Structure-Based Virtual Screening for Drug Discovery: a Problem-Centric Review (original) (raw)
- 15k Accesses
- 411 Citations
- 7 Altmetric
- Explore all metrics
Abstract
Structure-based virtual screening (SBVS) has been widely applied in early-stage drug discovery. From a problem-centric perspective, we reviewed the recent advances and applications in SBVS with a special focus on docking-based virtual screening. We emphasized the researchers’ practical efforts in real projects by understanding the ligand-target binding interactions as a premise. We also highlighted the recent progress in developing target-biased scoring functions by optimizing current generic scoring functions toward certain target classes, as well as in developing novel ones by means of machine learning techniques.
Similar content being viewed by others
INTRODUCTION
The discovery of innovative leads with potential interaction to specific targets is of central importance to the early-stage drug discovery. This is conventionally achieved by wet-lab high-throughput screening (HTS), an established technology adopted by pharmaceutical industry. On the other hand, the high cost and low hit rate associated with HTS have stimulated the development of computational alternatives and the broad application of the cheaper and faster screening in silico ([1](/article/10.1208/s12248-012-9322-0#ref-CR1 "Ripphausen P, Nisius B, Peltason L, Bajorath Jr. Quo vadis, virtual screening? A comprehensive survey of prospective applications. J Med Chem. 2010;53(24):8461–7. doi: 10.1021/jm101020z
."),[2](/article/10.1208/s12248-012-9322-0#ref-CR2 "Clark DE. What has virtual screening ever done for drug discovery? Expert Opin Drug Discov. 2008;3:841–51. doi:
10.1517/17460441.3.8.841
.")). The completion of the Human Genome Project has revealed a wealth of attractive druggable targets ([3](/article/10.1208/s12248-012-9322-0#ref-CR3 "Hopkins AL, Groom CR. The druggable genome. Nat Rev Drug Discov. 2002;1(9):727–30. doi:
10.1038/nrd892
.")). Meanwhile, structure biology advances in X-ray crystallography and nuclear magnetic resonance spectroscopy have further opened doors to structure-based virtual screening (SBVS) by offering in-depth structural details of these targets as well as their interactions with ligands ([4](/article/10.1208/s12248-012-9322-0#ref-CR4 "Villoutreix BO, Eudes R, Miteva MA. Structure-based virtual ligand screening: recent success stories. Comb Chem High Throughput Screen. 2009;12(10):1000–16."),[5](/article/10.1208/s12248-012-9322-0#ref-CR5 "Ghosh S, Nie A, An J, Huang Z. Structure-based virtual screening of chemical libraries for drug discovery. Curr Opin Chem Biol. 2006;10(3):194–202. doi:
10.1016/j.cbpa.2006.04.002
.")).There have been a mounting number of success stories reported by use of SBVS (4,[6](/article/10.1208/s12248-012-9322-0#ref-CR6 "Seifert MHJ, Lang M. Essential factors for successful virtual screening. Mini Rev Med Chem. 2007;8:63–72. doi: 10.2174/138955708783331540
.")), among which docking-based virtual screening (DBVS) is arguably the most widely applied one in practice ([7](/article/10.1208/s12248-012-9322-0#ref-CR7 "Tuccinardi T. Docking-based virtual screening: recent developments. Comb Chem High Throughput Screen. 2009;12(3):303–14.")). Here, we reviewed the recent advances and applications in SBVS from a problem-centric perspective with a focus on DBVS, such as the practical aspects about enriching screening library before docking, considering target flexibility, metal ions, water molecules, and other key ligand–target interactions and environmental factors during docking and improving pose/compound selection after docking. We emphasized the importance of profound knowledge of the targets and/or their interactions with ligands to a successful project. We also highlighted the recent progress in developing target-biased scoring function and the trend in applying machine learning techniques to build scoring functions. As the area of DBVS is often actively reviewed, we confined our survey to the primary publications since 2007 within a 5-year time frame.DOCKING-BASED VIRTUAL SCREENING
The basic inputs of a typical DBVS workflow are a target structure, either experimentally solved or computationally modeled, and a compound library of small molecules available via purchase or synthesis (Fig. 1). Often, both the target and the compound library require preparations, such as assigning proper tautomeric, stereoisomeric, and protonation states ([8](/article/10.1208/s12248-012-9322-0#ref-CR8 "Rapp CS, Schonbrun C, Jacobson MP, Kalyanaraman C, Huang N. Automated site preparation in physics-based rescoring of receptor ligand complexes. Proteins: Struct, Funct, Bioinf. 2009;77(1):52–61. doi: 10.1002/prot.22415
."),[9](/article/10.1208/s12248-012-9322-0#ref-CR9 "ten Brink T, Exner T. pKa based protonation states and microspecies for protein–ligand docking. J Comput Aided Mol Des. 2010;24(11):935–42. doi:
10.1007/s10822-010-9385-x
.")). Each compound in the library is virtually docked into the target binding site through a docking program, which computationally models the ligand–target interaction to achieve an optimal complementarity of steric and physicochemical properties. A mathematical algorithm (referred to as “scoring function”) is then used to evaluate the fitness between the docked compound and the target. This is often followed by a post-processing step, in which compounds were ranked and selected on the basis of calculated binding scores and/or other criteria, and usually only a small group of top-ranked compounds will be chosen as candidates for later experimental assays. During the past decades, a large number of docking programs have been developed ([10](/article/10.1208/s12248-012-9322-0#ref-CR10 "Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, et al. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J Comput Chem. 1998;19(14):1639–62. doi:
10.1002/(sici)1096-987x(19981115)19:14<1639::aid-jcc10>3.0.co;2-b
.")–[18](/article/10.1208/s12248-012-9322-0#ref-CR18 "Zsoldos Z, Szabo I, Szabo Z, Peter Johnson A. Software tools for structure based rational drug design. J Mol Struct (THEOCHEM). 2003;666-667:659–65. doi:
10.1016/j.theochem.2003.08.105
.")). Among the most popular ones are AutoDock, Dock, FlexX, Glide, Gold, Surflex, ICM, LigandFit, and eHiTS, to name only a few (Table [I](/article/10.1208/s12248-012-9322-0#Tab1)).Fig. 1
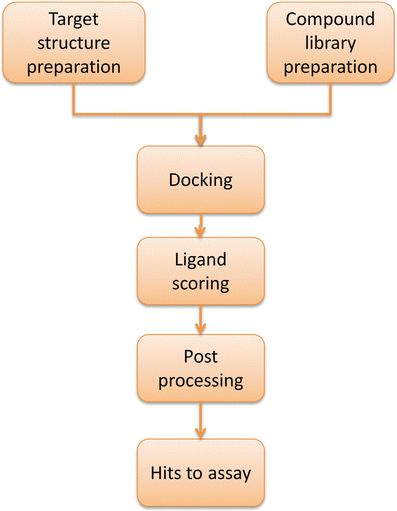
Typical workflow of a docking-based virtual screening (DBVS)
Table I Examples of Widely Used Docking Programs
Substantial process in DBVS requires a deep knowledge of the nature of the designated target system and/or the ligand–target binding mechanism ([6](/article/10.1208/s12248-012-9322-0#ref-CR6 "Seifert MHJ, Lang M. Essential factors for successful virtual screening. Mini Rev Med Chem. 2007;8:63–72. doi: 10.2174/138955708783331540
.")). It thus seems more appropriate in many applications to view DBVS from a problem-centric than a method-centric perspective ([19](/article/10.1208/s12248-012-9322-0#ref-CR19 "Schneider G. Virtual screening: an endless staircase? Nat Rev Drug Discov. 2010;9:273–6. doi:
10.1038/nrd3139
.")). In this work, we provided a review by focusing on the knowledge-based practices and efforts that were adopted by researchers throughout the workflow of DBVS (Fig. [1](/article/10.1208/s12248-012-9322-0#Fig1)). General advances in the ligand conformational sampling algorithms of docking programs have been extensively reviewed elsewhere ([7](/article/10.1208/s12248-012-9322-0#ref-CR7 "Tuccinardi T. Docking-based virtual screening: recent developments. Comb Chem High Throughput Screen. 2009;12(3):303–14."),[20](/article/10.1208/s12248-012-9322-0#ref-CR20 "Pujadas G, Vaque M, Ardevol A, Blade C, Salvado MJ, Blay M, et al. Protein–ligand docking: a review of recent advances and future perspectives. Curr Pharmaceut Anal. 2008;4:1–19. doi:
10.2174/157341208783497597
.")–[24](/article/10.1208/s12248-012-9322-0#ref-CR24 "Dias R, de Azevedo Jr WF. Molecular docking algorithms. Curr Drug Targets. 2008;9:1040–7. doi:
10.2174/138945008786949432
.")) and were thus not covered here.Enriching Compound Library before Docking
It is well accepted that the content and quality of a compound library have pivotal effects on the success of a DBVS project ([25](/article/10.1208/s12248-012-9322-0#ref-CR25 "Cummings MD, Maxwell AC, DesJarlais RL. Processing of small molecule databases for automated docking. Med Chem. 2007;3:107–13. doi: 10.2174/157340607779317481
.")). Table [II](/article/10.1208/s12248-012-9322-0#Tab2) summarizes an incomplete list of public and commercial chemical databases that are commonly screened in real practices. These databases often contain a vast amount of small-molecule compounds varying from several tens of thousands to several millions. Despite the increasing power of modern computers, a blind docking with all library compounds often leads to a waste of time and computer resource. Moreover, it will impose a great burden on later compound selection. Therefore, it would be always wise to remove undesirable compounds and select only relevant ones from a library before the cost-intensive docking. A common strategy is to apply fast physicochemical filters inspired by the rule of five ([26](/article/10.1208/s12248-012-9322-0#ref-CR26 "Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 1997;23(1–3):3–25. doi:
10.1016/s0169-409x(96)00423-1
