Getting Started (original) (raw)
Before You Start
- In the following example, we demonstrate key package functionality using the test data set that is included in the package
- You can follow along because all test data and associated CSV input files are contained in the package.
- Additional examples are also available under Example Vignettes
Components of the test dataset
- Paired-end short read amplicon data
- Files: S1_R1.fastq.gz, S1_R2.fastq.gz, S2_R1.fastq.gz, S2_R1.fastq.gz
- The files must be zipped and end in either R1.fastq.gz or_R2.fastq.gz_ and each sample must have both R1 and R2 files.
- New row for each unique sample
- Samples entered twice if samples contain two pooled metabarcodes, as in the test data template
- New row for each unique barcode and associated primer sequence
- Optional
Cutadaptand**DADA2** parameters
- Taxonomy databases
- UNITE fungal database (abridged version)
- oomyceteDB
Format of the paired-end read files
- The package takes forward and reverse Illumina short read sequence data.
- Format of file names To avoid errors, the only characters that are acceptable in sample names are letters and numbers. Characters can be separated by underscores, but no other symbols. The files must end with the suffix R1.fastq.gz orR2.fastq.gz.
Format of metadata file
- The format of the CSV file named metadata.csv is simple.
- A template is here.
- The only two required columns (with these headers) are:
- Column 1. sample_name
- Column 2. primer_info (applicable options: ‘rps10’, ‘its’, ‘r16S’, ‘other1’, other2’)
Additional columns
- Other columns should be pasted after these two columns.
- They can be referenced later during the analysis steps and save a step of loading metadata later.
Notes
- S1 and S2 are come from a rhododendron rhizobiome dataset and are a random subset of the full set of reads.
- S1 and S2 are included twice in the ‘metadata.csv’ sheet. This is because these two samples contain pooled metabarcodes (ITS1 and_rps10_).
- To demultiplex and run both analyses in tandem, include the same sample twice under sample_name, and then change the primer_name.
Here is what the metadata.csv looks like for this test dataset:
| sample_name | primer_name | organism |
|---|---|---|
| S1 | rps10 | Cry |
| S2 | rps10 | Cin |
| S1 | its | Cry |
| S2 | its | Cin |
Format of primer and parameters file
Primer sequence information and user-defined parameters are placed inprimerinfo_params.csv
- To simplify how functions are called, user will provide parameters within this input file.
- We recommend using the template linked here.
- Remember to add any additional optional**
DADA2** parameters you want to use. - The only three required columns (with these headers) are:
- Column 1. sample_name
- Column 2. forward (primer sequence)
- Column 3. reverse(primer sequence)
Example template for primerinfo_params.csv
| primer_name | forward | reverse | already_trimmed | minCutadaptlength | multithread | verbose | maxN | maxEE_forward | maxEE_reverse | truncLen_forward | truncLen_reverse | truncQ | minLen | maxLen | minQ | trimLeft | trimRight | rm.lowcomplex | minOverlap | maxMismatch | min_asv_length |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| rps10 | GTTGGTTAGAGYARAAGACT | ATRYYTAGAAAGAYTYGAACT | FALSE | 100 | TRUE | FALSE | 1.00E+05 | 5 | 5 | 0 | 0 | 5 | 150 | Inf | 0 | 0 | 0 | 0 | 15 | 0 | 50 |
| its | CTTGGTCATTTAGAGGAAGTAA | GCTGCGTTCTTCATCGATGC | FALSE | 50 | TRUE | FALSE | 1.00E+05 | 5 | 5 | 0 | 0 | 5 | 50 | Inf | 0 | 0 | 0 | 0 | 15 | 0 | 50 |
For more info on parameter specifics, see Documentation
Reference Database Format
- For now, the package is compatible with the following databases:
- oomycetedb from: https://grunwaldlab.github.io/OomyceteDB/
- SILVA 16S database with species assignments: https://www.arb-silva.de/
- UNITE fungal database from https://unite.ut.ee/repository.php
- A user can select up to two other databases, but will first need to reformat headers exactly like the SILVA database headers.
* See more in the ‘Documentation’ tab.
Databases are downloaded from these sources and then placed in the from the user-specified data folder where raw data files and csv files are located.
Additional Notes
- Computer specifications may be a limiting factor.
- If you are using the SILVA or UNITE databases for taxonomic assignment steps, an ordinary personal computer (unless it has sufficient RAM) may not have enough memory for the taxonomic assignment steps, even with few samples.
