reported to involve both ABA-dependent and independent pathways [41]. In the ABA-independent pathway, the transcription factor of DREB1 (DRE-binding protein) has also implicated in dehydration stress signaling in Arabidopsis [42]. In our research, one gene (Contig9406_All) related to LlDREB1 was identified, with a fold change of 0.31. We presume it would trigger and induce the expression of LlCOR12 and LlDRE2 (Contig13202_All, Contig12185_All), in combination with the up-regulation of the _LlCIS_-element (Contig8665) by a change of 1.51-fold. One of the big gene families that have been investigated in our study not only includes the LlAP2/EREBP (Contig10652_All) transcription factor, but also cold genes such as LlERF2, LlERF3, LlERF5, LlERF10 and LlmTERF (Contig18905_All, Contig772_All, Contig15936_All, Contig26562_All, Contig28555_All). On the other hand, the ABA-dependent pathway has genes related to transcription factors; here, LlNAC and LlBZIP (Contig20596_All, Contig12014_All) in L. lancifolium were differentially expressed with fold changes of 0.46 and 1.56, respectively, subsequently motivating the expression of the LlNAC1 and LlZIP proteins after cold exposure for 16 h. Moreover, we also obtained a large amount of information on the MYB family from L. lancifolium Illumina sequencing, including the chief transcription factors Ll1R-MYB1, LlR2R3-MYB and LlMYBR (Contig22140_All, Contig18508_All, Contig1641_All), which showed fold changes of 1.01, 6.30, and 1.85, respectively, and stimulated the identified cold gene LlMYB-DNAbinding protein up-regulation with a 2.92-fold change (Contig11131_All). Therefore, it is likely that the LlDREB1, LlCBL, LlAP2/EREBP, LlNAC, LlBZIP and LlR2R3-MYB genes of L. lancifolium show transcription patterns under cold stress similar to those under other abiotic stresses, which are believed to activate the transcription of specific target genes in ABA signaling in guard cells (Figure 10).
Whereas the cells of prokaryotes and unicellular eukaryotes are largely autonomous, the behavior of each individual cell in multicellular plants must be carefully regulated to meet the needs of the organism as a whole. This is accomplished by a variety of signaling molecules that are secreted or expressed on the surface of one cell and bind to receptors expressed by other cells, thereby integrating and coordinating the functions of the many individual cells that make up the complex organisms [37]. In this case, cells receive and respond to signals from a cold environment, and then cold signals are received by sensors on the membrane and the second signal molecules carry them from the cytoplasm to the nucleus by the catalytic protein kinase regulatory subunit. Within the nucleus, protein kinase C and PP2C induce the Ca2+ pathway and ABA signal transduction, respectively, and various transcription factors such as LlDREB1, LlAP2/EREBP, LlNAC, LlBZIP and LlR2R3-MYB recruit coactivators for the transcription of inducible genes including LlLEA LlERF, LlDRE2, LlNAC1, LlZIP and LlMYB-DNAbinding protein, which are regulators of cellular cold tolerance and metabolic activity in short-term cold stress, and LlFAD3, Llβ-amylase genes, LlP5CS and LlCLS, which enhance adaptation processes that involve changes in the expression of transcripts related to cellular osmoprotectants, and carbohydrate metabolism during the long-term cold stress. Such regulation of gene expression plays important roles in controlling the physiological cold response, cellular morphology, proliferation, survival, and differentiation of a wide variety of plant cells, as well as being implicated in learning and memory (Figure 10).
To further study the mechanism of the cold response and acclimation of L. lancifolium, we will select specific genes and verify their functions and expression by fluorescence in situ hybridization and genetic modification technology.
Conclusions
To the best of our knowledge in L. Lancifolium,, interest is further heightened by the fact that stress-regulated genes are stimulated and reacted during the short-term cold response, along with the physiological and biochemical changes during the long-term cold acclimation. The genome-wide transcriptome and physiological analysis presented in this study has expanded our knowledge of this process by identifying differentially expressed genes involved in cold regulation of carbohydrate metabolism, leaf structure and three model signaling pathways, the Ca2+ and ABA-dependent/independent pathways. Importantly, the high-resolution expression patterns presented here further our understanding of the molecular mechanisms involved in cold resistance and signal regulation for bulb flower breeding.