.")) or ligand-based similarity search seeded with known active ligands ([27](/article/10.1208/s12248-012-9322-0#ref-CR27 "Perez-Pineiro R, Burgos A, Jones DC, Andrew LC, Rodriguez H, Suarez M, et al. Development of a novel virtual screening cascade protocol to identify potential trypanothione reductase inhibitors. J Med Chem. 2009;52(6):1670–80. doi:
10.1021/jm801306g
.")).Table II Commonly Screened Chemical Databases
A more object-oriented and efficient approach might be designing a focused library for specific targets. For example, Gozalbes et al. have enriched a kinase-targeted compound library using kinase-specific filters, which were derived from systematic docking and scoring of 123 diverse ligands against three kinases with known crystal structures ([28](/article/10.1208/s12248-012-9322-0#ref-CR28 "Gozalbes R, Simon L, Froloff N, Sartori E, Monteils C, Baudelle R. Development and experimental validation of a docking strategy for the generation of kinase-targeted libraries. J Med Chem. 2008;51(11):3124–32. doi: 10.1021/jm701367r
.")). For each kinase, the filter is constructed in two steps. First, the highest score given by a certain scoring function among all docking poses of a known ligand is used as the score for this ligand. Second, the lowest score among all known ligands is selected as the threshold for the current scoring function. Combining all thresholds from six scoring functions comprises the final filter. This method was validated by testing 60 compounds, which were split evenly into two groups including those passed all the thresholds and the rest. An overall 6.7-fold higher hit rate was obtained for the first group. Likewise, Sage _et al_. ([29](/article/10.1208/s12248-012-9322-0#ref-CR29 "Sage C, Wang R, Jones G. G-protein coupled receptors virtual screening using genetic algorithm focused chemical space. J Chem Inf Model. 2011. doi:
10.1021/ci200043z
.")) have introduced the GA-focused descriptor active space (GAFDAS) method to design a focused chemical space for G-protein coupled receptors by selecting target-specific descriptors through genetic algorithm. Though their method was validated in the context of ligand-based virtual screening, it could be applied in SBVS to design enriched library as well.Structural details from observed ligand–target complexes are useful to derive pharmacophoric filters, which may be used for enriching a library with compounds that satisfy specific geometric and/or physicochemical constraints. For instance, Kireev et al. ([30](/article/10.1208/s12248-012-9322-0#ref-CR30 "Kireev D, Wigle TJ, Norris-Drouin J, Herold JM, Janzen WP, Frye SV. Identification of non-peptide malignant brain tumor (MBT) repeat antagonists by virtual screening of commercially available compounds. J Med Chem. 2010;53(21):7625–31. doi: 10.1021/jm1007374
.")) have applied the Discovery Studio software to construct a pharmacophore model including a hydrogen bond donor (HBD), a hydrogen bond acceptor (HBA), and an amine cation involved in an ionic bond with the Asp355 residue that are observed in the crystal structure of L3MBTL1 protein in complex with H4K20me2 ligand. With these pharmacophoric constraints, the original 5,888,263 compounds were dramatically reduced to 20,078 compounds, which were subsequently subject to docking analysis. Similarly, Lee _et al_. have constructed two pharmacophore models for vascular endothelial growth factor kinase 2 (VEGFR2) using a crystal complex structure and validated them with 15 known VEGFR2 inhibitors ([31](/article/10.1208/s12248-012-9322-0#ref-CR31 "Lee K, Jeong K-W, Lee Y, Song JY, Kim MS, Lee GS, et al. Pharmacophore modeling and virtual screening studies for new VEGFR-2 kinase inhibitors. Eur J Med Chem. 2010;45(11):5420–7. doi:
10.1016/j.ejmech.2010.09.002
.")). In their study, a set of 59,600 compounds was narrowed down to 16,000 and 19,100 compounds using the above two pharmacophore models as queries, respectively. In the absence of experimental structure of target, a homology model can also be indicative for analyzing the key ligand–target interactions. For example, in an attempt to discover novel inhibitors of protein arginine methyltransferase 1 (PRMT1), Heinke _et al_. have defined a structure-based pharmacophore model based on a homology structure of PRMT1 in complex with _S_\-adenosylhomocysteine ([32](/article/10.1208/s12248-012-9322-0#ref-CR32 "Heinke R, Spannhoff A, Meier R, Trojer P, Bauer I, Jung M, et al. Virtual screening and biological characterization of novel histone arginine methyltransferase PRMT1 inhibitors. Chem Med Chem. 2009;4(1):69–77. doi:
10.1002/cmdc.200800301
.")). The 6,232 compounds that matched the pharmacophoric features (one HBD, one HBA, and two hydrophobic/aromatic constraints) were enriched from the initial 189,000 compounds for subsequent docking study.Understanding Ligand–Target Interaction and Environmental Factors During Docking
Target Flexibility
Molecular targets are dynamic in their physiological environment, which are often crucial for various biological functions. The target binding pocket often adapts upon ligand binding to fit the ligands through various conformational changes ranging from small side-chain flip to large loop shift. Nevertheless, the experimentally solved target structures or ligand–target complex structures are basically static snapshots. Though previous works have shown that proper consideration of target flexibility can improve DBVS results ([33](/article/10.1208/s12248-012-9322-0#ref-CR33 "Rueda M, Bottegoni G, Abagyan R. Consistent improvement of cross-docking results using binding site ensembles generated with elastic network normal modes. J Chem Inf Model. 2009;49(3):716–25. doi: 10.1021/ci8003732
.")), it still represents one of the greatest challenges for current docking programs ([34](/article/10.1208/s12248-012-9322-0#ref-CR34 "Cavasotto CN, Singh N. Docking and high throughput docking: successes and the challenge of protein flexibility. Curr Comput-Aided Drug Des. 2008;4:221–34. doi:
10.2174/157340908785747474
.")) and becomes a hot issue in recent DBVS studies ([35](/article/10.1208/s12248-012-9322-0#ref-CR35 "Cozzini P, Kellogg GE, Spyrakis F, Abraham DJ, Costantino G, Emerson A, et al. Target flexibility: an emerging consideration in drug discovery and design. J Med Chem. 2008;51(20):6237–55. doi:
10.1021/jm800562d
.")–[39](/article/10.1208/s12248-012-9322-0#ref-CR39 "Lin J-H. Accommodating protein flexibility for structure-based drug design. Curr Top Med Chem. 2011;11:171–8. doi:
10.2174/156802611794863580
.")).Ensemble docking that takes advantage of multiple target conformers has emerged as a partial solution to account for target flexibility in docking. The MultiCopyMD method developed by Okamoto et al. can generate a target ensemble through molecular dynamics (MD) with multiple ligands in the target binding site simultaneously ([40](/article/10.1208/s12248-012-9322-0#ref-CR40 "Okamoto M, Takayama K, Shimizu T, Ishida K, Takahashi O, Furuya T. Identification of death-associated protein kinases inhibitors using structure-based virtual screening. J Med Chem. 2009;52(22):7323–7. doi: 10.1021/jm901191q
.")). Applying this target ensemble in their SBVS for novel inhibitors of death-associated protein kinase (DAPK), they discovered a highly potent (IC50 \= 69 nM) and selective inhibitor for DAPK1\. To select appropriate target conformers, Rueda _et al_. have suggested a simple recipe by choosing the target conformers co-crystallized with the largest ligands ([41](/article/10.1208/s12248-012-9322-0#ref-CR41 "Rueda M, Bottegoni G, Abagyan R. Recipes for the selection of experimental protein conformations for virtual screening. J Chem Inf Model. 2009;50(1):186–93. doi:
10.1021/ci9003943
.")), providing higher selectivity and better results than randomly picked ones when combined in ensemble. Using cyclin-dependent kinase 2 (CDK2) as a test example, Sperandio _et al_. have demonstrated normal mode analysis as an effective tool to select relevant target conformations with diverse binding sites ([42](/article/10.1208/s12248-012-9322-0#ref-CR42 "Sperandio O, Mouawad L, Pinto E, Villoutreix B, Perahia D, Miteva M. How to choose relevant multiple receptor conformations for virtual screening: a test case of Cdk2 and normal mode analysis. Eur Biophys J. 2010;39(9):1365–72. doi:
10.1007/s00249-010-0592-0
.")). Generally in ensemble docking, an individual docking run is required for each target conformation, which is thus computationally inefficient. To address this issue, Bottegoni _et al_. have proposed a 4D docking approach that allows fast and accurate account of target conformational ensembles in a single docking simulation ([43](/article/10.1208/s12248-012-9322-0#ref-CR43 "Bottegoni G, Kufareva I, Totrov M, Abagyan R. Four-dimensional docking: a fast and accurate account of discrete receptor flexibility in ligand docking. J Med Chem. 2008;52(2):397–406. doi:
10.1021/jm8009958
.")). This is achieved by merging 3D grids from optimally superimposed multiple target conformers into a single 4D object.Metal Ions
Some targets, such as metalloproteins, contain transition metal ions in their binding sites. The binding of ligand to these targets can be substantially distinct from other target types since such metal ions often coordinate ligand polar atoms, which may help to place and orient the ligand correctly in the binding sites. However, it is nontrivial to take metal ions into account accurately in current docking/scoring algorithms. The neglection of them would inevitably lead to underestimation of the metal–ligand interaction or even incorrectly docked ligands. Therefore, increasing attentions are being paid to metal ions in recent DBVS.
