Fanconi Anemia: Background, Etiology, Epidemiology (original) (raw)
Overview
Background
Fanconi anemia is the most frequently reported of the rare inherited bone marrow failure syndromes (IBMFSs). In 1927, Guido Fanconi first reported three brothers with macrocytosis, pancytopenia, and physical abnormalities. [1, 2]
In the early 1960s, several groups observed that cultured cells from patients with Fanconi anemia had increased numbers of chromosome breaks; later, the breakage rate was found to be specifically increased by the addition of deoxyribonucleic acid (DNA) cross-linkers, such as diepoxybutane (DEB) or mitomycin C (MMC). This led to the identification of patients with Fanconi anemia and aplastic anemia without birth defects and the diagnosis of Fanconi anemia in patients without aplastic anemia but with abnormal physical findings. (See Etiology.)
Furthermore, in cultured Fanconi anemia cells, cell cycle arrest in gap 2/mitosis (G2/M) occurs at lower concentrations of clastogens than in normal cells. This observation has led to flow cytometry–based screening tests used at some centers. (See Workup.)
The advent of molecular diagnostics has further improved the specificity of Fanconi anemia diagnosis.
Fanconi anemia accounts for approximately 25% of the cases of aplastic anemia seen at large referral centers. Approximately 25% of known patients with Fanconi anemia do not have major birth defects. (See Physical Examination.)
Birth defects associated with Fanconi anemia are demonstrated in the images below.

A 3-year-old patient with Fanconi anemia. Note the multiple birth defects, including short stature, microcephaly, microphthalmia, epicanthal folds, dangling thumbs, site of ureteral reimplantation, congenital dislocated hips, and rocker bottom feet. (Alter BP, Young NS. The bone marrow failure syndromes. In: Nathan DG, Oski FA, eds. Hematology of Infancy and Childhood, 4th ed. Philadelphia, PA: WB Saunders, Inc, 1993: 216-316.)
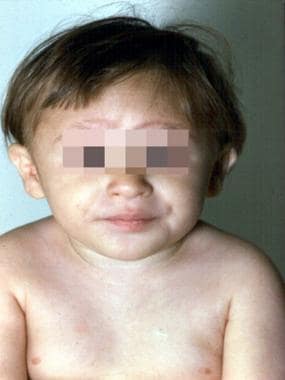
The 3-year-old patient with Fanconi anemia seen in the previous image. (Alter BP, Young NS. The bone marrow failure syndromes. In: Nathan DG, Oski FA, eds. Hematology of Infancy and Childhood, 4th ed. Philadelphia, PA: WB Saunders, Inc, 1993: 216-316.)
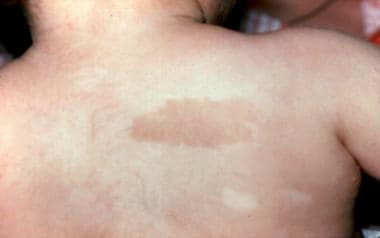
Café au lait spot and hypopigmented area in a 3-year-old patient with Fanconi anemia. Same patient as in the previous images. (Alter BP, Young NS. The bone marrow failure syndromes. In: Nathan DG, Oski FA, eds. Hematology of Infancy and Childhood, 4th ed. Philadelphia, PA: WB Saunders, Inc, 1993: 216-316.)
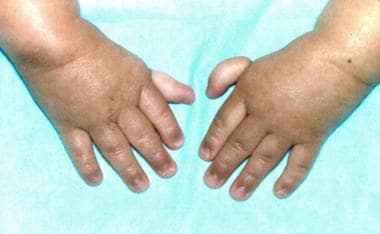
Thumbs attached by threads on a 3-year-old patient with Fanconi anemia (same patient as in the previous images). (Alter BP, Young NS. The bone marrow failure syndromes. In: Nathan DG, Oski FA, eds. Hematology of Infancy and Childhood, 4th ed. Philadelphia, PA: WB Saunders, Inc, 1993: 216-316.)
Go to Pediatric Chronic Anemia, Anemia of Prematurity, Donath-Landsteiner Hemolytic Anemia, Pediatric Acute Anemia, and Megaloblastic Anemia for complete information on these topics.
Complications of Fanconi anemia
Possible complications of Fanconi anemia include hemorrhages, infections, leukemia, myelodysplastic syndrome, and liver tumors and other cancers. [3, 4] (See Prognosis.)