- The test databases and reads are subsetted and the following example should run on a personal computer with at least 16 GB of RAM.
- If computer crashes during the taxonomic assignment step, you will need to switch to a computer with sufficient memory.
- You must ensure that you have enough storage to save intermediate files in a temporary directory (default) or user-specified directory before proceeding.
Installation
Dependencies:
First install Cutadapt program following the instructions here: https://cutadapt.readthedocs.io/en/stable/installation.html
Let’s locate where the **Cutadapt**executable is.
You must do this from a Terminal window:
#If you installed with pip or pipx, or homebrew, run this command from a Terminal window
which cutadapt
cutadapt --versionIf you followed the cutadapt installation instructions to create a conda environment called Cutadapt (change to whatever you named your environment), to install it in.
Open up a Terminal window and type these commands:
#Run commands from a Terminal window
conda activate cutadapt
which cutadapt
cutadapt --versionSecond, make sure the following R packages are installed:
demulticoder(Available through CRAN)DADA2(Latest version is 3.20)- To install, follow these instructions: https://www.bioconductor.org/packages/release/bioc/html/dada2.html
phyloseqmetacoder(Available through CRAN)
Loading packages
Reorganize data tables and set-up data directory structure
The sample names, primer sequences, and other metadata are reorganized in preparation for running**Cutadapt** to remove primers.
output<-prepare_reads(
data_directory = system.file("extdata", package = "demulticoder"),
output_directory = tempdir(), # Change to your desired output directory path or leave as is",
overwrite_existing = TRUE)
#> Existing files found in the output directory. Overwriting existing files.
#> Rows: 2 Columns: 25
#> ── Column specification ────────────────────────────────────────────────────────
#> Delimiter: ","
#> chr (3): primer_name, forward, reverse
#> dbl (18): minCutadaptlength, maxN, maxEE_forward, maxEE_reverse, truncLen_fo...
#> lgl (4): already_trimmed, count_all_samples, multithread, verbose
#>
#> ℹ Use `spec()` to retrieve the full column specification for this data.
#> ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
#> Rows: 2 Columns: 25
#> ── Column specification ────────────────────────────────────────────────────────
#> Delimiter: ","
#> chr (3): primer_name, forward, reverse
#> dbl (18): minCutadaptlength, maxN, maxEE_forward, maxEE_reverse, truncLen_fo...
#> lgl (4): already_trimmed, count_all_samples, multithread, verbose
#>
#> ℹ Use `spec()` to retrieve the full column specification for this data.
#> ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
#> Rows: 4 Columns: 3
#> ── Column specification ────────────────────────────────────────────────────────
#> Delimiter: ","
#> chr (3): sample_name, primer_name, organism
#>
#> ℹ Use `spec()` to retrieve the full column specification for this data.
#> ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
#> Creating output directory: /tmp/RtmpUTBhYG/demulticoder_run/prefiltered_sequences
Remove primers with Cutadapt
Before running Cutadapt, please ensure that you have installed it
cut_trim(
output,
cutadapt_path = "/usr/bin/cutadapt", #CHANGE LOCATION TO YOUR LOCAL INSTALLATION
overwrite_existing = TRUE)
#> Running cutadapt 3.5 for its sequence data
#> Running cutadapt 3.5 for rps10 sequence data
ASV inference step
Raw reads will be merged and ASVs will be inferred
make_asv_abund_matrix(
output,
overwrite_existing = TRUE)
#> 80608 total bases in 307 reads from 2 samples will be used for learning the error rates.
#> Sample 1 - 163 reads in 84 unique sequences.
#> Sample 2 - 144 reads in 96 unique sequences.
#> 82114 total bases in 307 reads from 2 samples will be used for learning the error rates.
#> Sample 1 - 163 reads in 128 unique sequences.