Röhrig et al. have studied the irons in heme proteins and demonstrated their importance for DBVS ([44](/article/10.1208/s12248-012-9322-0#ref-CR44 "Röhrig UF, Grosdidier A, Zoete V, Michielin O. Docking to heme proteins. J Comput Chem. 2009;30(14):2305–15. doi: 10.1002/jcc.21244
.")). Two docking runs were performed in parallel by using a test set of 50 heme-containing complexes with iron–ligand contact. In one standard docking using EADock, a success rate of only 28% was achieved, clearly indicating the underestimation of the role of iron–ligand interactions. They then introduced the Morse-like metal binding potentials into EADock, which were fitted to reproduce density functional theory calculations. As a result, the success rate was doubled to 62%. To evaluate the reliability of the chosen docking protocol for screening potent cytochrome P450 aromatase inhibitors (AIs), Caporuscio _et al_. investigated a set of known imidazole and triazole AIs and found that the Glide docking program failed to predict a correct binding mode in all cases where the azole nitrogen coordinates the heme iron ([45](/article/10.1208/s12248-012-9322-0#ref-CR45 "Caporuscio F, Rastelli G, Imbriano C, Del Rio A. Structure-based design of potent aromatase inhibitors by high-throughput docking. J Med Chem. 2011;54:4006–17. doi:
10.1021/jm2000689
.")). This observation inspired them to set up a metal constraint in Glide, which requires that a ligand atom lies within a certain region of the binding site in order to interact with specific target functionalities. Their structure-based design efforts eventually resulted in several novel AIs with IC50 activity in the range of 21.7 μM to 9.4 nM.Missing parameters of zinc ions is another common barrier for docking many metalloenzymes including histone deacetylases (HDACs). In seek of novel HDAC inhibitors, Park et al. derived potential parameters for zinc ions following a standard procedure ([46](/article/10.1208/s12248-012-9322-0#ref-CR46 "Park H, Kim S, Kim YE, Lim S-J. A structure-based virtual screening approach toward the discovery of histone deacetylase inhibitors: identification of promising zinc-chelating groups. Chem Med Chem. 2010;5(4):591–7. doi: 10.1002/cmdc.200900500
.")), in which geometry optimization of a simplified structural model was conducted for the active-site zinc ion cluster in complex with a hydroxamate-based inhibitor at the B3LYP/6–31 G\*\* theory level. With these zinc parameters, they discovered six novel HDAC inhibitors with IC50 value ranging from 1 to 100 μM.Water Molecules
There is a recognition that active-site water molecules play an important role in ligand-target binding ([47](/article/10.1208/s12248-012-9322-0#ref-CR47 "Thilagavathi R, Mancera RL. Ligand–protein cross-docking with water molecules. J Chem Inf Model. 2010;50(3):415–21. doi: 10.1021/ci900345h
.")). Such water molecules can significantly contribute enthalpically and entropically to ligand–target binding. The most known role of water molecules is to mediate the ligand–target interaction by forming hydrogen bonds at the interface between the ligand and the target. On the other hand, the presence (or absence) and the location of water molecules may vary largely among ligands ([48](/article/10.1208/s12248-012-9322-0#ref-CR48 "Santos R, Hritz J, Oostenbrink C. Role of water in molecular docking simulations of cytochrome P450 2D6. J Chem Inf Model. 2009;50(1):146–54. doi:
10.1021/ci900293e
.")). Despite their critical role, accounting for water molecules accurately in docking is a long-standing challenge. Several very recent studies directly targeted this issue.Abel and coworkers have developed a unique approach WaterMap ([49](/article/10.1208/s12248-012-9322-0#ref-CR49 "Abel R, Young T, Farid R, Berne BJ, Friesner RA. Role of the active-site solvent in the thermodynamics of factor Xa ligand binding. J Am Chem Soc. 2008;130(9):2817–31. doi: 10.1021/ja0771033
.")) to account for the contribution of the displacement of water molecules by ligand to binding free energy. It first identifies “hydration sites” in the active site by clustering the trajectories from MD simulation of a solvated target with explicit water molecules. Inhomogeneous solvation theory is then applied to compute the thermodynamic properties of these active-site solvents including enthalpic and entropic changes. A displaced solvent functional is derived to estimate the relative binding free energies of a series of congeneric ligands based on their measured free energies by displacing active-site water molecules. This feature has made WaterMap particularly suitable for (and thus also limited to) lead optimization by providing insightful guidance to medicinal chemistry. More recently, WaterMap has been augmented by the introduction of an additional term attributable to the occupation of the dry regions in the target active site by ligand atoms ([50](/article/10.1208/s12248-012-9322-0#ref-CR50 "Wang L, Berne BJ, Friesner RA. Ligand binding to protein-binding pockets with wet and dry regions. Proc Natl Acad Sci. 2011;108(4):1326–30. doi:
10.1073/pnas.1016793108
.")).Lie et al. have proposed a very interesting approach that attached water molecules to ligand during docking ([51](/article/10.1208/s12248-012-9322-0#ref-CR51 "Lie MA, Thomsen R, Pedersen CNS, Schiøtt B, Christensen MH. Molecular docking with ligand attached water molecules. J Chem Inf Model. 2011;51(4):909–17. doi: 10.1021/ci100510m
.")). In their method, ligand polar atoms are solvated with maximum number of water molecules, which are then retained or displaced depending on energy contributions during docking simulation. The novelty of their method is that each water molecule is treated as a flexible on/off part of the ligand, instead of being a static part of the target. In such a manner, water molecules are sampled with the same flexibility as the ligand itself. Their method has been evaluated with considerable improvement by using 12 structurally diverse complexes, where several water molecules bridge the ligand and the target.Rossato et al. have introduced a directional approach, AcquaAlta, to consider the solvation of ligand–target complexes ([52](/article/10.1208/s12248-012-9322-0#ref-CR52 "Rossato G, Ernst B, Vedani A, Smieško M. AcquaAlta: a directional approach to the solvation of ligand–protein complexes. J Chem Inf Model. 2011. doi: 10.1021/ci200150p
.")). Through an extensive analysis of the Cambridge Structural Database, they derived a geometric criteria defining interactions of water molecules with ligand and target. They also evaluated the propensity of ligand hydration through _ab initio_ calculations. AcquaAlta has been validated with 20 crystal structures and reproduced 76% of the positions of water molecules that were experimentally observed.Other Key Interactions
Understanding of the interactions essential for ligand–target binding is critical to the success of lead discovery and optimization. For example, in a recent attempt to identify novel inhibitors of trihydroxynaphthalene reductase (3HNR) ([53](/article/10.1208/s12248-012-9322-0#ref-CR53 "Brunskole Švegelj M, Turk S, Brus B, Lanišnik Rižner T, Stojan J, Gobec S. Novel inhibitors of trihydroxynaphthalene reductase with antifungal activity identified by ligand-based and structure-based virtual screening. J Chem Inf Model. 2011;51(7):1716–24. doi: 10.1021/ci2001499
.")), the authors first overlaid the known 3HNR inhibitors and then constructed a pharmacophore model that consists of several key interaction points within the active site: H-bonds with Ser149, Tyr163, Met200, and Tyr201 and π-stacking with Tyr208\. In accordance to these interactions, the docking experiment was conducted in such a way that it only considered docking solutions that predicted π-stacking with Tyr208 and an optional H-bond with Ser149\. The most potent hit compound they found exhibited a _K_ _i_ of 5.3 ± 0.3 μM against 3HNR.As revealed by the crystal structures of kinases in complex with ATP-competing inhibitors, such inhibitors typically form at least one hydrogen bond with backbone amide or carbonyl groups in the hinge region. Therefore, introducing relevant constraints with the hinge region for the molecules docked into the ATP sites of kinases would improve the chance of finding active compounds. This has been practiced by Ravindranathan et al. in the hit discovery of fibroblast growth factor receptor 1 (FGFR1) ([54](/article/10.1208/s12248-012-9322-0#ref-CR54 "Ravindranathan KP, Mandiyan V, Ekkati AR, Bae JH, Schlessinger J, Jorgensen WL. Discovery of novel fibroblast growth factor receptor 1 kinase inhibitors by structure-based virtual screening. J Med Chem. 2010;53(4):1662–72. doi: 10.1021/jm901386e
.")). Among the 23 purchasable compounds suggested by a virtual screening experiment against 2.2 million compounds, two were identified to inhibit FGFR1 kinase with medium potency (IC50 \= 23 and 50 μM, respectively).For certain target or ligand system, specifically designed methods may be more efficient. For example, Lang et al. recently have optimized DOCK 6 for docking small molecules to RNA targets ([55](/article/10.1208/s12248-012-9322-0#ref-CR55 "Lang PT, Brozell SR, Mukherjee S, Pettersen EF, Meng EC, Thomas V, et al. DOCK 6: combining techniques to model RNA–small molecule complexes. RNA. 2009;15(6):1–12. doi: 10.1261/rna.1563609
.")) and obtained a success rate of 70% for the ligands with less than seven rotatable bonds at the 2-Å heavy-atom root-mean-squared deviation threshold. The BALLDock/SLICK developed by Kerzmann is a ligand-specific docking approach for docking carbohydrate or carbohydrate-like compounds, which are often problematic for standard docking programs ([56](/article/10.1208/s12248-012-9322-0#ref-CR56 "Kerzmann A, Fuhrmann J, Kohlbacher O, Neumann D. BALLDock/SLICK: a new method for protein–carbohydrate docking. J Chem Inf Model. 2008;48(8):1616–25. doi:
10.1021/ci800103u
.")).Improving Pose/Compound Selection After Docking
Due to the poor performance of current scoring functions in estimating binding affinity and hence in ranking docked ligands, it is recognized that compound selection based on calculated scores is not sufficient and visual inspection is often necessary. However, a practical concern arises if one needs to manually inspect thousands of docking poses. Therefore, huge efforts have been devoted to automating this procedure based on the indications gained from ligand–target interactions ([57](/article/10.1208/s12248-012-9322-0#ref-CR57 "Waszkowycz B. Towards improving compound selection in structure-based virtual screening. Drug Discov Today. 2008;13(5–6):219–26. doi: 10.1016/j.drudis.2007.12.002
.")).The molecular interaction fingerprints (IFPs), which are simple bit strings that encode 3D information about ligand–target interactions into 1D binary vector, have been extended by Marcou and Rognan as a post-docking filter to prioritize the most relevant poses of low molecular weight fragments ([58](/article/10.1208/s12248-012-9322-0#ref-CR58 "Marcou G, Rognan D. Optimizing fragment and scaffold docking by use of molecular interaction fingerprints. J Chem Inf Model. 2007;47(1):195–207. doi: 10.1021/ci600342e