From literature reviews, it is estimated that 9% of patients develop leukemia, of which 95% are acute myeloid leukemia (usually rare in children), with a relative risk for acute myeloid leukemia of approximately 500-fold. The majority of cases develop between ages 15 and 35 years, with a cumulative incidence of 13% by age 50 years.
Myelodysplastic syndrome was reported in 7% of patients (>100 patients); many of these patients did not develop leukemia but died from complications of impaired marrow function. The risk of myelodysplastic syndrome in Fanconi anemia is increased about 5000-fold.
Liver tumors occurred in more than 45 patients, 43 of which were associated with androgen use, often in the context of aplastic anemia or other tumors, and were not usually malignant (although two thirds were histologically hepatomas, and the rest were adenomas).
There is a marked increase in solid tumors. In order of frequency, these tumors were tumors of the oropharynx, esophagus, vulva/vagina, brain, skin (nonmelanoma), cervix, breast, kidney, lung, lymph nodes (lymphoma), stomach, and colon, followed by osteogenic sarcoma and retinoblastoma. The relative risk of all cancers was approximately 40-fold, with a cumulative incidence of 30% by age 50 years. The risk of head and neck squamous cell carcinoma is 600-fold and for vulvar/vaginal squamous cell carcinoma approximately 3000-fold. A large number of oral cancers have been reported in patients with Fanconi anemia following bone marrow transplantation.
Due to the high sensitivity to chemotherapeutic agents, which damage DNA, the outcome for patients with Fanconi anemia and cancer is quite poor.
Patients with Fanconi anemia in the FANCD1/BRCA2 (the highest cancer risk genotype), N/PALB2, and J/BRIP1 groups (monoallelic breast cancer predisposition genes) have inordinately high rates of acute myeloid leukemia, brain tumors (medulloblastoma), and Wilms tumor, with a cumulative incidence of at least 1 of these cancers of 95% by age 5 years.
Congenital anomalies
The vast majority (75%) of individuals with Fanconi anemia have at least one physical anomaly. The most common are short stature and cutaneous, skeletal, craniofacial, and genitourinary anomalies. Additionally, approximately 5% of patients with Fanconi anemia have at least 3 of the defining features of VATER, or VACTERL, association (vertebral anomalies, anal atresia, cardiovascular anomalies, tracheoesophageal fistula, renal and/or radial anomalies, limb defects). Furthermore, individuals with an expanded phenotype, VACTERL-H (the highest incidence in the FANCD1/BRCA2 genotype), regardless of hematologic status, must be evaluated for Fanconi anemia. [6]
Other anomalies include developmental delay, hearing loss, congenital heart disease, and central nervous system (CNS) anomalies (arterial malformation, stenosis of the internal carotid, and small pituitary gland). (The clinical presentation of Fanconi anemia is discussed under Physical Examination.)
A study by Altintas et al looking at the Fanconi anemia/BRCA DNA repair pathways found that the Fanconi anemia phenotype was less severe in individuals with variants in the upstream complex pathway, with these patients lacking the features of VACTERL-H and/or PHENO (Pigmentation, small-Head, small-Eyes, Neurologic, Otologic, Short stature). The VACTERL-H phenotype was associated with the ID complex pathway. The investigators also found better survival in the upstream complex patients than in individuals with variants in the ID complex or downstream complex. [8]
![]()
Etiology
Fanconi anemia is an autosomal recessive disease in more than 99% of patients (FANCB is X-linked recessive); each patient with Fanconi anemia is homozygous or doubly heterozygous for mutations in one of the 15 genes known to be responsible for the disorder. The cloned genes are FANCA, B, C, D1/BRCA2, D2, E, F, G/XRCC9, I, J/BRIP1, L, M, N/PALB2, O/RAD51C, and P/SLX4. Although most are unique genes, several were previously known, including FANCD1 (BRCA2), FANCG (XRCC9), FANCJ (BRPI1/BACH1), FANCN (PALB2), FANCO (RED51C), and (SLX4). Heterozygotes for BRCA2 and possibly BACH1 and PALB2 are at increased risk for breast and other cancers.
The first 13 Fanconi anemia proteins have discrete functions, with A, B, C, E, F, G, L, and M appearing to form a nuclear complex, which leads to ubiquitination of the I and D2 proteins. The latter is involved in DNA damage response mechanisms in cooperation with FANCD1, FANCJ, and FANCN, as well as BRCA1, RAD51, Mre11, and other proteins.
