GitHub - OpenGene/fastplong: Ultra-fast preprocessing and quality control for long-read sequencing data (original) (raw)
fastplong
Ultrafast preprocessing and quality control for long reads (Nanopore, PacBio, Cyclone, etc.).
If you're searching for tools to preprocess short reads (Illumina, MGI, etc.), please use fastp
fastplong supports batch processing of multiple FASTQ files in a folder, see - batch processing
- simple usage
- examples of report
- get fastplong
- input and output
- fastplong workflow
- filtering
- adapters
- per read cutting by quality score
- break read to subreads by discarding low quality regions
- mask low quality regions with N
- global trimming
- output splitting
- batch processing
- all options
simple usage
fastplong -i in.fq -o out.fq
Both input and output can be gzip compressed. By default, the HTML report is saved to fastplong.html (can be specified with -h option), and the JSON report is saved to fastplong.json (can be specified with -j option).
examples of report
fastplong creates reports in both HTML and JSON format.
- HTML report: http://opengene.org/fastplong/fastplong.html
- JSON report: http://opengene.org/fastplong/fastplong.json
get fastplong
install with Bioconda
conda install -c bioconda fastplong
download the latest prebuilt binary for Linux users
This binary was compiled on CentOS, and tested on CentOS/Ubuntu
download the latest build
wget http://opengene.org/fastplong/fastplong chmod a+x ./fastplong
or download specified version, i.e. fastplong v0.2.2
wget http://opengene.org/fastplong/fastplong.0.2.2 mv fastplong.0.2.2 fastplong chmod a+x ./fastplong
or compile from source
fastplong depends on libdeflate and isa-l for fast decompression and compression of zipped data, and depends on libhwy for SIMD acceleration. It's recommended to install all of them via Anaconda:
conda install conda-forge::libdeflate
conda install conda-forge::isa-l
conda install conda-forge::libhwy
You can also try to install them with other package management systems like apt/yum on Linux, or brew on MacOS. Otherwise you can compile them from source (https://github.com/intel/isa-l, https://github.com/ebiggers/libdeflate, and https://github.com/google/highway)
download and build fastplong
get source (you can also use browser to download from master or releases)
git clone https://github.com/OpenGene/fastplong.git
build
cd fastplong make -j
test
make test
Install
sudo make install
input and output
Specify input by -i or --in, and specify output by -o or --out.
- if you don't specify the output file names, no output files will be written, but the QC will still be done for both data before and after filtering.
- the output will be gzip-compressed if its file name ends with
.gz
output to STDOUT
fastplong supports streaming the passing-filter reads to STDOUT, so that it can be passed to other compressors like bzip2, or be passed to aligners like minimap2 or bowtie2.
- specify
--stdoutto enable this mode to stream output to STDOUT
input from STDIN
- specify
--stdinif you want to read the STDIN for processing.
store the reads that fail the filters
- give
--failed_outto specify the file name to store the failed reads. - if one read failed and is written to
--failed_out, itsfailure reasonwill be appended to its read name. For example,failed_quality_filter,failed_too_shortetc.
process only part of the data
If you don't want to process all the data, you can specify --reads_to_process to limit the reads to be processed. This is useful if you want to have a fast preview of the data quality, or you want to create a subset of the filtered data.
do not overwrite exiting files
You can enable the option --dont_overwrite to protect the existing files not to be overwritten by fastplong. In this case, fastplong will report an error and quit if it finds any of the output files (read, json report, html report) already exists before.