#> Sample 2 - 144 reads in 119 unique sequences.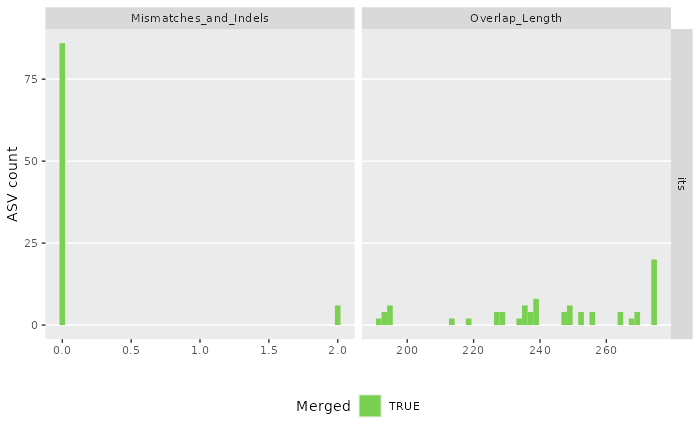
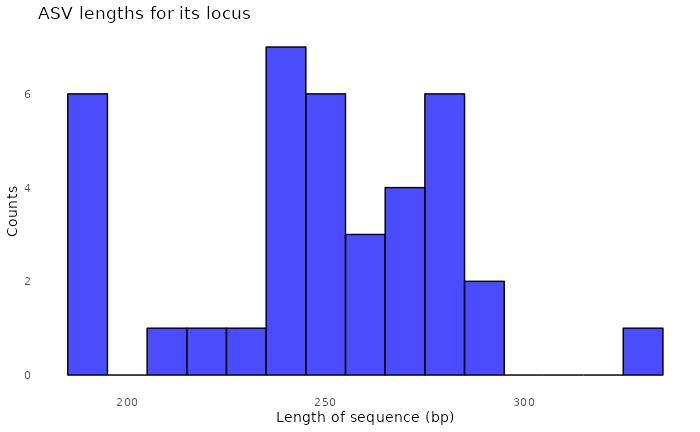
#> 91897 total bases in 327 reads from 2 samples will be used for learning the error rates.
#> Sample 1 - 145 reads in 107 unique sequences.
#> Sample 2 - 182 reads in 133 unique sequences.
#> 91567 total bases in 327 reads from 2 samples will be used for learning the error rates.
#> Sample 1 - 145 reads in 114 unique sequences.
#> Sample 2 - 182 reads in 170 unique sequences.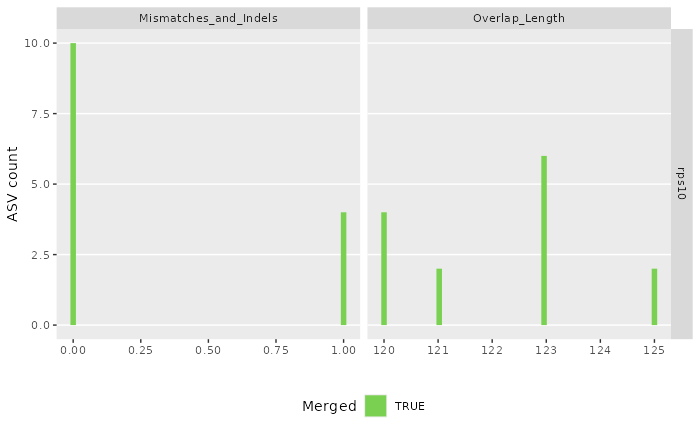
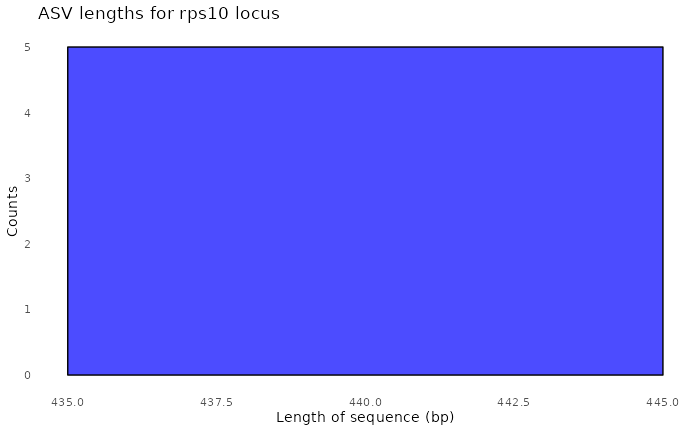
#> $its
#> [1] "/tmp/RtmpUTBhYG/demulticoder_run/asvabund_matrixDADA2_its.RData"
#>
#> $rps10
#> [1] "/tmp/RtmpUTBhYG/demulticoder_run/asvabund_matrixDADA2_rps10.RData"Taxonomic assignment step
Using the core assignTaxonomy function from**DADA2**, taxonomic assignments will be given to ASVs.
assign_tax(
output,
asv_abund_matrix,
retrieve_files=TRUE,
overwrite_existing = TRUE)
#> samplename_barcode input filtered denoisedF denoisedR merged nonchim
#> 1 S1_its 299 163 146 141 132 132
#> 2 S2_its 235 144 113 99 99 99
#> samplename_barcode input filtered denoisedF denoisedR merged nonchim
#> 1 S1_rps10 196 145 145 145 145 145
#> 2 S2_rps10 253 182 181 181 181 181Format of output matrices
- The default output is a CSV file per metabarcode with the inferred ASVs and sequences, taxonomic assignments, and bootstrap supports provided by
DADA2. These data can then be used as input for downstream steps. - To view these files, locate the files with the prefixfinal_asv_abundance_matrix in your specified output directory.