.")). In their study, IFPs were evaluated with four popular docking tools (FlexX, Glide, Gold, and Surflex) for extracting the scaffolds of true CDK2 inhibitors. They observed that scoring by the Tanimoto similarity of IFPs to a given reference was statistically superior to conventional scoring functions in placing the low molecular weight fragment in the CDK2 binding site.Based on the assumption that active compounds should have specific contacts with their target to display activity and also to tackle the inefficiency of traditional clustering of docking poses, Bouvier et al. have proposed the Automatic analysis of Poses using Self-Organizing Map (AuPosSOM) method for pose ranking with careful analysis of interatomic contacts between the docked ligand and the target ([59](/article/10.1208/s12248-012-9322-0#ref-CR59 "Bouvier G, Evrard-Todeschi N, Girault J-P, Bertho G. Automatic clustering of docking poses in virtual screening process using self-organizing map. Bioinformatics. 2010;26(1):53–60. doi: 10.1093/bioinformatics/btp623
.")). They have demonstrated that it is possible to differentiate active compounds from inactive ones using only mean protein contacts’ footprints calculated from the multiple conformations given by docking software.Protein-specific structural filtration has been introduced by Novikov et al. to improve the performance of DBVS ([60](/article/10.1208/s12248-012-9322-0#ref-CR60 "Novikov F, Stroylov V, Stroganov O, Chilov G. Improving performance of docking-based virtual screening by structural filtration. J Mol Model. 2010;16(7):1223–30. doi: 10.1007/s00894-009-0633-8
.")). The filter was defined by a set of crucial ligand–target interactions that are structurally conserved in the available ligand-bound target structures. The application of this method achieved a substantial improvement of enrichment factor ranging from several folds to several hundreds folds against a set of ten diverse protein targets. The authors demonstrated that the structural filtration had effectively repaired the deficiencies of scoring functions, resulting in a considerably lower false positive rate.Wei et al. have demonstrated that binding energy landscape analysis could help to discriminate true hits from high-scoring decoys in virtual screening ([61](/article/10.1208/s12248-012-9322-0#ref-CR61 "Wei D, Zheng H, Su N, Deng M, Lai L. Binding energy landscape analysis helps to discriminate true hits from high-scoring decoys in virtual screening. J Chem Inf Model. 2010;50(10):1855–64. doi: 10.1021/ci900463u
.")). In their work, two parameters (_i.e._, the energy gap and the number of local binding wells in the landscape) were used to account for the kinetic accessibility. With a linear combination of the two parameters, they obtained, in a five-fold cross-validation, the areas under the receiver operator characteristic curves (AUC) of 0.878 for neuraminidase and 0.776 for cyclooxygenase 2 (COX2), respectively. In a more independent test using the directory of useful decoys (DUD) set, the enrichment ratio given by these two parameters when combined with docking scores was improved to 200–300% as compared to that using scoring function alone.SCORING FUNCTIONS
Scoring function is at the heart of molecular docking by assisting a docking program to efficiently explore the binding space of a ligand. It is also responsible for evaluating the binding affinity once the correct binding pose is identified. Therefore, the predictability of scoring functions has a significant impact on the productivity of DBVS.
A multitude of scoring functions have been reported in the past decades ([10](/article/10.1208/s12248-012-9322-0#ref-CR10 "Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, et al. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J Comput Chem. 1998;19(14):1639–62. doi: 10.1002/(sici)1096-987x(19981115)19:14<1639::aid-jcc10>3.0.co;2-b
.")–[15](/article/10.1208/s12248-012-9322-0#ref-CR15 "Jain AN. Surflex: fully automatic flexible molecular docking using a molecular similarity-based search engine. J Med Chem. 2003;46(4):499–511. doi:
10.1021/jm020406h
."),[62](/article/10.1208/s12248-012-9322-0#ref-CR62 "Böhm H-J. Prediction of binding constants of protein ligands: a fast method for the prioritization of hits obtained from de novo design or 3D database search programs. J Comput Aided Mol Des. 1998;12(4):309. doi:
10.1023/a:1007999920146
.")–[71](/article/10.1208/s12248-012-9322-0#ref-CR71 "Huang S-Y, Zou X. Inclusion of solvation and entropy in the knowledge-based scoring function for protein–ligand interactions. J Chem Inf Model. 2010;50(2):262–73. doi:
10.1021/ci9002987
.")) (Table [III](/article/10.1208/s12248-012-9322-0#Tab3)), and new ones are still emerging. Current scoring functions, as reviewed in other works ([23](/article/10.1208/s12248-012-9322-0#ref-CR23 "Moitessier N, Englebienne P, Lee D, Lawandi J, Corbeil CR. Towards the development of universal, fast and highly accurate docking/scoring methods: a long way to go. Br J Pharmacol. 2008;153(S1):S7–S26. doi:
10.1038/sj.bjp.0707515
."),[72](/article/10.1208/s12248-012-9322-0#ref-CR72 "Huang S-Y, Grinter SZ, Zou X. Scoring functions and their evaluation methods for protein-ligand docking: recent advances and future directions. Phys Chem Chem Phys. 2010;12(40):12899–908. doi:
10.1039/c0cp00151a
.")), can be roughly classified into three types: (a) Force field-based scoring functions employ classic force field to compute the noncovalent ligand–target interactions, such as van der Waals and electrostatic energies. They are often augmented by a GB/SA or PB/SA term in order to account for solvation effects. (b) Empirical scoring functions calculate the overall binding free energy from several energetic terms, including hydrogen bond interaction and hydrophobic interaction. The weighting factors of all terms are calibrated from a set of known complexes with experimentally determined structures and binding affinities. (c) Knowledge-based scoring functions compute the ligand–target interactions as a sum of distance-dependent statistical potentials between the ligand and the target. It is notable that the deduction of such potentials needs only the structural information of ligand–target complexes, which is being accumulated rapidly due to structural biology advances.Table III Examples of Current Scoring Functions
The performance of various scoring functions has been investigated by several comparative studies ([73](/article/10.1208/s12248-012-9322-0#ref-CR73 "Cheng T, Li X, Li Y, Liu Z, Wang R. Comparative assessment of scoring functions on a diverse test set. J Chem Inf Model. 2009;49(4):1079–93. doi: 10.1021/ci9000053
.")–[77](/article/10.1208/s12248-012-9322-0#ref-CR77 "Wang R, Lu Y, Wang S. Comparative evaluation of 11 scoring functions for molecular docking. J Med Chem. 2003;46(12):2287–303. doi:
10.1021/jm0203783
.")), with respect to the ability of reproducing known binding pose, predicting binding affinity and rank-ordering a compound library. The state-of-the-art scoring functions are at different levels of accuracy, and it is clear that no single scoring function consistently outperforms others in all cases. It is concluded from previous comparative studies that today’s scoring functions are often capable of identifying the correct binding pose of a ligand, while binding affinity prediction with high accuracy is still far from reach ([73](/article/10.1208/s12248-012-9322-0#ref-CR73 "Cheng T, Li X, Li Y, Liu Z, Wang R. Comparative assessment of scoring functions on a diverse test set. J Chem Inf Model. 2009;49(4):1079–93. doi:
10.1021/ci9000053
.")). Therefore, considerable efforts have been made to improve the performance of current scoring functions. Common strategies include adding additional factors to account for solvation and entropic effects ([71](/article/10.1208/s12248-012-9322-0#ref-CR71 "Huang S-Y, Zou X. Inclusion of solvation and entropy in the knowledge-based scoring function for protein–ligand interactions. J Chem Inf Model. 2010;50(2):262–73. doi:
10.1021/ci9002987
.")), deriving more accurate energy terms by high-level quantum calculations ([78](/article/10.1208/s12248-012-9322-0#ref-CR78 "Raub S, Steffen A, Kämper A, Marian CM. AIScore: chemically diverse empirical scoring function employing quantum chemical binding energies of hydrogen-bonded complexes. J Chem Inf Model. 2008;48(7):1492–510. doi:
10.1021/ci7004669
.")), and consensus scoring by combination of multiple scoring functions ([79](/article/10.1208/s12248-012-9322-0#ref-CR79 "Charifson PS, Corkery JJ, Murcko MA, Walters WP. Consensus scoring: a method for obtaining improved hit rates from docking databases of three-dimensional structures into proteins. J Med Chem. 1999;42(25):5100–9. doi:
10.1021/jm990352k
."),[80](/article/10.1208/s12248-012-9322-0#ref-CR80 "Wang R, Wang S. How does consensus scoring work for virtual library screening? An idealized computer experiment. J Chem Inf Comput Sci. 2001;41(5):1422–6. doi:
10.1021/ci010025x
.")). In this review, we highlighted the recent progress in developing target-biased scoring functions as well as those employed machine learning techniques.Target-Biased Scoring Functions
Most of the today’s scoring functions are generic models derived from the large-scale experimental data of ligand–target complexes and are presumably applicable to all sorts of target classes. However, previous comparative studies have revealed that a universally accurate scoring function is still out of reach. A practical remedy to this might be developing target-biased alternatives for specific targets or tasks ([81](/article/10.1208/s12248-012-9322-0#ref-CR81 "Seifert MHJ. Targeted scoring functions for virtual screening. Drug Discov Today. 2009;14(11–12):562–9. doi: 10.1016/j.drudis.2009.03.013
.")).Target-Biased Scoring Functions Derived by Re-parameterization
The most straightforward way to obtain a target-biased scoring function is, probably, to re-calibrate an existing all-purpose scoring function directly on certain target classes. For example, DrugScore-RNA ([82](/article/10.1208/s12248-012-9322-0#ref-CR82 "Pfeffer P, Gohlke H. DrugScoreRNA: knowledge-based scoring function to predict RNA–ligand interactions. J Chem Inf Model. 2007;47(5):1868–76. doi: 10.1021/ci700134p
.")) adopts the same framework as DrugScore ([69](/article/10.1208/s12248-012-9322-0#ref-CR69 "Gohlke H, Hendlich M, Klebe G. Knowledge-based scoring function to predict protein–ligand interactions. J Mol Biol. 2000;295(2):337–56. doi:
10.1006/jmbi.1999.3371
.")) but is derived from 670 crystal structures of nucleic acid–ligand and nucleic acid–protein complexes. Similar idea has been implemented in the kinase family-specific potential of mean force (kinase-PMF) ([68](/article/10.1208/s12248-012-9322-0#ref-CR68 "Xue M, Zheng M, Xiong B, Li Y, Jiang H, Shen J. Knowledge-based scoring functions in drug design. 1. Developing a target-specific method for kinase−ligand interactions. J Chem Inf Model. 2010;50(8):1378–86. doi:
10.1021/ci100182c
.")), a kinase-targeted scoring function adjusted from the original PMF04 ([67](/article/10.1208/s12248-012-9322-0#ref-CR67 "Muegge I. PMF scoring revisited. J Med Chem. 2005;49(20):5895–902. doi:
10.1021/jm050038s
.")).Tweaking the parameters in original scoring functions toward specific targets is also a prevalent strategy to derive target-biased scoring functions. For example, Teramoto and Fukunishi have applied a supervised scoring model to tailor the FlexX scoring function (_F_-score), which outperformed its former version on three of the five tested targets ([83](/article/10.1208/s12248-012-9322-0#ref-CR83 "Teramoto R, Fukunishi H. Supervised scoring models with docked ligand conformations for structure-based virtual screening. J Chem Inf Model. 2007;47(5):1858–67. doi: 10.1021/ci700116z