split the output to multiple files for parallel processing
See output splitting
fastplong workflow
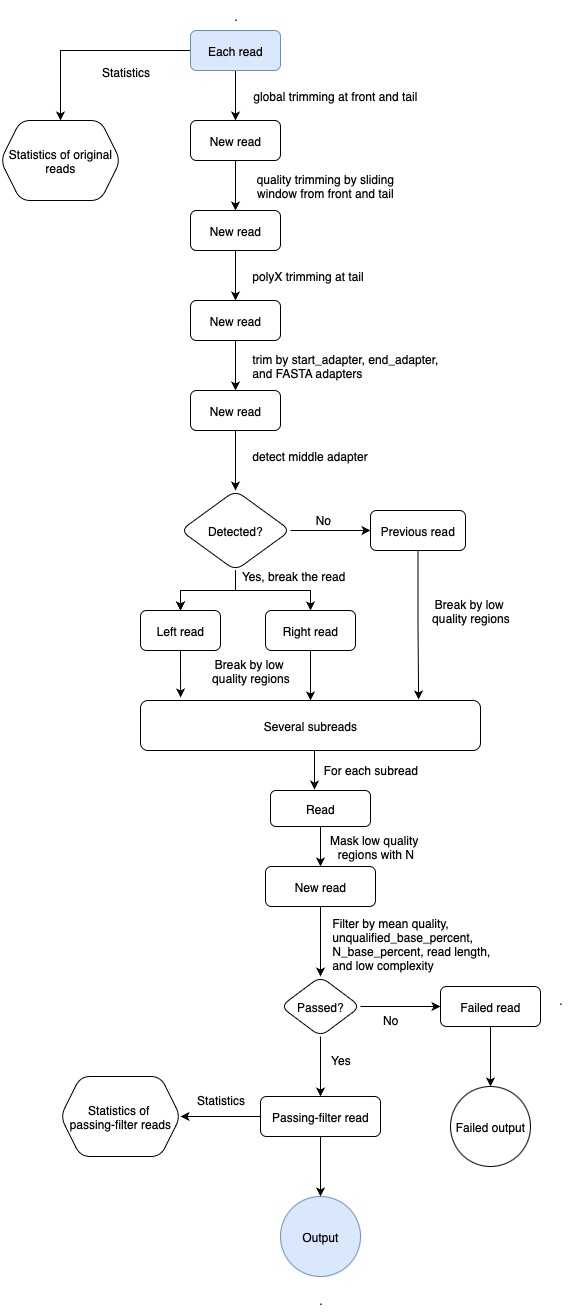
filtering
Multiple filters have been implemented.
quality filter
Quality filtering is enabled by default, but you can disable it by -Q or disable_quality_filtering.
fastplong supports filtering by limiting the N base number (--n_base_limit, disabled by default) and N base percentage (-n, --n_percent_limit, enabled by default). For example, to limit the N base no more than 100, and no more than 20%, you can use the command:
fastplong -i in.fq -o out.fq --n_base_limit 100 --n_percent_limit 20
To filter reads by its percentage of unqualified bases, two options should be provided:
-q, --qualified_quality_phredthe quality value that a base is qualified. Default 15 means phred quality >=Q15 is qualified.-u, --unqualified_percent_limithow many percents of bases are allowed to be unqualified (0~100). Default 40 means 40%
You can also filter reads by its average quality score
-m, --mean_qualif one read's average quality score <avg_qual, then this read is discarded. Default 0 means no requirement (int [=0])
length filter
Length filtering is enabled by default, but you can disable it by -L or --disable_length_filtering. The minimum length requirement is specified with -l or --length_required.
You can specify --length_limit to discard the reads longer than length_limit. The default value 0 means no limitation.
Other filter
New filters are being implemented. If you have a new idea or new request, please file an issue.
adapters
fastplong trims adapter in both read start and read end. Adapter trimming is enabled by default, but you can disable it by -A or --disable_adapter_trimming.
fastplong -i in.fq -o out.fq -s AAGGATTCATTCCCACGGTAACAC -e GTGTTACCGTGGGAATGAATCCTT
- If the adapter sequences are known, it's recommended to specify
-s, --start_adapterfor read start adapter sequence, and-e, --end_adapterfor read end adapter sequence as well. - If
--end_adapteris not specified but--start_adapteris specified, then fastplong will use the reverse complement sequence ofstart_adapterto beend_adapter. - You can also specify
-a, --adapter_fastato give a FASTA file to tellfastplongto trim multiple adapters in this FASTA file. Here is a sample of such adapter FASTA file:
>Adapter 1
AGATCGGAAGAGCACACGTCTGAACTCCAGTCA
>Adapter 2
AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGT
>polyA
AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA
- The adapter sequence in the FASTA file should be at least 6bp long, otherwise it will be skipped. And you can give whatever you want to trim, rather than regular sequencing adapters (i.e. polyA).
- If all these adapter options (
start_adapter,end_adapterandadapter_fasta) are not specified,fastplongwill try to detect the read start and read end adapters automatically. The detected adapter sequences may be a bit shorter or longer than the real ones. And there is a certain probability of misidentification, especially when most reads don't have adapters (it won't cause too bad result in this case). - fastplong calculates edit distance when detecting adapters. You can specify the
-d, --distance_thresholdto adjust the mismatch tolerance of adapter comparing. The default value is 0.25, which means allowing 25% mismatch ratio (i.e. allow 10 distance for 40bp adapter). Suggest to increase this value when the data is much noisy (high error rate), and decrease this value when the data is with high quality (low error rate). - to make a cleaner trimming, fastplong will trim a little more bases connected to the adapters. This option can be specified by
--trimming_extension, with a default value of 10.
