Ebola Example (original) (raw)
Ebola: a super-spreading SEIR model
In this example we define a SEIR super-spreading model for Ebola and apply it to phylogenies estimated by Dudas et.al. from whole genome sequences collected during the 2014-2015 Western African epidemic.
A typical SEIR model has the following form:
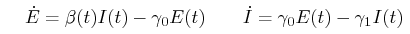
where γ₀ is the rate at which infected hosts progress from an uninfectious exposed state (E) to an infectious state (I), and γ₁ is the rate at which infectious hosts die or recover. In a typical mass-action model, the per-capita transmission rate β(t) is a function directly proportional to S/(S+E+I+R). Instead we:
- define β(t) as a linear function at+b, mainly to demonstrate the flexibility of the framework and package.
- model super-spreading by introducing two infectious compartments Il (low risk) and Ih (high risk), with per-capita transmission rates β(t) and τβ(t) respectively, and transmission risk ratio τ > 1.
The (Ebola) SEIR super-spreading model has the following form:
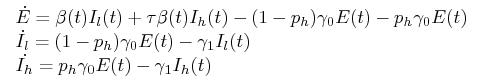
where ph specifies the fraction of the exposed/infectious flow that goes (from E) to Ih, with the remainder going to Il. Note that the parametric description of β(t) allows us to dispense with the specifications of S(t) and R(t).
The corresponding death (μ), birth (F) and migration (G) matrices are:
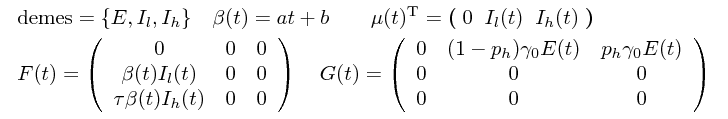
The population model above can be easily specified as a PopModelODE PhyDyn/BEAST object:
beta = max(0.0, at + b) gamma0E*(1-p_h) gamma0Ep_h betaIl betaIhtau gamma1Il gamma1Ih gamma1(Il + Ih)
We apply the SEIR super-spreading model to one of the Ebola virus phylogenies estimated from data collected during the 2014-2015 epidemic in Western Africa (Dudas et.al.). The resulting XML Beast/PhyDyn analysis file can be found here. The PhyDyn population model presented above is completed with the following:
The analysis:
- assumes incubation and infectiousness periods of 11.4 and 5 days respectively, and fixes ph at 20%.
- estimates: the slope a and intercept b of β(t), the transmission risk ratio tau, and the initial number of infected E.
- uses a fixed tree. The tree was taken from one of the phylogenies estimated by Dudas and downsampled to 400 tips, with node annotations corresponding to time/dates in years.
- does not include Date or Type traits: deme information is extracted directly from the taxa names while dates are taken from the newick tree newick annotations.
- does not have a
t1parameter; this means that t1 is set to the height of the tree.