Methods
Ethics
This research did not involve any human subjects, human material, or human data. The field study did not involve any endangered or protected species.
Plant material and total RNA isolation
The materials used in these experiments were derived from _L. lancifolium_’s and Oriental hybrids cultivated in the nursery of Beijing Forestry University (BJFU) (116.3°E, 40.0°N) under the following growth conditions: 70% relative humidity, 25°C:18°C, day:night temperatures, with watering every 3 days. The bulbs were collected and stored at 4°C for one month. Then, the bulbs were cultivated under aseptic conditions to induce leaf formation. After 4 weeks of asepsis condition, we divided the plantlets into two groups; the control sample (CK0h) and the cold-treated sample (CT). Fresh leaves, stems, bulbs, and roots were subjected to a 4°C cold treatment for 2 h, 8 h, 16 h, 24 h, 48 h, 4 d, 7 d, 14 d, or 20 d. At each time point, samples were collected and stored at -80°C until RNA extraction. Total RNA was extracted from the tissues using an RNAisomate RNA Easyspin Isolation System (Aidlab Biotech, Beijing, China) according to the manufacturer’s instructions. The quality of RNA was verified using a 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). The RNA concentration was at least 160 μg/mm3 in all samples. To prepare cDNA, we used a pooled RNA mixture containing 60 μg RNA from each sample.
cDNA library preparation and transcriptome sequencing
Illumina sequencing was conducted using the Solexa mRNA-Seq platform at the Shanghai manufacturer’s instructions (Illumina, San Diego, CA, USA). Briefly, we used magnetic beads with oligo(dT) to isolate poly(A) mRNA after isolating total RNA from L. lancifolium leaves in the control (0 h) and after 2 h and 16 h of cold treatment. Second-strand cDNA was synthesized using appropriate buffers, dNTPs, RNase H, and DNA polymerase I. ShoBiotechnology Corporation (SBC), Shanghai, China (http://www.ebioservice.com) according to the rt fragments were depurated with a QiaQuick PCR extraction kit (Qiagen, Hilden, Germany) and resolved with an elution buffer for end repair and by addition of poly(A). For PCR amplification, we selected suitable fragments as templates based on the results of agarose gel electrophoresis. The library was sequenced using an Illumina HiSeq™ 2000. Because raw reads produced from sequencing machines contain low-quality reads that negatively affect subsequent bioinformatics analyses, we discarded reads with adapters, those with more than 5% unknown nucleotides, and those of low quality (≤ 20% of the bases with a quality score (Q) ≤ 10) using an in-house Perl script. The average proportion of clean reads in each sample was 89.6%–91.7%. The clean reads were used for further analyses.
Analysis of Illumina transcriptome sequencing results
De novo assembly was carried out using scaffolding contig methods with CLC Genomics Workbench (version: 5.5) with the default parameters, and a minimum contig length of ≥400. The assembled de novo sequences were designated as primary unigenes. After assessing the different K-mer sizes, we found that 29-mer yielded the best assemblies and so this size class was selected to construct the de Bruijn graph. Primary unigenes from UniGene of three samples’ were assembled using CAP3 ES, yielding final unigenes. Assembled final unigenes were used for BLASTx searches (E-value <1e-5) against the UniProt database (date: 2013.04) and the Swiss-Prot protein database (date: 2013.05) (http://www.expasy.ch/sprot), which has the largest and most particular protein annotation database (approximately including 24,889,084). To functionally annotate sequences, we used Blast2GO program (Conesaet et al., 2005) to assign gene ontology (GO) terms (http://www.geneontology.org). Also, to predict and classify possible functions, 13705 unigene sequences were aligned to 25 Clusters of Orthologous Groups (COGs) in the COG database (http://www.ncbi.nlm.nih.gov/COG). Kyoto Encyclopedia of Genes and Genomes Pathway (KEGG; http://www.genome.jp/kegg) annotations were carried out according to the KEGG database using BLASTx (E-value threshold 10-5).