per read cutting by quality score
fastplong supports per read sliding window cutting by evaluating the mean quality scores in the sliding window. fastplong supports 2 different operations, and you enable one or both:
-5, --cut_frontmove a sliding window from front (5') to tail, drop the bases in the window if its mean quality is below cut_mean_quality, stop otherwise. Default is disabled. The leading N bases are also trimmed. Usecut_front_window_sizeto set the widnow size, andcut_front_mean_qualityto set the mean quality threshold. If the window size is 1, this is similar as the TrimmomaticLEADINGmethod.-3, --cut_tailmove a sliding window from tail (3') to front, drop the bases in the window if its mean quality is below cut_mean_quality, stop otherwise. Default is disabled. The trailing N bases are also trimmed. Usecut_tail_window_sizeto set the widnow size, andcut_tail_mean_qualityto set the mean quality threshold. If the window size is 1, this is similar as the TrimmomaticTRAILINGmethod.
break read to subreads by discarding low quality regions
fastplong can detect low quality regions by moving a sliding window through the read. Specify -b or --break to enable this feature. You can adjust the sliding window size by --break_window_size, and adjust the mean quality requirement by break_mean_quality.
The subreads will have names like @r1-original-name, @r2-original-name..., and each subread will be quality checked and filtered separately.
WARNING: This may result in significant data loss. If you want to keep more data, please lower the mean quality requirement or improve the sliding window size.
mask low quality regions with N
fastplong can detect low quality regions and replace the bases in these regions with N base. Specify -N or --mask to enable this feature. You can adjust the sliding window size by --mask_window_size, and adjust the mean quality requirement by mask_mean_quality.
WARNING: This may cause many reads failed to pass the N base limit filter. If you want to keep more data, you can lower the mean quality requirement, improve the sliding window size or adjust the N base percent limit --n_percent_limit.
It's not suggested to enable both --break and --mask together. But if they are enabled together, fastplong will break the reads first, and then mask the subreads.
global trimming
fastplong supports global trimming, which means trim all reads in the front or the tail. This function is useful since sometimes you want to drop some cycles of a sequencing run.
For example, the last cycle is uaually with low quality, and it can be dropped with -t 1 or --trim_tail=1 option.
- The front/tail trimming settings are given with
-f, --trim_frontand-t, --trim_tail.
output splitting
For parallel processing of FASTQ files (i.e. alignment in parallel), fastplong supports splitting the output into multiple files. The splitting can work with two different modes: by limiting file number or by limiting lines of each file. These two modes cannot be enabled together.
The file names of these split files will have a sequential number prefix, adding to the original file name specified by --out1 or --out2, and the width of the prefix is controlled by the --split_prefix_digits option. For example, --split_prefix_digits=4, --out1=out.fq, --split=3, then the output files will be 0001.out.fq,0002.out.fq,0003.out.fq
splitting by limiting file number
Specify --split to specify how many files you want to have. fastplong evaluates the read number of a FASTQ by reading its first ~1M reads. This evaluation is not accurate so the file sizes of the last several files can be a little differnt (a bit bigger or smaller). For best performance, it is suggested to specify the file number to be a multiple of the thread number.
splitting by limiting the lines of each file
Specify --split_by_lines to limit the lines of each file. The last files may have smaller sizes since usually the input file cannot be perfectly divided. The actual file lines may be a little greater than the value specified by --split_by_lines since fastplong reads and writes data by blocks (a block = 1000 reads).
batch processing
parallel.py is a script to preprocess all FASTQ files within a folder in parallel. It will automatically couple the paired-end FASTQ files.
This script will generate an overall.html to present an aggregate summary for all processed FASTQ files.
example
python parallel.py -i /path/to/input/folder -o /path/to/output/folder -r /path/to/reports/folder -a '--cut_front --cut_tail'
which means to
. process all the FASTQ data in /path/to/input/folder
. using fastplong in PATH
. the arguments --cut_front and --cut_tail will be passed to fastplong, to apply sliding window quality trimming from front and tail
. output all clean data to /path/to/output/folder
. output all HTML and JSON reports to /path/to/reports/folder
See python parallel.py -h for details.
all options
usage: fastplong -i -o [options...]
fastplong: ultra-fast FASTQ preprocessing and quality control for long reads
usage: ./fastplong [options] ...
options:
-i, --in read input file name (string [=])
-o, --out read output file name (string [=])
--failed_out specify the file to store reads that cannot pass the filters. (string [=])
-z, --compression compression level for gzip output (1 ~ 9). 1 is fastest, 9 is smallest, default is 4. (int [=4])
--stdin input from STDIN.
--stdout stream passing-filters reads to STDOUT. This option will result in interleaved FASTQ output for paired-end output. Disabled by default.
--reads_to_process specify how many reads/pairs to be processed. Default 0 means process all reads. (int [=0])
--dont_overwrite don't overwrite existing files. Overwritting is allowed by default.
-V, --verbose output verbose log information (i.e. when every 1M reads are processed).
