Tutorial SIR2 Trajectory (original) (raw)
A Structured SIR model
The population model specification is the most important input to a PhyDyn analysis. Before setting up the parameters for a MCMC BEAST2 analysis, it is recommended to define and test the population model Ordinary Differential Equations (ODEs) beforehand in order to identify conceptual and specification errors eg. errors related to PhyDyn's syntax.
The files used in this tutorial can be found in the examples/SIR2 directory of the PhyDyn repository.
The Model
As running example, let’s consider a two-deme SIR model where S denotes the number of hosts that are susceptible, and I0 and I1 denote early and chronic infection stages, respectively. Upon infection, hosts enter the state I0 of average duration 1/γ0 and transmit infection at a rate β0. Infected hosts in I0 progress to state I1 of average duration 1/γ1 with transmission rate β1. The natural birth rate for S is b. The equations that describe the system are as follows:
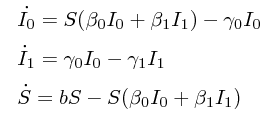
The birth (F), migration (G) and death matrices for this model are:
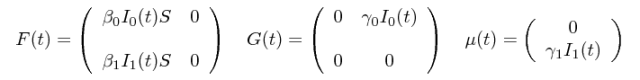
The rest of this tutorial shows how to specify our two-deme SIR model as BEAST2/PhyDyn XML files.
PopModelODE: From formulae to XML
In PhyDyn, population models are specified using the PopModelODE class. A PopModelODE object is usually linked to a STreeLikelihoodODE object which can then be used by a full BEAST2 analysis as, for example, a tree prior. At this point of the analysis workflow, our main concern is to test our population model specification. The recommended way to do this and consequently, the recommended way to start any PhyDyn analysis, is to use the TrajectoryOut class.
BEAST2/PhyDyn take XML files as input. XML files for BEAST2 tree sampling analyses are quite complex and difficult to read and edit, even for an expert user. Fortunately, PhyDyn population models are relatively easy to specify. The instructions below show how to write the PhyDyn XML specification of our sample two-deme SIR model.
- Download the sir2TrajectoryTemplate.xml to your local directory. Make a copy and re-name it to sir2Trajectory.xml.
> cp sir2TrajectoryTemplate.xml sir2Trajectory.xml
The XML file contains the skeleton of a use-case of the TrajectoryOut class, tailored to our example and with place-holders for our model specification.
- Open sir2Trajectory.xml in a text editor. Replace
--insert-equations--with:
k0 = beta0S k1 = beta1S k0I0 beta1SI1 gamma0I0 0 gamma1I1 bS - (beta0I0+ beta1I1)*S
Note the one-to-one correspondence between the matrixeq XML entries and the matrix equations above, with equation types birth, migration and death matching entries in F, G and mu. Non-demes, such as S, use a special equation type: nondeme.
We have also added a couple of definitions, k0 and k1. Definitions are executed before the evaluation of the derivatives defined by the matrix equations. They are used to avoid code repetition and simplify the formulas. Above, they are included just as an example - they add no value to the specification.
The model is 'closed' by providing values to its free-variables: model parameters (usually rates), (non)deme initial values (at time t0), and the initial and final times of the trajectory (t0 and t1).
Replace
--insert-model-parameters--with:Replace
--insert-initial-values--with:
This (TrajectoryParameters) part of the specification defines the initial (t0) and final (t1) times of the population trajectory, as well as the method used to solve the ODE equations (classicrk, the default). Classic Runge-Kutta is a fixed step method; the parameter integrationSteps determines the number of (evenly-spaced) of points used in the time-series storing the solution (including the values of the F,G and death matrices).
The TrajectoryOut class takes as input a PopModelODE specification (with id twodemes) and, upon execution, creates a csv file (file=sir2.csv parameter) with the trajectory points generated by the solution of the population model ODE system.
- Run the trajectory two deme model with BEAST2:
> beast sir2Trajectory.xml
The execution prints back a text version of the population model. We can check that our model has been parsed correctly by comparing this output with the intended ODE expressions.
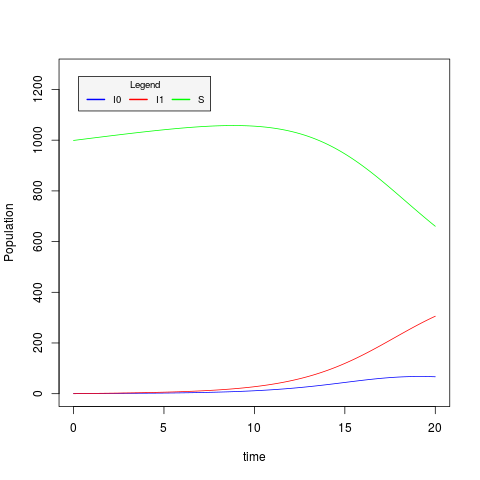
The plotSIR2.R script plots the values of S,I0 and I1 stored in the csv file generated by the previous command:
> Rscript plotSIR2.R sir2.csv
The resulting trajectory plot file is sir2.csv.png:
Simplifying the XML
Editing the PopModelODE XML object can get cumbersome for bigger examples. In response to this, the last versions PhyDyn provide a more succinct way of entering the population model matrix formulas into the XML. Recall the execution of the trajectory PhyDyn XML:
> beast sir2Trajectory.xml
Upon execution, PhyDyn prints a text version of the model to standard input. The first part of the output should looks like this:
model-name = twodeme;
definitions = {
k0 = beta0*S;
k1 = beta1*S;
}
equations = {
F(I0,I0) = k0*I0;
F(I1,I0) = beta1*S*I1;
G(I0,I1) = gamma0*I0;
D(I0) = 0;
D(I1) = gamma1*I1;
dot(S) = b*S-(beta0*I0+beta1*I1)*S;
}
The model formulas, definitions and equations, are entered as lists of semi-colon-separated assignments, where the right-hand-side correspond to the arithmetical expressions used in the original XML. In some cases, the printed expressions may contain extra parenthesis that should make make more explicit the parsing rules of PhyDyn's syntax.
The left-hand-side of the matrix equations correspond to:
- Entries to the birth
F(<source-deme>,<target-deme>), migrationG(<source-deme>,<target-deme>)andD(<deme>)matrices. - Non-deme derivatives
dot(<nondeme>)
The text output has been defined such that it can be parsed back into their corresponding matrix equations. The new XML format replaces the <definition> and <matrixeq> components with two XML elements: <definitions> and <matrixeqs>.
Copy your
k0 = beta0*S; k1 = beta1*S; F(I0,I0) = k0*I0; F(I1,I0) = k1*I1; G(I0,I1) = gamma0*I0; D(I0) = 0; D(I1) = gamma1*I1; dot(S) = b*S-(beta0*I0+beta1*I1)*S;sir2Trajectory.xmlfile tosir2TrajectoryFinal.xml. Open the new file in a text editor. Go to thespec=PopModelODEcomponent and replace all the<definition>and<matrixeq>entries with:Go the
finalparameter in theTrajectoryOutcomponent and replacesir2.csvwithsir2Final.csv. The resulting file should look like this.Save and run the file using BEAST2. The printed text version of the model should be the same as the one obtained from the previous run. The generated
sir2Final.csvshould be identical tosir2.csv.
Generate a new plot with the plotSIR2.R script and compare your results.