Bioinformatics for functional annotation of differential gene expression
A rigorous algorithm to identify differentially expressed genes was developed based on the method of Audic et al. (1997). The false discovery rate (FDR) was used to determine the threshold of the P-value in multiple tests and analyses. We used an FDR of < 0.001 and the absolute value of log2 (ratio) ≥ 2 as thresholds to define significantly different gene expression [43]. For further analyses, we used an additional criterion, which involved using only differentially expressed genes (DEGs) with a minimum of a four-fold change in expression.
Transcription factors analysis
Transcription factors were predicted according to protein sequences obtained from CDS predictions. We used HMM search to search for plant transcription factor domains (http://plntfdb.bio.uni-potsdam.de/v3.0/) and classified unigenes according to gene family information.
Real-time quantitative PCR verification
Total RNA was isolated from the leaf, stem, bulbs, and roots of lily plants subjected to 4°C cold treatments, as described above. First-strand cDNA synthesis was performed using Superscript II reverse transcriptase (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions, using 1 μg total RNA and oligo(dT) primers. qRT-PCR was performed using a Rotor-Gene 3000 real-time PCR detection system (Qiagen) using SYBR® qPCR Mix (Toyobo, Tokyo, Japan) according to the manufacturer’s protocol. The primers used in this study were designed with Beacon Designer (Premier, Palo Alto, CA, USA) and are listed in Table 1. Real-time PCRs was carried out using prepared cDNA (80 μg) with each set of primers and probe and iQ™ SYBR® Green Supermix (Cat. No.170-8882, Bio-Rad, Hercules, CA, USA). The PCR cycling conditions were as follows:95°C (30 s), 60°C (30 s), and 72°C (15 s). All reactions were performed in biological triplicates. Relative mRNA levels were calculated using the 2-△△Ct method [44] against the internal reference gene TIP1, with expression in CT 0 h used as the internal control. The sequences of primers used for QRT-PCR are listed in Table 4.
Heat-map generation
A heat-map of legume-specific genes and the genes with the highest transcript levels was generated using the heat-map function in the gplots CRAN library. After excluding legume-specific genes that did not have a RPKM normalized log2-transformed transcription count greater than zero in at least one tissue, 315 genes remained. The LSGs were taken from the Glyma1.01 gene set. The genes with the highest transcript levels were determined based on the sum of raw counts in all tissues. Boxes were added to reveal clusters of genes with similarly expression in specific tissues. (Additional file 1: Table S1) showed additional details indicating the gene represented by each cell in the heat-map.
Leaf structural characteristics
To investigate the internal anatomy of leaves, sections were cut through the leaf midrib. The proximal halves of individual leaves were fixed in 0.3 mg/cm3 paraformaldehyde, 5% ethanoic acid, and 50% ethanol, and then dehydrated in a graded ethanol (50%–95%) series. Sections (1 μm thick) were cut with a micrometre (Ultracut UCT, Leica Microsystems,Welzlar, Germany), stained with toluidine, and imaged with a microscope and imaging system (Optiphot 2 with DS-L1, Nikon, Tokyo, Japan). The cut surface was mid-way between the midrib and margin, near the widest point of the leaf.
Carbohydrate and electrical conductivity analysis
Total soluble proteins, soluble sugars, starch, and malondialdehyde (MDA) content were determined using leaf tissue from plants subjected to 1 to 20 days of cold treatment. The leaf tissue was collected and stored at -80°C. Carbohydrate content and electrical conductivity were measured as described by Gilmour [45]. Soluble sugars were analyzed using the phenol-sulfuric acid method. Soluble proteins were determined using the Coomassie brilliant blue colorimetric method. Starch was quantified using the anthrone-sulfuric acid method and MDA content was determined using the thiobarbituric acid method. Absorbance was measured using a plate reader (POLARstar OPTIMA, BMG Labtech, Offenburg, Germany). Electrical conductivity was measured by the bath method using a desktop meter (EC3175-307, JENCO, San Diego, CA, USA).
Availability of supporting data
The data sets supporting the results of this article are available in the [NCBI GenBank] repository, [unique persistent identifier (KJ467617, KJ467618, KJ467619, KJ467620, KJ467621, KJ467622, KJ467623, KJ467624, KJ489024, KJ489025, KJ489026,) and hyperlink to datasets in http://www.ncbi.nlm.nih.gov/genbank/].