- In this example, it will be in the home directory withindemulticoder_test.
- You can then open your matrices using Excel
- We recommend checking the column with the ASV sequences to make sure there aren’t truncated sequences *It is important to also review the column showing the taxonomic assignments of each ASV and their associated bootstrap supports.
We can also load the matrices into our R environment and view then from R Studio.
We do that here with the rps10 matrix
rps10_matrix<-read.csv("~/demulticoder_test/final_asv_abundance_matrix_rps10.csv", header=TRUE)
head(rps10_matrix)
#> asv_id
#> 1 asv_1
#> 2 asv_2
#> 3 asv_3
#> 4 asv_4
#> 5 asv_5
#> sequence
#> 1 GAAAATCTTTGTGTCGGTGGTTCAAATCCACCTCCAGACAAATTAATTTTTTAAAACTTATGTTTATATTAAGAATTACATTTAAATCTATTAAAAAAATAAATAACATTAAACCTATTTTATTAAATCAAAAAAATTTAAATAAATTTAATAATATTAAAATAAATGGAATATTTAAAACAAAAAATAAAAATAAAATTTTTACAGTATTAAAATCACCACATGTTAATAAAAAAGCACGTGAACATTTTATTTATAAAAATTTTACACAAAAAGTTGAAATTAAATGTTTAAATATTTTTCAATTATTAAATTTTTTAATTTTGATTAAAAAAATCTTAACAGAAAATTTTATTATTACTACAAAAATAATTAAACAATAATTAATATATGCTTATAGCTTAATGGATAAAGCATTAGATTGCGGATCTACAAAATGAA
#> 2 GAAAATCTTTGTGTCGGTGGTTCAAGTCCGCCTCCAAACATCTTATAAAAATGTATGTATATTTTACGAATTAGTTTTAAATCAGTAGAAAAAATAAATGAATTAAAAAAAAATATCTCAAAAATAAAAAAAATATATAATTTTAAAAATTTAAAAGTAAATGGTATTTTTAGAACAAAAAATAAAAATAAAATTTTTACATTATTAAAATCACCCCATGTTAATAAAAAATCACGTGAACATTTTATATATAAAAATTATATTCAAAAAATTGATTTAAATTTTTCAAATTTTTTTCAATTATTAAATTTTTTAGTAATTTTAAAAAAAATATTATCTAAAGATTTTTTAATCAATATCAAAATTATTAAAAAAAATAAAAAAAATATGCTTATAGCTTAATGGATAAAGCGTTAGATTGCGGATCTATAAAATGAA
#> 3 GAAAATCTTTGTGTCGGTGGTTCAAGTCCACCTCCAGACAAAAATAATAACTATGTATATTTTAAGAATAACTTTTAAATCAATACAAAAAATAAATAATATTAAAAAAAATTTATTAAAATTAAAAAAAATAAATAAATTTAAAAATATTCAAATAAAAGGAATATTTAAAATAAAAGATAAAAATAAAATTTTTACTTTATTAAAATCACCTCACGTAAATAAAAAATCTCGTGAACATTTTATTTATAAAAATTATACTCAAAAAATAGATGTAAAATTTTCAAATATTATTGAATTATTTAATTTTATAATACTTATTAAAAAAGTTTTAACTAAAAATTTTATAATAAATTTTAAAATTATAAAATATAATAAAAAAAAAATGCTTATAGCTTAATGGATAAAGCGTTAGATTGCGGATCTATAAAATGAA
#> 4 GAAAATCTTTGTGTCGGTGGTTCAAATCCGCCTCCAAACAAAAATATAAATTATTTTATATGTATATTTTAAGAATAAGTTTTAAATCTGTATCAAAAATAGATAAAATTAAAAAAGAATTATCAAGATTAAAAAAAATATATAATTTTAAAAATATAACAATTAATGGTATTTTTAAAACAAAAAATAAAGATAAAATTTTTACATTATTAAAATCACCTCATGTTAATAAAAAATCTCGCGAACATTTTATTTATAAAAATTATGTACAAAAAATAGATTTAAATTTTATTAATATTTTTCAATTAATAAATTTTTTAATAATTTTAAAAAAAAAATTATCAAAAAATGTATTAATAAATGTAAAAATAATTAAAAAATAAAAAAATATGCTTATAGCTTAATGGATAAAGCGTTAGATTGCGGATCTATAAAATGAA