The widely variant Fanconi anemia phenotype may depend not on the specific gene involved but on whether the mutation is null or leads to a partially functional gene product. (A study by Ramírez et al found that “patients with some mutant [Fanconi anemia] protein expression experienced better hematologic evolution and, consequently, survived for longer than patients with mutations resulting in total protein absence.” [9] ) The specific role of Fanconi anemia gene mutations in the pathogenesis of birth defects, bone marrow failure, or oncogenesis is not yet clear. The designation of the extremely rare proteins O and P remains controversial.
![]()
Epidemiology
Fanconi anemia has been reported in persons of all races. However, owing to founder effects, the heterozygote frequency is greater in South African Afrikaners, [10] sub-Saharan Blacks, and Spanish Roma [11] than in the overall world population, leading to expected birth rates in these subpopulations of around one case per 40,000 births. Among Ashkenazi Jews in the United States, the carrier frequency is approximately 1 case per 90 people, with a projected birth rate of one case per 30,000 people. [12]
The male-to-female ratio in the literature cases is 1.2:1, although equal numbers are expected in a disorder with over 99% autosomal recessive inheritance.
Fanconi anemia has been diagnosed in patients from birth to middle age, with a median age of 7 years. Individuals with birth defects are diagnosed at younger ages than are persons without birth defects.
![]()
Prognosis
Treatment of aplastic anemia with medications, supportive use of blood products, and stem cell transplantation increases the life expectancy beyond the projected median of approximately age 30 years.
Cancer prevention strategies, in particular the avoidance of smoking, and screening to identify early malignancies may reduce the mortality rate from cancer. With regard to the first serious adverse event, patients with a large number of birth defects are at higher risk for early onset severe aplastic anemia, while those with fewer anomalies are more likely to develop leukemia or a solid tumor as young adults.
Although many patients with Fanconi anemia are short and have skeletal anomalies, intelligence is usually normal, and education and career planning should be encouraged.
Mortality/morbidity
Regarding mortality and morbidity, [13] major adverse events for patients with Fanconi anemia are aplastic anemia (usually severe), leukemia, and solid tumors. The projected median survival from all causes for more than 2000 cases reported in the literature improved in the 21st century; from 1927-1999 and 2000-2009, median survival ages were 21 years and 29 years, respectively.
A later study, from the National Cancer Institute, found that out of 200 patients with Fanconi anemia, the median survival age was 37 years. Survival varied by genotype; for example, patients with FANCA variants had a median survival age of 43.4 years, while those with FANCD1/BRCA2 variants had a median survival age of just 4.3 years. [14]
Bone marrow failure usually presents in childhood, with petechiae, bruising, and hemorrhages due to thrombocytopenia; pallor and fatigue from anemia; and infections due to neutropenia. The annual hazard rate for severe aplastic anemia reached 5% per year by age 10 years and was less than 1% per year in adults, with a cumulative incidence of 50% by age 50 years.
Leukemia usually presents primarily in teens and young adults, reaching a hazard rate of 1% per year, with a cumulative incidence of 10% by age 50 years. About one third of cases of Fanconi anemia and leukemia in the literature did not have a prior diagnosis of Fanconi anemia, as well as a preceding phase of aplastic anemia. More than 100 cases in the literature were reported to have myelodysplastic syndrome.
The hazard rate for solid tumors rises steadily to greater than 10% per year by age 45 years, with a cumulative incidence of 25% by age 50 years, often without prior hematologic disease. As for acute myeloid leukemia, about one third of reported cases presented with a tumor and were subsequently diagnosed as Fanconi anemia.
A positive correlation between absent or abnormal radii and other congenital anomalies and bone marrow failure has been noted. The relative hazard of bone marrow failure and leukemia is higher in FANCG, compared with FANCA, and in FANCC, compared with FANCA. Patients with homozygous null mutations in FANCA have a higher risk of leukemia than do those with allelic mutations. Patients with biallelic mutations in BRCA2/FANCD1 have an extraordinarily high risk of acute myeloid leukemia, brain tumors (medulloblastoma), and Wilms tumors, with an approximately 95% chance of developing one of these tumors by age 5 years. Genetic background (Japanese vs Ashkenazi Jewish) and specific allelic mutations in FANCC can modulate the phenotype.