And also, the other data sets supporting the results of this article are included within the article and its additional files TXTS3.
Author’s contributions
JMW carried out the molecular genetic studies, participated in the sequence alignment and drafted the manuscript. YY carried out the immunoassays. XHL participated in the sequence alignment. JMW, JH participated in the design of the study and performed the statistical analysis. JHG, QW conceived of the study, and participated in its design and coordination and helped to draft the manuscript. All authors read and approved the final manuscript.
Abbreviations
BLAST:
Basic local alignment search tool’s Control 0 h
CT:
Cold treatment
COG:
Cluster of orthologous group
DEGs:
Differentially expressed geneses Expressed sequence tag
GO:
Gene ontology
KEGG:
Kyoto encyclopedia of genes and genomes
MDA:
Malondialdehyde
qRT-PCR:
Real-time quantitative reverse transcription PCR
SP:
Swiss-Prot protein database
SOM:
Self-organizing map
TF:
Transcription factor.
References
- Bray EA, Bailey-Serres J, Weretilnyk E: Responses to abiotic stresses. Chapter 22. Responses to Abiotic Stresses. Edited by: Gruissem W, Buchannan B, Jones R. 2000, Rockville, MD: American Society of Plant Physiologists, 1158-1249.
Google Scholar - Kreps JA, Wu Y, Chang H-S, Zhu T, Wang X, Harper JF: Transcriptome changes for Arabidopsis in response to salt, osmotic, and cold stress. Plant Physiol. 2002, 130: 2129-2141. 10.1104/pp.008532.
Article CAS PubMed Central PubMed Google Scholar - Cheng C, Yun K-Y, Ressom HW, Mohanty B, Bajic VB, Jia Y, Yun SJ, Reyes BGD: An early response regulatory cluster induced by low temperature and hydrogen peroxide in seedlings of chilling- tolerant japonica rice. BMC Genomics. 2007, 8 (7): 175-
Article PubMed Central PubMed Google Scholar - Christov NK, Yoneyama S, Shimamoto Y, Imai R: Differential expression of wheat genes during cold acclimation. Cytol Genet. 2007, 41 (1): 142-150.
Article Google Scholar - Cao PX, Song J, Zhou CJ, Weng ML, Liu J, Wang FX, Zhao F, Feng DQ, Wang B: Characterization of multiple cold induced genes from Ammopiptanthus mongolicus and functional analyses of gene AmEBP1. Plant Mol Biol. 2009, 69 (6): 529-539.
Article CAS PubMed Google Scholar - Allen GJ, Chu SP, Schumacher K, Shimazaki CT, Vafeados D, Kemper A, Hawke SD, Tallman G, Tsien RY, Harper JF: Alteration of stimulus-specific guard cell calcium oscillations and stomatal closing in Arabidopsis det3 mutant. Science. 2000, 289: 2338-2342. 10.1126/science.289.5488.2338.
Article CAS PubMed Google Scholar - Seki M, Narusaka M, Abe H, Kasμga M, Yamaguchi-Shinozaki K, Carninci P, Hayashizaki Y, Shinozaki K: Monitoring the expression pattern of 1300 Arabidopsis genes under droμght and cold stresses by using a full-length cDNA microarray. Plant Cell. 2011, 13 (6): 61-72.
Google Scholar - Thomashow MF: Freezing tolerance genes and regulatory mechanisms. Ann Rev Plant Physiol. Plant Mol Biol. 1999, 50: 571-599.
CAS Google Scholar - Fowler S, Thomashow MF: Arabidopsis transcriptome profiling indicates that multiple regulatory pathways are activated during cold acclimation in addition to the CBF cold response pathway. Plant Cell. 2002, 14: 1675-1690. 10.1105/tpc.003483.
Article CAS PubMed Central PubMed Google Scholar - Zhang G, Guo G, Hu X, Zhang Y, Li Q, Li R, Zhuang R, Lu Z, He Z, Fang X, Chen L: Deep RNA sequencing at single base-pair resolution reveals high complexity of the rice transcriptome. Genome Res. 2010, 20 (2): 646-654.