.")). The TOP approach suggested by Seifert ([84](/article/10.1208/s12248-012-9322-0#ref-CR84 "Seifert MHJ. Optimizing the signal-to-noise ratio of scoring functions for protein–ligand docking. J Chem Inf Model. 2008;48(3):602–12. doi:
10.1021/ci700345n
.")) have employed iterative taboo search to optimize the scoring function in ProPose and the original Böhm scoring function against three targets, including CDK2, estrogen receptor, and COX2\. By adding negative data of ligands that are known not to bind particular target, Pham and Jain have tuned the scoring function in Surflex-Dock and observed substantially enhanced screening enrichment for HIV protease and poly(ADP-ribose) polymerase ([85](/article/10.1208/s12248-012-9322-0#ref-CR85 "Pham T, Jain A. Customizing scoring functions for docking. J Comput Aided Mol Des. 2008;22(5):269–86. doi:
10.1007/s10822-008-9174-y
.")). An augmented Flo+ scoring function has been developed by Catana and Stouten using N-way partial least squares (PLS) ([86](/article/10.1208/s12248-012-9322-0#ref-CR86 "Catana C, Stouten PFW. Novel, customizable scoring functions, parameterized using N-PLS, for structure-based drug discovery. J Chem Inf Model. 2007;47(1):85–91. doi:
10.1021/ci600357t
.")), which significantly improved the correlation between observed and calculated p_K_ _i_ values from _R_ 2 \= 0.5 to 0.8 on a relatively diverse set of ligand–target complexes spanning seven protein families. Therefore, it would be attractive if scoring functions offer extendable or customizable features.Target-Biased Scoring Functions Require no Re-parameterization
The above-mentioned target-biased scoring functions typically require re-parameterization or special treatment of established scoring functions. Too often, existing scoring functions are available to end-users as black boxes, hence it is not readily possible to adjust their parameters by any optimization algorithm. Several approaches have been proposed to address this issue. One of the earliest examples is the MultiScore that employs the raw scores from eight scoring functions to characterize the observed p_K_ i ([87](/article/10.1208/s12248-012-9322-0#ref-CR87 "Terp GE, Johansen BN, Christensen IT, Jørgensen FS. A new concept for multidimensional selection of ligand conformations (MultiSelect) and multidimensional scoring (MultiScore) of protein–ligand binding affinities. J Med Chem. 2001;44(14):2333–43. doi: 10.1021/jm001090l
.")), which has been found to work better for matrix metalloproteinases. The implied idea is slightly different from that of consensus scoring ([79](/article/10.1208/s12248-012-9322-0#ref-CR79 "Charifson PS, Corkery JJ, Murcko MA, Walters WP. Consensus scoring: a method for obtaining improved hit rates from docking databases of three-dimensional structures into proteins. J Med Chem. 1999;42(25):5100–9. doi:
10.1021/jm990352k
."),[80](/article/10.1208/s12248-012-9322-0#ref-CR80 "Wang R, Wang S. How does consensus scoring work for virtual library screening? An idealized computer experiment. J Chem Inf Comput Sci. 2001;41(5):1422–6. doi:
10.1021/ci010025x
.")) in that it assumes uneven contributions from individual scoring functions. In a similar way, the AutoShim method has incorporated the original Flo+ score as well as additional target-specific pharmacophore points (shims) as descriptors in PLS analysis ([88](/article/10.1208/s12248-012-9322-0#ref-CR88 "Martin EJ, Sullivan DC. AutoShim: empirically corrected scoring functions for quantitative docking with a crystal structure and IC50 training data. J Chem Inf Model. 2008;48(4):861–72. doi:
10.1021/ci7004548
.")). More recently, Cheng _et al_. have proposed a knowledge-guided strategy (KGS) based on the similarity principle aiming to improve the accuracy of binding affinity prediction of current scoring functions ([89](/article/10.1208/s12248-012-9322-0#ref-CR89 "Cheng T, Liu Z, Wang R. A knowledge-guided strategy for improving the accuracy of scoring functions in binding affinity prediction. BMC Bioinf. 2010;11(1):193. doi:
10.1186/1471-2105-11-193
.")). The KGS strategy computes the binding affinity of a query ligand–target complex based on the known binding affinity of an appropriate reference complex, which is required to share a similar pattern of key ligand–target interactions to that of the query complex of interest. The KGS strategy has been validated with both observed and docked ligand–target complex structures. Moreover, it can in principle work in concert with any scoring method, and its application is not limited to specific classes of ligand–target complexes.Machine Learning and Scoring Functions
Machine learning techniques are powerful to construct and optimize predictive models. In recent years, there is an increasing interest in developing novel scoring functions by means of machine learning ([90](/article/10.1208/s12248-012-9322-0#ref-CR90 "Hecht D, Fogel GB. Computational intelligence methods for docking scores. Curr Comput-Aided Drug Des. 2009;5:56–68. doi: 10.2174/157340909787580863
.")). A notable feature is that they take into account the commonly observed ligand–target binding interactions in an implicit manner, which obviates the need of explicitly modeling the error-prone interactions, including solvation and entropic effects. Moreover, machine learning techniques such as neural networks (NN), support vector machines (SVM), and random forest (RF) are able to account for the nonlinear dependence among the various interactions involved in ligand–target binding. As a result, despite being less concrete on the physicochemical basis, they often demonstrated a superior or at least comparable performance to that of classic scoring functions in binding affinity estimation.The NNScore scoring function developed by Durrant and McCammon is based on NN ([91](/article/10.1208/s12248-012-9322-0#ref-CR91 "Durrant JD, McCammon JA. NNScore: a neural-network-based scoring function for the characterization of protein–ligand complexes. J Chem Inf Model. 2010;50(10):1865–71. doi: 10.1021/ci100244v
.")), which attempts to computationally simulate the microscopic organization of human brain. The input layer consists of 194 neurodes that are related to ligand–target interactions. Kinnings _et al_. ([92](/article/10.1208/s12248-012-9322-0#ref-CR92 "Kinnings SL, Liu N, Tonge PJ, Jackson RM, Xie L, Bourne PE. A machine learning-based method to improve docking scoring functions and its application to drug repurposing. J Chem Inf Model. 2011;51(2):408–19. doi:
10.1021/ci100369f
.")) have applied SVM to train a new scoring function for identifying inhibitors of _Mycobacterium tuberculosis_ InhA, using the individual energy terms as descriptors obtained directly from the built-in scoring function of eHiTS. Amini _et al_. have introduced the support vector inductive logic programming as a general approach to develop system-specific scoring functions ([93](/article/10.1208/s12248-012-9322-0#ref-CR93 "Amini A, Shrimpton PJ, Muggleton SH, Sternberg MJE. A general approach for developing system-specific functions to score protein–ligand docked complexes using support vector inductive logic programming. Proteins: Struct, Funct, Bioinf. 2007;69(4):823–31. doi:
10.1002/prot.21782
.")). The descriptors they used are the distances from each fragment’s central ligand atom to target atoms. In the development of PHOENIX scoring function, Tang _et al_. have adopted an indirect idea ([94](/article/10.1208/s12248-012-9322-0#ref-CR94 "Tang YT, Marshall GR. PHOENIX: a scoring function for affinity prediction derived using high-resolution crystal structures and calorimetry measurements. J Chem Inf Model. 2011;51(2):214–28. doi:
10.1021/ci100257s
.")). They first modeled independently enthalpy (Δ_H_) and the change of entropy (_T_Δ_S_) by fitting relevant descriptors to experimentally measured calorimetric data through PLS and then calculated the binding free energy (Δ_G_) according to thermodynamic cycle.Similar to the idea of using occurrence count of ligand–target atom pair as geometric descriptor to generate a scoring function ([95](/article/10.1208/s12248-012-9322-0#ref-CR95 "Deng W, Breneman C, Embrechts MJ. Predicting protein–ligand binding affinities using novel geometrical descriptors and machine-learning methods. J Chem Inf Comput Sci. 2004;44(2):699–703. doi: 10.1021/ci034246+
.")), Li _et al_. ([96](/article/10.1208/s12248-012-9322-0#ref-CR96 "Li L, Khanna M, Jo I, Wang F, Ashpole NM, Hudmon A, et al. Target-specific support vector machine scoring in structure-based virtual screening: computational validation, in vitro testing in kinases, and effects on lung cancer cell proliferation. J Chem Inf Model. 2011;51(4):755–9. doi:
10.1021/ci100490w
.")) have developed a target-specific scoring method, SVM-SP, by using SVM. SVM-SP employs 135 atom pair potentials as descriptors that are derived in the same way as traditional knowledge-based scoring functions. The effectiveness of SVM-SP has been strongly supported by the discovery of three novel micromolar hits against epidermal growth factor receptor. The recently released RF-score by Ballester and Mitchell ([97](/article/10.1208/s12248-012-9322-0#ref-CR97 "Ballester PJ, Mitchell JBO. A machine learning approach to predicting protein–ligand binding affinity with applications to molecular docking. Bioinformatics. 2010;26(9):1169–75. doi:
10.1093/bioinformatics/btq112
.")) has been built with RF, where a set of descriptors are introduced based on the count of a particular ligand–target atom pair within a certain distance range. Despite the relatively coarse definition of ligand–target atom pairs, which considers only atomic number with no concern about distance dependence, RF-score strikingly outperformed all 16 state-of-the-art scoring functions in a recent benchmark ([73](/article/10.1208/s12248-012-9322-0#ref-CR73 "Cheng T, Li X, Li Y, Liu Z, Wang R. Comparative assessment of scoring functions on a diverse test set. J Chem Inf Model. 2009;49(4):1079–93. doi:
10.1021/ci9000053
.")).SUMMARY
SBVS becomes routine in both pharmaceutical companies and academic groups for early-stage drug discovery. In this work, we reviewed the recent advances and applications in DBVS from a problem-centric perspective with an emphasis on the integration of available knowledge adopted by researchers in real practice. It is found that enriching a screening library for a specific target before docking can improve both computational efficiency and hit rate. Also, effective consideration of key ligand-target interactions and other environmental factors during docking, such as target flexibility, metal ions, and water molecules, can give enhanced DBVS performance. In addition, post-docking processing techniques that automate the selection of appropriate poses/compounds not only greatly alleviate the human intervention of docking outputs but also improve the final outcome simultaneously. Developing target-biased scoring functions represents a trend in tweaking current all-purpose alternatives toward specific target classes. Recent development of scoring function also observed an increasing use of machine learning techniques, which have an intrinsic non-linear feature and can implicitly account for some really challenging ligand–target interactions such as solvation and entropic effects.