The risk of liver tumors is increased 400-fold, the risk of leukemia is about 500-fold, and head and neck cancers are increased approximately 600-fold. The risk of esophageal cancer is increased 2000-fold, and the risk of vulvar/vaginal cancer is increased 3000-fold. In competing risk analyses, the cumulative incidence of solid tumors reaches 30% by age 45 years and does not level off. Although bone marrow failure and leukemia, which may be treated or prevented by hematopoietic stem cell transplantation or gene therapy, are the concerns in treating children and adolescents, solid tumors remain the major threat to older patients with Fanconi anemia.
In a retrospective analysis of 145 patients with Fanconi anemia, 9 patients evolved to leukemia, and 14 developed 18 solid tumors. [6] The ratio of observed-to-expected cancers for all cancer diagnoses or for solid tumors was 40, and the ratio was 600 for leukemia. The cumulative incidence of leukemia, death from marrow failure, death from a solid tumor, and having a stem cell transplant (not necessarily a favorable outcome) was 10%, 11%, 29%, and 43%, respectively. Note that the risk of head and neck squamous cell carcinomas appeared to be higher in patients who had undergone bone marrow transplantation. [7, 15]
A study by Sauter et al suggested that the prevalence of oral human papillomavirus (HPV) is greater in persons with Fanconi anemia. The report found the oral HPV rate to be 11.1% in 126 patients with Fanconi anemia, versus 2.5% in 162 unaffected first-degree family members. More specifically, the oral HPV rate in sexually active persons with Fanconi anemia was 17.7%, versus 2.4% in family members, while in sexually inactive individuals with Fanconi anemia the prevalence of HPV was 8.7%, versus 2.9% in siblings. [16]
A study by Sathyanarayana et al suggested that in patients with Fanconi anemia, greater age is positively correlated with the incidence of chronic kidney disease. [17]
![]()
Patient Education
Educate patients and their families regarding behaviors with risk of bleeding as well as maintenance of hygiene to reduce infections. Emphasize the need to comply with medications and transfusions. Educate patients and their families about cancer prevention (eg, smoking, drinking, diet, lifestyle) and cancer screening (eg, bone marrow, oropharyngeal, and gynecologic examinations).
The genetic basis of Fanconi anemia needs to be explained, and apparently unaffected siblings should be tested for Fanconi anemia homozygosity. Provide genetic counseling to parents, caregivers, and other carriers or potential carriers with regard to the risk of recurrence. Discuss phenotypic variability within a family.
![]()
- Rayi A, Hozayen S. Chromosome Instability Syndromes. StatPearls. 2022 Sep 19. [QxMD MEDLINE Link]. [Full Text].
- Keefe P, Bokhari SRA. Fanconi Syndrome. StatPearls. 2023 Jul 4. [QxMD MEDLINE Link]. [Full Text].
- Katzenellenbogen RA, Carter JJ, Stern JE, et al. Skin and mucosal human papillomavirus seroprevalence in persons with fanconi anemia. Clin Vaccine Immunol. 2015 Apr. 22 (4):413-20. [QxMD MEDLINE Link].
- Alter BP. Fanconi anemia and the development of leukemia. Best Pract Res Clin Haematol. 2014 Sep-Dec. 27 (3-4):214-21. [QxMD MEDLINE Link]. [Full Text].
- Alter BP, Kupfer G. Fanconi anemia. Pagon RA, Bird TD, Dolan CR, et al, eds. Gene Reviews. Seattle, Wash: University of Washington; 2013.
- Rosenberg PS, Greene MH, Alter BP. Cancer incidence in persons with Fanconi anemia. Blood. 2003 Feb 1. 101(3):822-6. [QxMD MEDLINE Link].
- Rosenberg PS, Alter BP, Ebell W. Cancer risks in Fanconi anemia: findings from the German Fanconi Anemia Registry. Haematologica. 2008 Apr. 93(4):511-7. [QxMD MEDLINE Link].
- Altintas B, Giri N, McReynolds LJ, Best A, Alter BP. Genotype-phenotype and outcome associations in patients with Fanconi anemia: The National Cancer Institute cohort. Haematologica. 2022 Apr 14. [QxMD MEDLINE Link].
- Ramirez MJ, Pujol R, Minguillon J, et al. Prognostic significance of mutation type and chromosome fragility in Fanconi anemia. Am J Hematol. 2025 Feb. 100 (2):272-84. [QxMD MEDLINE Link]. [Full Text].