Article CAS PubMed Central PubMed Google Scholar - Raherison E, Rigault P, Caron S, Poulin P, Boyle B, Verta JP, Giguere I, Bomal C, Bohlmann J, MacKay J: Transcriptome profiling in conifers and the PiceaGenExpress database show patterns of diversification within gene families and interspecific conservation in vascular gene expression. BMC Genomics. 2012, 13: 434-449. 10.1186/1471-2164-13-434.
Article CAS PubMed Central PubMed Google Scholar - Qiu Q, Ma T, Hu Q, Liu B, Wu Y, Zhou H, Wang Q, Wang J, Liu J: Genome- scale transcriptome analysis of the desert poplar, Populus euphratica. Tree Physiol. 2011, 31 (4): 452-461. 10.1093/treephys/tpr015.
Article PubMed Google Scholar - Xu DL, Long H, Liang JJ, Zhang J, Chen X, Li JL, Pan ZF, Deng GB, Yu MQ: De novo assembly and characterization of the root transcriptome of Aegilops variabilis during an interaction with the cereal cyst nematode. BMC Genomics. 2012, 13: 133-137. 10.1186/1471-2164-13-133.
Article CAS PubMed Central PubMed Google Scholar - Cong HQ, Li ZY, Xu L: Characterizing developmental and inducible differentiation between juvenile and adult plants of Aechmea fasciata treated with ethylene by transcriptomic analysis. Plant Growth Regul. 2013, 69: 247-257. 10.1007/s10725-012-9767-2.
Article CAS Google Scholar - Trick M, Long Y, Meng J, Bancroft I: Single nucleotide polymorphism (SNP) discovery in the polyploid Brassica napus using Solexa transcriptome sequencing. Plant Biotechnol J. 2009, 7 (4): 334-346. 10.1111/j.1467-7652.2008.00396.x.
Article CAS PubMed Google Scholar - Li P, Ponnala L, Gandotra N, Wang L, Si Y, Tausta SL, Kebrom TH, Provart N, Patel R, Myers CR, Reidel EJ, Turgeon R, Liu P, Sun Q, Nelson T, Brutnell TP: The developmental dynamics of the maize leaf transcriptome. Nat Genet. 2010, 42 (12): 1060-1067. 10.1038/ng.703.
Article CAS PubMed Google Scholar - Zhang J, Liang S, Duan J, Wang J, Chen S, Cheng Z, Zhang Q, Liang X, Li Y: De novo assembly and Characterisation of the Transcriptome during seed development, and generation of genic-SSR markers in Peanut (Arachis hypogaea L.). BMC Genomics. 2012, 13: 90-10.1186/1471-2164-13-90.
Article CAS PubMed Central PubMed Google Scholar - Gahlan P, Singh HR, Shankar R, Sharma N, Kumari A, Chawla V, Ahuja PS, Kumar S: De novo sequencing and characterization of Picrorhiza kurrooa transcriptome at two temperatures showed major transcriptome adjustments. BMC Genomics. 2012, 13: 126-10.1186/1471-2164-13-126.
Article CAS PubMed Central PubMed Google Scholar - Pilar M, Gregogy T, Mccollum MP: Transcriptome profiling of grapefruit flavedo following exposure to low temperature and conditioning treatments uncovers principal molecular components involved in chilling tolerance and susceptibility. Plant Cell Environ. 2008, 31: 752-768. 10.1111/j.1365-3040.2008.01793.x.
Article Google Scholar - Pang T, Ye CY, Xia XL, Yin WL: De novo sequencing and transcriptome analysis of the desert shrub, Ammopiptanthus mongolicus, during cold acclimation using IIIimina/Solexa. BMC Genomic. 2013, 14: 488-10.1186/1471-2164-14-488.
Article CAS Google Scholar - Chen JH, Song YP: Genome-Wide analysis of gene expression in response to droμght stress in Populus simomnii. Plant Mol Biol Rep. 2013, 31: 946-996. 10.1007/s11105-013-0563-6.
Article CAS Google Scholar - Kawamura Y, Uemura M: Mass spectrometric approach for identifying putative plasma membrane proteins of Arabidopsis leaves associated with cold acclimation. Plant J. 2003, 36: 141-154. 10.1046/j.1365-313X.2003.01864.x.