Despite the listed advances here, current improvements in DBVS over state-of-the-art, in large part, only serve as patches or temporary remedies to existing methods, which often rely on expertise knowledge and thus may have limited applications in real practice. A universally accurate and reliable solution is still far from reach in the near future. Revolutionary innovations are definitely in urgent need and thus highly encouraged to address the fundamental challenges such as target flexibility and water molecules.
REFERENCES
- Ripphausen P, Nisius B, Peltason L, Bajorath Jr. Quo vadis, virtual screening? A comprehensive survey of prospective applications. J Med Chem. 2010;53(24):8461–7. doi:10.1021/jm101020z.
Article PubMed CAS Google Scholar - Clark DE. What has virtual screening ever done for drug discovery? Expert Opin Drug Discov. 2008;3:841–51. doi:10.1517/17460441.3.8.841.
Article CAS Google Scholar - Hopkins AL, Groom CR. The druggable genome. Nat Rev Drug Discov. 2002;1(9):727–30. doi:10.1038/nrd892.
Article PubMed CAS Google Scholar - Villoutreix BO, Eudes R, Miteva MA. Structure-based virtual ligand screening: recent success stories. Comb Chem High Throughput Screen. 2009;12(10):1000–16.
Article PubMed CAS Google Scholar - Ghosh S, Nie A, An J, Huang Z. Structure-based virtual screening of chemical libraries for drug discovery. Curr Opin Chem Biol. 2006;10(3):194–202. doi:10.1016/j.cbpa.2006.04.002.
Article PubMed CAS Google Scholar - Seifert MHJ, Lang M. Essential factors for successful virtual screening. Mini Rev Med Chem. 2007;8:63–72. doi:10.2174/138955708783331540.
Article Google Scholar - Tuccinardi T. Docking-based virtual screening: recent developments. Comb Chem High Throughput Screen. 2009;12(3):303–14.
Article PubMed CAS Google Scholar - Rapp CS, Schonbrun C, Jacobson MP, Kalyanaraman C, Huang N. Automated site preparation in physics-based rescoring of receptor ligand complexes. Proteins: Struct, Funct, Bioinf. 2009;77(1):52–61. doi:10.1002/prot.22415.
Article CAS Google Scholar - ten Brink T, Exner T. pKa based protonation states and microspecies for protein–ligand docking. J Comput Aided Mol Des. 2010;24(11):935–42. doi:10.1007/s10822-010-9385-x.
Article PubMed CAS Google Scholar - Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, et al. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J Comput Chem. 1998;19(14):1639–62. doi:10.1002/(sici)1096-987x(19981115)19:14<1639::aid-jcc10>3.0.co;2-b.
Article CAS Google Scholar - Ewing TJ, Makino S, Skillman AG, Kuntz ID. DOCK 4.0: search strategies for automated molecular docking of flexible molecule databases. J Comput Aided Mol Des. 2001;15:411–28. doi:10.1023/A:1011115820450.
Article PubMed CAS Google Scholar - Rarey M, Kramer B, Lengauer T, Klebe G. A fast flexible docking method using an incremental construction algorithm. J Mol Biol. 1996;261(3):470–89. doi:10.1006/jmbi.1996.0477.
Article PubMed CAS Google Scholar - Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, et al. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem. 2004;47:1739–49. doi:10.1021/jm0306430.
Article PubMed CAS Google Scholar - Jones G, Willett P, Glen RC, Leach AR, Taylor R. Development and validation of a genetic algorithm for flexible docking. J Mol Biol. 1997;267(3):727–48. doi:10.1006/jmbi.1996.0897.
Article PubMed CAS Google Scholar - Jain AN. Surflex: fully automatic flexible molecular docking using a molecular similarity-based search engine. J Med Chem. 2003;46(4):499–511. doi:10.1021/jm020406h.
Article PubMed CAS Google Scholar - Abagyan R, Totrov M, Kuznetsov D. ICM: a new method for protein modeling and design: applications to docking and structure prediction from the distorted native conformation. J Comput Chem. 1994;15:488–506. doi:10.1002/jcc.540150503.
Article CAS Google Scholar - Venkatachalam CM, Jiang X, Oldfield T, Waldman M. LigandFit: a novel method for the shape-directed rapid docking of ligands to protein active sites. J Mol Graph Model. 2003;21:289–307. doi:10.1016/S1093-3263(02)00164-X.
Article PubMed CAS Google Scholar - Zsoldos Z, Szabo I, Szabo Z, Peter Johnson A. Software tools for structure based rational drug design. J Mol Struct (THEOCHEM). 2003;666-667:659–65. doi:10.1016/j.theochem.2003.08.105.
Article CAS Google Scholar - Schneider G. Virtual screening: an endless staircase? Nat Rev Drug Discov. 2010;9:273–6. doi:10.1038/nrd3139.
Article PubMed CAS Google Scholar - Pujadas G, Vaque M, Ardevol A, Blade C, Salvado MJ, Blay M, et al. Protein–ligand docking: a review of recent advances and future perspectives. Curr Pharmaceut Anal. 2008;4:1–19. doi:10.2174/157341208783497597.
Article Google Scholar - Meng X-Y, Zhang H-X, Mezei M, Cui M. Molecular docking: a powerful approach for structure-based drug discovery. Curr Comput-Aided Drug Des. 2011;7:146–57. doi:10.2174/157340911795677602.
PubMed CAS Google Scholar - Yuriev E, Agostino M, Ramsland Pa. Challenges and advances in computational docking: 2009 in review. J Mol Recognit. 2010;24(2):149–64. doi:10.1002/jmr.1077.
Article PubMed Google Scholar - Moitessier N, Englebienne P, Lee D, Lawandi J, Corbeil CR. Towards the development of universal, fast and highly accurate docking/scoring methods: a long way to go. Br J Pharmacol. 2008;153(S1):S7–S26. doi:10.1038/sj.bjp.0707515.
Article PubMed CAS Google Scholar - Dias R, de Azevedo Jr WF. Molecular docking algorithms. Curr Drug Targets. 2008;9:1040–7. doi:10.2174/138945008786949432.
Article PubMed CAS Google Scholar - Cummings MD, Maxwell AC, DesJarlais RL. Processing of small molecule databases for automated docking. Med Chem. 2007;3:107–13. doi:10.2174/157340607779317481.
Article PubMed CAS Google Scholar - Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 1997;23(1–3):3–25. doi:10.1016/s0169-409x(96)00423-1.
Article CAS Google Scholar - Perez-Pineiro R, Burgos A, Jones DC, Andrew LC, Rodriguez H, Suarez M, et al. Development of a novel virtual screening cascade protocol to identify potential trypanothione reductase inhibitors. J Med Chem. 2009;52(6):1670–80. doi:10.1021/jm801306g.
Article PubMed CAS Google Scholar - Gozalbes R, Simon L, Froloff N, Sartori E, Monteils C, Baudelle R. Development and experimental validation of a docking strategy for the generation of kinase-targeted libraries. J Med Chem. 2008;51(11):3124–32. doi:10.1021/jm701367r.