- Tipping AJ, Pearson T, Morgan NV, et al. Molecular and genealogical evidence for a founder effect in Fanconi anemia families of the Afrikaner population of South Africa. Proc Natl Acad Sci U S A. 2001 May 8. 98(10):5734-9. [QxMD MEDLINE Link]. [Full Text].
- Callén E, Casado JA, Tischkowitz MD, et al. A common founder mutation in FANCA underlies the world's highest prevalence of Fanconi anemia in Gypsy families from Spain. Blood. 2005 Mar 1. 105(5):1946-9. [QxMD MEDLINE Link].
- Verlander PC, Kaporis A, Liu Q, Zhang Q, Seligsohn U, Auerbach AD. Carrier frequency of the IVS4 + 4 A-->T mutation of the Fanconi anemia gene FAC in the Ashkenazi Jewish population. Blood. 1995 Dec 1. 86(11):4034-8. [QxMD MEDLINE Link].
- Shimamura A, Alter BP. Pathophysiology and management of inherited bone marrow failure syndromes. Blood Rev. 2010 May. 24(3):101-22. [QxMD MEDLINE Link]. [Full Text].
- Altintas B, Giri N, McReynolds LJ, Best A, Alter BP. Genotype-phenotype and outcome associations in patients with Fanconi anemia: the National Cancer Institute cohort. Haematologica. 2023 Jan 1. 108 (1):69-82. [QxMD MEDLINE Link]. [Full Text].
- Rosenberg PS, Socié G, Alter BP, Gluckman E. Risk of head and neck squamous cell cancer and death in patients with Fanconi anemia who did and did not receive transplants. Blood. 2005 Jan 1. 105(1):67-73. [QxMD MEDLINE Link].
- Sauter SL, Wells SI, Zhang X, et al. Oral human papillomavirus is common in individuals with Fanconi anemia. Cancer Epidemiol Biomarkers Prev. 2015 May. 24 (5):864-72. [QxMD MEDLINE Link].
- Sathyanarayana V, Lee B, Wright NB, et al. Patterns and frequency of renal abnormalities in Fanconi anaemia: implications for long-term management. Pediatr Nephrol. 2018 Apr 12. [QxMD MEDLINE Link].
- Alter BP, Rosenberg PS, Brody LC. Clinical and molecular features associated with biallelic mutations in FANCD1/BRCA2. J Med Genet. 2007 Jan. 44(1):1-9. [QxMD MEDLINE Link]. [Full Text].
- Alter BP, Rosenberg PS. VACTERL-H Association and Fanconi Anemia. Mol Syndromol. 2013 Feb. 4(1-2):87-93. [QxMD MEDLINE Link]. [Full Text].
- Dalle JH. HSCT for Fanconi anemia in children: factors that influence early and late results. Bone Marrow Transplant. 2008 Oct. 42 Suppl 2:S51-3. [QxMD MEDLINE Link].
- Pasquini R, Carreras J, Pasquini MC, et al. HLA-matched sibling hematopoietic stem cell transplantation for fanconi anemia: comparison of irradiation and nonirradiation containing conditioning regimens. Biol Blood Marrow Transplant. 2008 Oct. 14(10):1141-7. [QxMD MEDLINE Link]. [Full Text].
- Mitchell R, Wagner JE, Hirsch B, DeFor TE, Zierhut H, MacMillan ML. Haematopoietic cell transplantation for acute leukaemia and advanced myelodysplastic syndrome in Fanconi anaemia. Br J Haematol. 2014 Feb. 164 (3):384-95. [QxMD MEDLINE Link]. [Full Text].
- Smetsers SE, Smiers FJ, Bresters D, Sonnevelt MC, Bierings MB. Four decades of stem cell transplantation for Fanconi anaemia in the Netherlands. Br J Haematol. 2016 Sep. 174 (6):952-61. [QxMD MEDLINE Link].
- Giardino S, de Latour RP, Aljurf M, et al. Outcome of patients with Fanconi anemia developing myelodysplasia and acute leukemia who received allogeneic hematopoietic stem cell transplantation: A retrospective analysis on behalf of EBMT group. Am J Hematol. 2020 Apr 8. [QxMD MEDLINE Link].
- Lum SH, Eikema DJ, Piepenbroek B, et al. Outcomes of hematopoietic stem cell transplantation in 813 pediatric patients with Fanconi anemia. Blood. 2024 Sep 19. 144 (12):1329-42. [QxMD MEDLINE Link].