Article CAS PubMed Google Scholar - Moellering ER, Muthan B, Benning C: Freezing tolerance in plants requires lipid remodeling at the outer chloroplast membrane. Science. 2010, 330: 226-228. 10.1126/science.1191803.
Article CAS PubMed Google Scholar - Degenkolbe T, Giavalisco P, Zuther E, Seiwert B, Hincha DK, Willmitzer L: Differential remodeling of the lipidome during cold acclimation in natural accessions of Arabidopsis thaliana. Plant J. 2012, 32: 972-982.
Article Google Scholar - Gibson S, Arondel V, Iba K, Somerville C: Cloning of a temperature-regulated gene encoding a chloroplast omega-3 desaturase from Arabidopsis thaliana. Plant Physiol. 1994, 106: 1615-1621. 10.1104/pp.106.4.1615.
Article CAS PubMed Central PubMed Google Scholar - Gorsuch PA, Pandey S, Atkin OK: Temporal heterogeneity of cold acclimation phenotypes in Arabidopsis leaves. Plant Cell Environ. 2010, 33: 44-258.
Article Google Scholar - Ashraf M, Foolad MR: Roles of glycine betaine and proline in improving plant abiotic stress resistance. Environ Exp Bot. 2007, 59: 206-216. 10.1016/j.envexpbot.2005.12.006.
Article CAS Google Scholar - Verbrμggen N, Hermans C: Proline accumulation in plants: a review. Amino Acids. 2008, 35: 753-759. 10.1007/s00726-008-0061-6.
Article Google Scholar - Tunnacliffe A, Wise MJ: The continuing conundrum of the LEA proteins. Naturwissenschaften. 2007, 94: 791-812. 10.1007/s00114-007-0254-y.
Article CAS PubMed Google Scholar - Rajesh S, Manickam A: Prediction of functions for two LEA proteins from mung bean. Bioinformation. 2007, 1: 133-138.
Article Google Scholar - Qiu ZB, Wan LC: The regulation of cambial activity in Chinese fir (Cunninghamia lanceolata) involves extensive transcriptome remodeling. New Phytologist. 2013, 199: 627-870. 10.1111/nph.12404.
Article Google Scholar - Xin Z: Cold comfort farm: the acclimation of plants to freezing temperatures. Plant Cell Environ. 2001, 23: 893-902.
Article Google Scholar - Porankiewicz J, Wang J, Clarke AK: New insights into the ATP-dependent Clp protease: Escherichia coli and beyond. Mol Microbiol. 1999, 32: 449-458. 10.1046/j.1365-2958.1999.01357.x.
Article CAS PubMed Google Scholar - Wang B, Guo G, Wang C, Lin Y, Wang X, Zhao M, Guo Y, He M, Zhang Y, Pan L: Survey of the transcriptome of Aspergillus oryzae via massively parallel mRNA sequencing. Nucleic Acids Res. 2010, 38: 5075-5087. 10.1093/nar/gkq256.
Article CAS PubMed Central PubMed Google Scholar - Srichuwonga S, Isonoa N, Mishimab T, Hisamatsua M: Structure of lintnerized starch is related to X-ray diffraction pattern and susceptibility to acid and enzyme hydrolysis of starch granules. Int J Biol Macromol. 2007, 37: 115-121.
Article Google Scholar - Gomperts B, Kramer IM, Tatham PER: Signal transcription. Beijing Sci Publ. 2007, 7: 145-156. ISBN 978-7-03-018217-3
Google Scholar - Cooper GM, Hausman RE: The cell: a molecular approach. Beijing Sci Publ. 2012, 15: 603-605. ISBN 978-7-03-031324-9
Google Scholar - Donald WH, Rebecca B: Regulation of Cardiac Na+, Ca2+ Exchange and KATP Potassium Channels by PIP2. Science. 1996, 273: 956-959. 10.1126/science.273.5277.956.
Article Google Scholar - Saijo Y, Hata S, Kyozuka J, Shimamoto K, Izui K: Over-expression of a single Ca2 + -dependent protein kinase confers both cold and salt/droμght tolerance on rice plants. Plant J. 2000, 23: 319-327. 10.1046/j.1365-313x.2000.00787.x.