#> 5 GAAAATCTTTGTGTCGGTGGTTCAAGTCCGCCTCCAAACATCTTATAAAAATGTATGTATATTTTACGAATTAGTTTTAAATCAGTAGAAAAAATAAATGAATTAAAAAAAAATATCTCAAAAATAAAAAAAATATATAATTTTAAAAATTTAAAAGTAAAATGGTATTTTTAGAACAAAAAATAAAAATAAAATTTTTACATTATTAAAATCACCCCATGTTAATAAAAAATCACGTGAACATTTTATATATAAAAATTATATTCAAAAAATTGATTTAAATTTTTCAAATTTTTTTCAATTATTAAATTTTTTAGTAATTTTAAAAAAAACATTATCTAAAGATTTTTTAATCAATATTAAAATTATTAAAAAAAATAAAAAAATATGCTTATAGCTTAATGGATAAAGCGTTAGATTGCGGATCTATAAAATGAA
#> dada2_tax
#> 1 Stramenopila--100--Kingdom;Oomycota--100--Phylum;Oomycetes--100--Class;Peronosporales--26--Order;Peronosporaceae--26--Family;Phytophthora--23--Genus;Phytophthora_sp.kununurra--13--Species;oomycetedb_650--13--Reference
#> 2 Stramenopila--100--Kingdom;Oomycota--100--Phylum;Oomycetes--100--Class;Pythiales--100--Order;Pythiaceae--100--Family;Pythium--100--Genus;Pythium_dissotocum--49--Species;oomycetedb_749--24--Reference
#> 3 Stramenopila--100--Kingdom;Oomycota--100--Phylum;Oomycetes--100--Class;Pythiales--100--Order;Pythiaceae--100--Family;Globisporangium--100--Genus;Globisporangium_cylindrosporum--22--Species;oomycetedb_41--22--Reference
#> 4 Stramenopila--100--Kingdom;Oomycota--100--Phylum;Oomycetes--100--Class;Pythiales--94--Order;Pythiaceae--94--Family;Pythium--89--Genus;Pythium_aphanidermatum--19--Species;oomycetedb_727--19--Reference
#> 5 Stramenopila--100--Kingdom;Oomycota--100--Phylum;Oomycetes--100--Class;Pythiales--100--Order;Pythiaceae--100--Family;Pythium--100--Genus;Pythium_oopapillum--67--Species;oomycetedb_771--67--Reference
#> S1_rps10 S2_rps10
#> 1 842 17
#> 2 578 220
#> 3 0 587
#> 4 0 516
#> 5 0 163We can also inspect the first rows of the ITS1 matrix
its_matrix<-read.csv("~/demulticoder_test/final_asv_abundance_matrix_its.csv", header=TRUE)
head(its_matrix)
#> asv_id
#> 1 asv_1
#> 2 asv_2
#> 3 asv_3
#> 4 asv_4
#> 5 asv_5
#> 6 asv_6
#> sequence
#> 1 AAAAAGTCGTAACAAGGTTTCCGTAGGTGAACCTGCGGAAGGATCATTAGTGAATCTTCAAAGTCGGCTCGTCAGATTGTGCTGGTGGAGACACATGTGCACGTCTACGAGTCGCAAACCCACACACCTGTGCATCTATGACTCTGAGTGCCGCTTTGCATGGCCCCTTGATTTGGGCCTGGCGCTCGAGTACTTTCACACACTCTCGAATGTAATGGAATGTCTTCTTGTGCATAACGTACAAACAGAAACAACTTTCAACAACGGATCTCTTGGCTCTC
#> 2 AAAAAGTCGTAACAAGGTTTCCGTAGGTGAACCTGCGGAAGGATCATTAGTGAATCTTCAAAGTCGGCTCGTCGGATTGTGCTGGTGGAGACACATGTGCACGTCTACGAGTCGCAAACCCACACACCTGTGCATCTATGACTCTGAGTGCCGCTTTGCATGGCCCCTTGATTTGGGCCTGGCGCTCGAGTACTTTCACACACTCTCGAATGTAATGGAATGTCTTGTTGTGCATAACGTACAAACAGAAACAACTTTCAACAACGGATCTCTTGGCTCTC