Article PubMed CAS Google Scholar - Sage C, Wang R, Jones G. G-protein coupled receptors virtual screening using genetic algorithm focused chemical space. J Chem Inf Model. 2011. doi:10.1021/ci200043z.
- Kireev D, Wigle TJ, Norris-Drouin J, Herold JM, Janzen WP, Frye SV. Identification of non-peptide malignant brain tumor (MBT) repeat antagonists by virtual screening of commercially available compounds. J Med Chem. 2010;53(21):7625–31. doi:10.1021/jm1007374.
Article PubMed CAS Google Scholar - Lee K, Jeong K-W, Lee Y, Song JY, Kim MS, Lee GS, et al. Pharmacophore modeling and virtual screening studies for new VEGFR-2 kinase inhibitors. Eur J Med Chem. 2010;45(11):5420–7. doi:10.1016/j.ejmech.2010.09.002.
Article PubMed CAS Google Scholar - Heinke R, Spannhoff A, Meier R, Trojer P, Bauer I, Jung M, et al. Virtual screening and biological characterization of novel histone arginine methyltransferase PRMT1 inhibitors. Chem Med Chem. 2009;4(1):69–77. doi:10.1002/cmdc.200800301.
PubMed CAS Google Scholar - Rueda M, Bottegoni G, Abagyan R. Consistent improvement of cross-docking results using binding site ensembles generated with elastic network normal modes. J Chem Inf Model. 2009;49(3):716–25. doi:10.1021/ci8003732.
Article PubMed CAS Google Scholar - Cavasotto CN, Singh N. Docking and high throughput docking: successes and the challenge of protein flexibility. Curr Comput-Aided Drug Des. 2008;4:221–34. doi:10.2174/157340908785747474.
Article CAS Google Scholar - Cozzini P, Kellogg GE, Spyrakis F, Abraham DJ, Costantino G, Emerson A, et al. Target flexibility: an emerging consideration in drug discovery and design. J Med Chem. 2008;51(20):6237–55. doi:10.1021/jm800562d.
Article PubMed CAS Google Scholar - Durrant JD, McCammon JA. Computer-aided drug-discovery techniques that account for receptor flexibility. Curr Opin Pharmacol. 2010;10(6):770–4. doi:10.1016/j.coph.2010.09.001.
Article PubMed CAS Google Scholar - Sotriffer CA. Accounting for induced-fit effects in docking: what is possible and what is not? Curr Top Med Chem. 2011;11:179–91. doi:10.2174/156802611794863544.
PubMed CAS Google Scholar - Lill MA. Efficient incorporation of protein flexibility and dynamics into molecular docking simulations. Biochemistry. 2011;50(28):6157–69. doi:10.1021/bi2004558.
Article PubMed CAS Google Scholar - Lin J-H. Accommodating protein flexibility for structure-based drug design. Curr Top Med Chem. 2011;11:171–8. doi:10.2174/156802611794863580.
PubMed CAS Google Scholar - Okamoto M, Takayama K, Shimizu T, Ishida K, Takahashi O, Furuya T. Identification of death-associated protein kinases inhibitors using structure-based virtual screening. J Med Chem. 2009;52(22):7323–7. doi:10.1021/jm901191q.
Article PubMed CAS Google Scholar - Rueda M, Bottegoni G, Abagyan R. Recipes for the selection of experimental protein conformations for virtual screening. J Chem Inf Model. 2009;50(1):186–93. doi:10.1021/ci9003943.
Article Google Scholar - Sperandio O, Mouawad L, Pinto E, Villoutreix B, Perahia D, Miteva M. How to choose relevant multiple receptor conformations for virtual screening: a test case of Cdk2 and normal mode analysis. Eur Biophys J. 2010;39(9):1365–72. doi:10.1007/s00249-010-0592-0.
Article PubMed CAS Google Scholar - Bottegoni G, Kufareva I, Totrov M, Abagyan R. Four-dimensional docking: a fast and accurate account of discrete receptor flexibility in ligand docking. J Med Chem. 2008;52(2):397–406. doi:10.1021/jm8009958.
Article Google Scholar - Röhrig UF, Grosdidier A, Zoete V, Michielin O. Docking to heme proteins. J Comput Chem. 2009;30(14):2305–15. doi:10.1002/jcc.21244.
PubMed Google Scholar - Caporuscio F, Rastelli G, Imbriano C, Del Rio A. Structure-based design of potent aromatase inhibitors by high-throughput docking. J Med Chem. 2011;54:4006–17. doi:10.1021/jm2000689.
Article PubMed CAS Google Scholar - Park H, Kim S, Kim YE, Lim S-J. A structure-based virtual screening approach toward the discovery of histone deacetylase inhibitors: identification of promising zinc-chelating groups. Chem Med Chem. 2010;5(4):591–7. doi:10.1002/cmdc.200900500.
PubMed CAS Google Scholar - Thilagavathi R, Mancera RL. Ligand–protein cross-docking with water molecules. J Chem Inf Model. 2010;50(3):415–21. doi:10.1021/ci900345h.
Article PubMed CAS Google Scholar - Santos R, Hritz J, Oostenbrink C. Role of water in molecular docking simulations of cytochrome P450 2D6. J Chem Inf Model. 2009;50(1):146–54. doi:10.1021/ci900293e.
Article Google Scholar - Abel R, Young T, Farid R, Berne BJ, Friesner RA. Role of the active-site solvent in the thermodynamics of factor Xa ligand binding. J Am Chem Soc. 2008;130(9):2817–31. doi:10.1021/ja0771033.
Article PubMed CAS Google Scholar - Wang L, Berne BJ, Friesner RA. Ligand binding to protein-binding pockets with wet and dry regions. Proc Natl Acad Sci. 2011;108(4):1326–30. doi:10.1073/pnas.1016793108.
Article PubMed CAS Google Scholar - Lie MA, Thomsen R, Pedersen CNS, Schiøtt B, Christensen MH. Molecular docking with ligand attached water molecules. J Chem Inf Model. 2011;51(4):909–17. doi:10.1021/ci100510m.
Article PubMed CAS Google Scholar - Rossato G, Ernst B, Vedani A, Smieško M. AcquaAlta: a directional approach to the solvation of ligand–protein complexes. J Chem Inf Model. 2011. doi:10.1021/ci200150p.
- Brunskole Švegelj M, Turk S, Brus B, Lanišnik Rižner T, Stojan J, Gobec S. Novel inhibitors of trihydroxynaphthalene reductase with antifungal activity identified by ligand-based and structure-based virtual screening. J Chem Inf Model. 2011;51(7):1716–24. doi:10.1021/ci2001499.
Article PubMed Google Scholar - Ravindranathan KP, Mandiyan V, Ekkati AR, Bae JH, Schlessinger J, Jorgensen WL. Discovery of novel fibroblast growth factor receptor 1 kinase inhibitors by structure-based virtual screening. J Med Chem. 2010;53(4):1662–72. doi:10.1021/jm901386e.
Article PubMed CAS Google Scholar - Lang PT, Brozell SR, Mukherjee S, Pettersen EF, Meng EC, Thomas V, et al. DOCK 6: combining techniques to model RNA–small molecule complexes. RNA. 2009;15(6):1–12. doi:10.1261/rna.1563609.
Article Google Scholar - Kerzmann A, Fuhrmann J, Kohlbacher O, Neumann D. BALLDock/SLICK: a new method for protein–carbohydrate docking. J Chem Inf Model. 2008;48(8):1616–25. doi:10.1021/ci800103u.
Article PubMed CAS Google Scholar - Waszkowycz B. Towards improving compound selection in structure-based virtual screening. Drug Discov Today. 2008;13(5–6):219–26. doi:10.1016/j.drudis.2007.12.002.
Article PubMed CAS Google Scholar - Marcou G, Rognan D. Optimizing fragment and scaffold docking by use of molecular interaction fingerprints. J Chem Inf Model. 2007;47(1):195–207. doi:10.1021/ci600342e.
Article PubMed CAS Google Scholar - Bouvier G, Evrard-Todeschi N, Girault J-P, Bertho G. Automatic clustering of docking poses in virtual screening process using self-organizing map. Bioinformatics. 2010;26(1):53–60. doi:10.1093/bioinformatics/btp623.
Article PubMed CAS Google Scholar - Novikov F, Stroylov V, Stroganov O, Chilov G. Improving performance of docking-based virtual screening by structural filtration. J Mol Model. 2010;16(7):1223–30. doi:10.1007/s00894-009-0633-8.
Article PubMed CAS Google Scholar - Wei D, Zheng H, Su N, Deng M, Lai L. Binding energy landscape analysis helps to discriminate true hits from high-scoring decoys in virtual screening. J Chem Inf Model. 2010;50(10):1855–64. doi:10.1021/ci900463u.
Article PubMed CAS Google Scholar - Böhm H-J. Prediction of binding constants of protein ligands: a fast method for the prioritization of hits obtained from de novo design or 3D database search programs. J Comput Aided Mol Des. 1998;12(4):309. doi:10.1023/a:1007999920146.
Article PubMed Google Scholar - Wang R, Lai L, Wang S. Further development and validation of empirical scoring functions for structure-based binding affinity prediction. J Comput Aided Mol Des. 2002;16(1):11–26. doi:10.1023/a:1016357811882.
Article PubMed CAS Google Scholar - Gehlhaar DK, Verkhivker GM, Rejto PA, Sherman CJ, Fogel DR, Fogel LJ, et al. Molecular recognition of the inhibitor AG-1343 by HIV-1 protease: conformationally flexible docking by evolutionary programming. Chem Biol. 1995;2(5):317–24. doi:10.1016/1074-5521(95)90050-0.
Article PubMed CAS Google Scholar - Eldridge MD, Murray CW, Auton TR, Paolini GV, Mee RP. Empirical scoring functions: I. The development of a fast empirical scoring function to estimate the binding affinity of ligands in receptor complexes. J Comput Aided Mol Des. 1997;11(5):425–45. doi:10.1023/a:1007996124545.