Article CAS PubMed Google Scholar - Ludwig AA, Romeis T, Jones JDG: CDPK-mediated signalling pathways: specificity and cross-talk. J Exp Bot. 2004, 55: 181-188.
Article CAS PubMed Google Scholar - Agarwal PK, Jha B: Transcription factors in plants and ABA dependent and independent abiotic stress signalling. Biol Plantarum. 2010, 54: 201-212. 10.1007/s10535-010-0038-7.
Article CAS Google Scholar - Yoshida R, Hobo T, Ichimura K, Mizoguchi T, Takahashi F, Aronso J, Ecker JR, Shinozaki K: ABA-activated SnRK2 protein kinase is required for dehydration stress signaling in Arabidopsis. Plant Cell Physiol. 2002, 43: 1473-1483. 10.1093/pcp/pcf188.
Article CAS PubMed Google Scholar - Benjamini Y, Yekutieli D: The control of the false discovery rate in multiple testing under dependency. Ann Stat. 2001, 29: 1165-1188. 10.1214/aos/1013699998.
Article Google Scholar - Gilmour SJ, Zarka DG, Stockinger EJ, Salazar MP, Houghton JM, Thomashow MF: Low temperature regulation of the Arabidopsis CBF family of AP2 transcriptional activators as an early step in cold-induced COR gene expression. Plant J. 1998, 16: 433-442. 10.1046/j.1365-313x.1998.00310.x.
Article CAS PubMed Google Scholar
Acknowledgements
This work was supported by the '863’ research program (Grant No. 2011AA10020804), China National Natural Science Foundation (Grant No. 31071815 and No. 31272204), and the D. Programs Foundation of the Ministry of Education of China (Grant No. 20110014110006). We thank Professor Guijun Yan for his insightful comments on the paper. We also thank Heyan Cao, Shunchao Xu, Jie Guan, Wenqi Li and Chengke Li, who studied in our laboratory.
Author information
Authors and Affiliations
- College of Landscape Architecture & China National Engineering Research Center for Floriculture, Beijing Forestry University, No.35 Qinghua East Road Haidian District, Beijing, 100083, China
Jingmao Wang, Xiaohua Liu, Jie Huang, Qing Wang, Jiahui Gu & Yingmin Lu - College of soil and water conservation, Beijing Forestry University, No.35 Qinghua East Road Haidian District, Beijing, 100083, China
Yang Yang
Authors
- Jingmao Wang
You can also search for this author inPubMed Google Scholar - Yang Yang
You can also search for this author inPubMed Google Scholar - Xiaohua Liu
You can also search for this author inPubMed Google Scholar - Jie Huang
You can also search for this author inPubMed Google Scholar - Qing Wang
You can also search for this author inPubMed Google Scholar - Jiahui Gu
You can also search for this author inPubMed Google Scholar - Yingmin Lu
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toYingmin Lu.
Additional information
Competing interests
The authors declare that they have no competing interests.
Electronic supplementary material
12864_2013_7041_MOESM1_ESM.doc
Additional file 1: Table S1. Differential expression genes in the heat-map.Heat-map of 65 differentially expressed genes involved in transcription factor, signal transport, stress kinase, defense/stress response, target protein compound in the cold response and acclimation of Lilium lancifolium. Table S2. The analysis of variance (ANOVA) of different physiology measurements for L. Lancifolium. (DOC 154 KB)
12864_2013_7041_MOESM2_ESM.txt
Additional file 2: TXT S3: The nucleotide sequences and NCBI GenBank accession numbers for the identified genes. (TXT 13 KB)
Authors’ original submitted files for images
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly credited. The Creative Commons Public Domain Dedication waiver (https://creativecommons.org/publicdomain/zero/1.0/ ) applies to the data made available in this article, unless otherwise stated.
About this article
Cite this article
Wang, J., Yang, Y., Liu, X. et al. Transcriptome profiling of the cold response and signaling pathways in Lilium lancifolium.BMC Genomics 15, 203 (2014). https://doi.org/10.1186/1471-2164-15-203
- Received: 18 October 2013
- Accepted: 07 March 2014
- Published: 17 March 2014
- DOI: https://doi.org/10.1186/1471-2164-15-203