-A, --disable_adapter_trimming adapter trimming is enabled by default. If this option is specified, adapter trimming is disabled
-s, --start_adapter the adapter sequence at read start (5'). (string [=auto])
-e, --end_adapter the adapter sequence at read end (3'). (string [=auto])
-a, --adapter_fasta specify a FASTA file to trim both read by all the sequences in this FASTA file (string [=])
-d, --distance_threshold threshold of sequence-adapter-distance/adapter-length (0.0 ~ 1.0), greater value means more adapters detected (double [=0.25])
--trimming_extension when an adapter is detected, extend the trimming to make cleaner trimming, default 10 means trimming 10 bases more (int [=10])
-f, --trim_front trimming how many bases in front for read, default is 0 (int [=0])
-t, --trim_tail trimming how many bases in tail for read, default is 0 (int [=0])
-x, --trim_poly_x enable polyX trimming in 3' ends.
--poly_x_min_len the minimum length to detect polyX in the read tail. 10 by default. (int [=10])
-5, --cut_front move a sliding window from front (5') to tail, drop the bases in the window if its mean quality < threshold, stop otherwise.
-3, --cut_tail move a sliding window from tail (3') to front, drop the bases in the window if its mean quality < threshold, stop otherwise.
-W, --cut_window_size the window size option shared by cut_front, cut_tail or cut_sliding. Range: 11000, default: 4 (int [=4])
-M, --cut_mean_quality the mean quality requirement option shared by cut_front, cut_tail or cut_sliding. Range: 136 default: 20 (Q20) (int [=20])
--cut_front_window_size the window size option of cut_front, default to cut_window_size if not specified (int [=4])
--cut_front_mean_quality the mean quality requirement option for cut_front, default to cut_mean_quality if not specified (int [=20])
--cut_tail_window_size the window size option of cut_tail, default to cut_window_size if not specified (int [=4])
--cut_tail_mean_quality the mean quality requirement option for cut_tail, default to cut_mean_quality if not specified (int [=20])
-N, --mask mask the low quality regions with N, these regions are detected by sliding window with mean quality < mask_mean_quality.
--mask_window_size the size of the sliding window to evaluate the mean quality for N masking(51000000), default: 50 (int [=50])
--mask_mean_quality the mean quality requirement for sliding window N masking (530), default: 10 (Q10) (int [=10])
-b, --break break the reads by discarding the low quality regions, these regions are detected by sliding window with mean quality < break_mean_quality.
--break_window_size the size of the sliding window to evaluate the mean quality for sliding window breaking(51000000), default: 100 (int [=100])
--break_mean_quality the mean quality requirement for sliding window breaking (530), default: 10 (Q10) (int [=10])
-Q, --disable_quality_filtering quality filtering is enabled by default. If this option is specified, quality filtering is disabled
-q, --qualified_quality_phred the quality value that a base is qualified. Default 15 means phred quality >=Q15 is qualified. (int [=15])
-u, --unqualified_percent_limit how many percents of bases are allowed to be unqualified (0100). Default 40 means 40% (int [=40])
--n_base_limit if number of N base is >n_base_limit, then this read is discarded (01000000). 0 means no N allowed, default 1000000 means no N limit (int [=1000000])
-n, --n_percent_limit if one read's N base percentage is >n_percent_limit, then this read is discarded (0100). Default 10 means 10% (int [=10])
-m, --mean_qual if one read's mean_qual quality score <mean_qual, then this read is discarded. Default 0 means no requirement (int [=0])
-L, --disable_length_filtering length filtering is enabled by default. If this option is specified, length filtering is disabled
-l, --length_required reads shorter than length_required will be discarded, default is 20. (int [=20])
--length_limit reads longer than length_limit will be discarded, default 0 means no limitation. (int [=0])
-y, --low_complexity_filter enable low complexity filter. The complexity is defined as the percentage of base that is different from its next base (base[i] != base[i+1]).
-Y, --complexity_threshold the threshold for low complexity filter (0100). Default is 30, which means 30% complexity is required. (int [=30])
-j, --json the json format report file name (string [=fastplong.json])
-h, --html the html format report file name (string [=fastplong.html])
-R, --report_title should be quoted with ' or ", default is "fastplong report" (string [=fastplong report])
-w, --thread worker thread number, default is 3 (int [=3])
--split split output by limiting total split file number with this option (2999), a sequential number prefix will be added to output name ( 0001.out.fq, 0002.out.fq...), disabled by default (int [=0])
--split_by_lines split output by limiting lines of each file with this option(>=1000), a sequential number prefix will be added to output name ( 0001.out.fq, 0002.out.fq...), disabled by default (long [=0])
--split_prefix_digits the digits for the sequential number padding (110), default is 4, so the filename will be padded as 0001.xxx, 0 to disable padding (int [=4])
-?, --help print this message