Article PubMed CAS Google Scholar - McMartin C, Bohacek RS. QXP: powerful, rapid computer algorithms for structure-based drug design. J Comput Aided Mol Des. 1997;11(4):333–44. doi:10.1023/a:1007907728892.
Article PubMed CAS Google Scholar - Muegge I. PMF scoring revisited. J Med Chem. 2005;49(20):5895–902. doi:10.1021/jm050038s.
Article Google Scholar - Xue M, Zheng M, Xiong B, Li Y, Jiang H, Shen J. Knowledge-based scoring functions in drug design. 1. Developing a target-specific method for kinase−ligand interactions. J Chem Inf Model. 2010;50(8):1378–86. doi:10.1021/ci100182c.
Article PubMed CAS Google Scholar - Gohlke H, Hendlich M, Klebe G. Knowledge-based scoring function to predict protein–ligand interactions. J Mol Biol. 2000;295(2):337–56. doi:10.1006/jmbi.1999.3371.
Article PubMed CAS Google Scholar - Mooij WTM, Verdonk ML. General and targeted statistical potentials for protein–ligand interactions. Proteins: Struct, Funct, Bioinf. 2005;61(2):272–87. doi:10.1002/prot.20588.
Article CAS Google Scholar - Huang S-Y, Zou X. Inclusion of solvation and entropy in the knowledge-based scoring function for protein–ligand interactions. J Chem Inf Model. 2010;50(2):262–73. doi:10.1021/ci9002987.
Article PubMed CAS Google Scholar - Huang S-Y, Grinter SZ, Zou X. Scoring functions and their evaluation methods for protein-ligand docking: recent advances and future directions. Phys Chem Chem Phys. 2010;12(40):12899–908. doi:10.1039/c0cp00151a.
Article PubMed CAS Google Scholar - Cheng T, Li X, Li Y, Liu Z, Wang R. Comparative assessment of scoring functions on a diverse test set. J Chem Inf Model. 2009;49(4):1079–93. doi:10.1021/ci9000053.
Article PubMed CAS Google Scholar - Warren GL, Andrews CW, Capelli A-M, Clarke B, LaLonde J, Lambert MH, et al. A critical assessment of docking programs and scoring functions. J Med Chem. 2006;49(20):5912–31. doi:10.1021/jm050362n.
Article PubMed CAS Google Scholar - Ferrara P, Gohlke H, Price DJ, Klebe G, Brooks CL. Assessing scoring functions for protein–ligand interactions. J Med Chem. 2004;47(12):3032–47. doi:10.1021/jm030489h.
Article PubMed CAS Google Scholar - Wang R, Lu Y, Fang X, Wang S. An extensive test of 14 scoring functions using the PDBbind refined set of 800 protein–ligand complexes. J Chem Inf Comput Sci. 2004;44(6):2114–25. doi:10.1021/ci049733j.
Article PubMed CAS Google Scholar - Wang R, Lu Y, Wang S. Comparative evaluation of 11 scoring functions for molecular docking. J Med Chem. 2003;46(12):2287–303. doi:10.1021/jm0203783.
Article PubMed CAS Google Scholar - Raub S, Steffen A, Kämper A, Marian CM. AIScore: chemically diverse empirical scoring function employing quantum chemical binding energies of hydrogen-bonded complexes. J Chem Inf Model. 2008;48(7):1492–510. doi:10.1021/ci7004669.
Article PubMed CAS Google Scholar - Charifson PS, Corkery JJ, Murcko MA, Walters WP. Consensus scoring: a method for obtaining improved hit rates from docking databases of three-dimensional structures into proteins. J Med Chem. 1999;42(25):5100–9. doi:10.1021/jm990352k.
Article PubMed CAS Google Scholar - Wang R, Wang S. How does consensus scoring work for virtual library screening? An idealized computer experiment. J Chem Inf Comput Sci. 2001;41(5):1422–6. doi:10.1021/ci010025x.
Article PubMed CAS Google Scholar - Seifert MHJ. Targeted scoring functions for virtual screening. Drug Discov Today. 2009;14(11–12):562–9. doi:10.1016/j.drudis.2009.03.013.
Article PubMed Google Scholar - Pfeffer P, Gohlke H. DrugScoreRNA: knowledge-based scoring function to predict RNA–ligand interactions. J Chem Inf Model. 2007;47(5):1868–76. doi:10.1021/ci700134p.
Article PubMed CAS Google Scholar - Teramoto R, Fukunishi H. Supervised scoring models with docked ligand conformations for structure-based virtual screening. J Chem Inf Model. 2007;47(5):1858–67. doi:10.1021/ci700116z.
Article PubMed CAS Google Scholar - Seifert MHJ. Optimizing the signal-to-noise ratio of scoring functions for protein–ligand docking. J Chem Inf Model. 2008;48(3):602–12. doi:10.1021/ci700345n.
Article PubMed CAS Google Scholar - Pham T, Jain A. Customizing scoring functions for docking. J Comput Aided Mol Des. 2008;22(5):269–86. doi:10.1007/s10822-008-9174-y.
Article PubMed CAS Google Scholar - Catana C, Stouten PFW. Novel, customizable scoring functions, parameterized using N-PLS, for structure-based drug discovery. J Chem Inf Model. 2007;47(1):85–91. doi:10.1021/ci600357t.
Article PubMed CAS Google Scholar - Terp GE, Johansen BN, Christensen IT, Jørgensen FS. A new concept for multidimensional selection of ligand conformations (MultiSelect) and multidimensional scoring (MultiScore) of protein–ligand binding affinities. J Med Chem. 2001;44(14):2333–43. doi:10.1021/jm001090l.
Article PubMed CAS Google Scholar - Martin EJ, Sullivan DC. AutoShim: empirically corrected scoring functions for quantitative docking with a crystal structure and IC50 training data. J Chem Inf Model. 2008;48(4):861–72. doi:10.1021/ci7004548.
Article PubMed CAS Google Scholar - Cheng T, Liu Z, Wang R. A knowledge-guided strategy for improving the accuracy of scoring functions in binding affinity prediction. BMC Bioinf. 2010;11(1):193. doi:10.1186/1471-2105-11-193.
Article Google Scholar - Hecht D, Fogel GB. Computational intelligence methods for docking scores. Curr Comput-Aided Drug Des. 2009;5:56–68. doi:10.2174/157340909787580863.
Article CAS Google Scholar - Durrant JD, McCammon JA. NNScore: a neural-network-based scoring function for the characterization of protein–ligand complexes. J Chem Inf Model. 2010;50(10):1865–71. doi:10.1021/ci100244v.
Article PubMed CAS Google Scholar - Kinnings SL, Liu N, Tonge PJ, Jackson RM, Xie L, Bourne PE. A machine learning-based method to improve docking scoring functions and its application to drug repurposing. J Chem Inf Model. 2011;51(2):408–19. doi:10.1021/ci100369f.
Article PubMed CAS Google Scholar - Amini A, Shrimpton PJ, Muggleton SH, Sternberg MJE. A general approach for developing system-specific functions to score protein–ligand docked complexes using support vector inductive logic programming. Proteins: Struct, Funct, Bioinf. 2007;69(4):823–31. doi:10.1002/prot.21782.
Article CAS Google Scholar - Tang YT, Marshall GR. PHOENIX: a scoring function for affinity prediction derived using high-resolution crystal structures and calorimetry measurements. J Chem Inf Model. 2011;51(2):214–28. doi:10.1021/ci100257s.
Article PubMed CAS Google Scholar - Deng W, Breneman C, Embrechts MJ. Predicting protein–ligand binding affinities using novel geometrical descriptors and machine-learning methods. J Chem Inf Comput Sci. 2004;44(2):699–703. doi:10.1021/ci034246+.
Article PubMed CAS Google Scholar - Li L, Khanna M, Jo I, Wang F, Ashpole NM, Hudmon A, et al. Target-specific support vector machine scoring in structure-based virtual screening: computational validation, in vitro testing in kinases, and effects on lung cancer cell proliferation. J Chem Inf Model. 2011;51(4):755–9. doi:10.1021/ci100490w.
Article PubMed CAS Google Scholar - Ballester PJ, Mitchell JBO. A machine learning approach to predicting protein–ligand binding affinity with applications to molecular docking. Bioinformatics. 2010;26(9):1169–75. doi:10.1093/bioinformatics/btq112.
Article PubMed CAS Google Scholar
ACKNOWLEDGMENTS
We thank the Intramural Research Program of the National Institutes of Health (NIH), National Library of Medicine (NLM) for funding support.
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
- National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, 8600 Rockville Pike, Bethesda, Maryland, 20894, USA
Tiejun Cheng, Qingliang Li, Zhigang Zhou, Yanli Wang & Stephen H. Bryant
Authors
- Tiejun Cheng
You can also search for this author inPubMed Google Scholar - Qingliang Li
You can also search for this author inPubMed Google Scholar - Zhigang Zhou
You can also search for this author inPubMed Google Scholar - Yanli Wang
You can also search for this author inPubMed Google Scholar - Stephen H. Bryant
You can also search for this author inPubMed Google Scholar
Corresponding authors
Correspondence toYanli Wang or Stephen H. Bryant.
Additional information
Guest Editor: Xiang-Qun Xie
Tiejun Cheng, Qingliang Li and Zhigang Zhou contributed equally to this work.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Cheng, T., Li, Q., Zhou, Z. et al. Structure-Based Virtual Screening for Drug Discovery: a Problem-Centric Review.AAPS J 14, 133–141 (2012). https://doi.org/10.1208/s12248-012-9322-0
- Received: 17 August 2011
- Accepted: 04 January 2012
- Published: 27 January 2012
- Issue Date: March 2012
- DOI: https://doi.org/10.1208/s12248-012-9322-0