- MacMillan ML, DeFor TE, Young JA, et al. Alternative donor hematopoietic cell transplantation for Fanconi anemia. Blood. 2015 Jun 11. 125 (24):3798-804. [QxMD MEDLINE Link].
- Wang Y, Zhou W, Alter BP, et al. Chromosomal Aberrations and Survival after Unrelated Donor Hematopoietic Stem Cell Transplant in Patients with Fanconi Anemia. Biol Blood Marrow Transplant. 2018 Jun 4. [QxMD MEDLINE Link].
- A 3-year-old patient with Fanconi anemia. Note the multiple birth defects, including short stature, microcephaly, microphthalmia, epicanthal folds, dangling thumbs, site of ureteral reimplantation, congenital dislocated hips, and rocker bottom feet. (Alter BP, Young NS. The bone marrow failure syndromes. In: Nathan DG, Oski FA, eds. Hematology of Infancy and Childhood, 4th ed. Philadelphia, PA: WB Saunders, Inc, 1993: 216-316.)
- The 3-year-old patient with Fanconi anemia seen in the previous image. (Alter BP, Young NS. The bone marrow failure syndromes. In: Nathan DG, Oski FA, eds. Hematology of Infancy and Childhood, 4th ed. Philadelphia, PA: WB Saunders, Inc, 1993: 216-316.)
- Café au lait spot and hypopigmented area in a 3-year-old patient with Fanconi anemia. Same patient as in the previous images. (Alter BP, Young NS. The bone marrow failure syndromes. In: Nathan DG, Oski FA, eds. Hematology of Infancy and Childhood, 4th ed. Philadelphia, PA: WB Saunders, Inc, 1993: 216-316.)
- Thumbs attached by threads on a 3-year-old patient with Fanconi anemia (same patient as in the previous images). (Alter BP, Young NS. The bone marrow failure syndromes. In: Nathan DG, Oski FA, eds. Hematology of Infancy and Childhood, 4th ed. Philadelphia, PA: WB Saunders, Inc, 1993: 216-316.)

Author
Jeffrey M Lipton, MD, PhD Professor of Pediatrics and Molecular Medicine, Hofstra North Shore-Long Island Jewish School of Medicine; Professor, Elmezzi Graduate School of Molecular Medicine; Director, Patient-Oriented Research, Feinstein Institute for Medical Research; Director, Pediatric Hematology/Oncology and Stem Cell Transplantation, Steven and Alexandra Cohen Children's Medical Center of New York
Jeffrey M Lipton, MD, PhD is a member of the following medical societies: Alpha Omega Alpha, American Pediatric Society, Children's Oncology Group, American Society of Hematology, American Society of Pediatric Hematology/Oncology, Society for Pediatric Research
Disclosure: Nothing to disclose.
Coauthor(s)
Blanche P Alter, MD, MPH, FAAP Senior Clinician, Clinical Genetics Branch, Division of Cancer Epidemiology and Genetics, National Cancer Institute; Adjunct Faculty, Medical Genetics Fellowship Program, National Human Genome Research Institute; Visiting Professor of Pediatrics, part time, Johns Hopkins School of Medicine; Adjunct Professor of Pediatrics, George Washington University School of Medicine and Health Sciences
Blanche P Alter, MD, MPH, FAAP is a member of the following medical societies: Alpha Omega Alpha, American Pediatric Society, American Society for Clinical Investigation, American Society of Hematology, Society for Pediatric Research
Disclosure: Nothing to disclose.
Specialty Editor Board
Mary L Windle, PharmD Adjunct Associate Professor, University of Nebraska Medical Center College of Pharmacy; Editor-in-Chief, Medscape Drug Reference
Disclosure: Nothing to disclose.
Chief Editor
Emmanuel C Besa, MD Professor Emeritus, Department of Medicine, Division of Hematologic Malignancies and Hematopoietic Stem Cell Transplantation, Kimmel Cancer Center, Jefferson Medical College of Thomas Jefferson University
Emmanuel C Besa, MD is a member of the following medical societies: American Association for Cancer Education, American Society of Clinical Oncology, American College of Clinical Pharmacology, American Federation for Medical Research, American Society of Hematology, New York Academy of Sciences
Disclosure: Nothing to disclose.
Additional Contributors
Acknowledgements
The authors acknowledge the support and encouragement of their patients, their families, and referring physicians. This research was supported (in part) by the Intramural Research Program of the NIH and the National Cancer Institute.