#> 3 AAAAAGTCGTAACAAGGTTTCCGTAGGTGAACCTGCGGAAGGATCATTAGTGAATCTTCAAAGTCGGCTCGTCGGATTGTGCTGGTGGAGACACATGTGCACGTCTACGAGTCGCAAACCCACACACCTGTGCATCTATGACTCTGAGTGCCGCTTTGCATGGCCCCTTGATTTGGGCCTGGCGCTCGAGTACTTTCACACACTCTCGAATGTAATGGAATGACTTGTTGTGCATAACGTACAAACAGAAACAACTTTCAACAACGGATCTCTTGGCTCTC
#> 4 AAGTCGTAACAAGGTTTCCGTAGGTGAACCTGCGGAAGGATCATTAACGAGTTCAACTGGGGTCTGTGGGGATTCGATGCTGGCCTCCGGGCATGTGCTCGTCCCCCGGACCTCACATCCACTCACAACCCCTGTGCATCATGAGAGGTGTGGTCCTCCGTCATGGGAGCAGCTCCCCTCCGGGCGTGTTTGTACACCCCGGGCGGTCGAGCCGGGACTGCACTTCGACGCCTTTACACAAACCTTTGAATCAGTGATGTAGAATGTCATGGCCAGCGATGGTCGAACTTTAAATACAACTTTCAACAACGGATCTCTTGGCTCTC
#> 5 AAAAAGTCGTAACAAGGTTTCCGTAGGTGAACCTGCGGAAGGATCATTAGTGAATCTTCAAAGTCGGCTCGTCGGATTGTGCTGGTGGAGACACATGTGCACATCTACGAGTCGCAAACCCACACACCTGTGCATCTATGACTCTGAGTGCCGCTTTGCATGGCCCCTTGATTTGGGCCTGGCGCTCGAGTACTTTCACACACTCTCGAATGTAATGGAATGTCTTGTTGTGCATAACGTACAAACAGAAACAACTTTCAACAACGGATCTCTTGGCTCTC
#> 6 AAGTCGTAACAAGGTTTCCGTAGGTGAACCTGCGGAAGGATCATTAAAAATGAAGCCGGGAAACCGGTCCTTCTAACCCTTGATTATCTTAATTTGTTGCTTTGGTGGGCCGCGCAAGCACCGGCTTCGGCTGGATCGTGCCCGCCAGAGGACCACAACTCTGTATCAAATGTCGTCTGAGTACTATATAATAGTTAAAACTTTCAACAACGGATCTCTTGGTTCTG
#> dada2_tax
#> 1 Fungi--100--Kingdom;Basidiomycota--100--Phylum;Agaricomycetes--100--Class;Sebacinales--100--Order;unidentified--98--Family;unidentified--98--Genus;Sebacinales_sp--98--Species
#> 2 Fungi--100--Kingdom;Basidiomycota--100--Phylum;Agaricomycetes--100--Class;Sebacinales--100--Order;unidentified--97--Family;unidentified--97--Genus;Sebacinales_sp--97--Species
#> 3 Fungi--100--Kingdom;Basidiomycota--100--Phylum;Agaricomycetes--100--Class;Sebacinales--100--Order;unidentified--99--Family;unidentified--99--Genus;Sebacinales_sp--99--Species
#> 4 Fungi--100--Kingdom;Basidiomycota--62--Phylum;Agaricomycetes--59--Class;Hymenochaetales--21--Order;Repetobasidiaceae--8--Family;Rickenella--8--Genus;Rickenella_sp--8--Species
#> 5 Fungi--100--Kingdom;Basidiomycota--100--Phylum;Agaricomycetes--100--Class;Sebacinales--100--Order;unidentified--96--Family;unidentified--96--Genus;Sebacinales_sp--96--Species
#> 6 Fungi--100--Kingdom;Ascomycota--100--Phylum;Leotiomycetes--98--Class;Helotiales--98--Order;unidentified--86--Family;unidentified--86--Genus;Helotiales_sp--86--Species
#> S1_its S2_its
#> 1 436 0
#> 2 405 0
#> 3 0 299
#> 4 209 65
#> 5 0 200
#> 6 16 116Reformat ASV matrix as taxmap and**phyloseq** objects after optional filtering of low abundance ASVs
Objects can now be used for downstream data analysis
Here we make heattrees using **taxmap**object.
First we make a heat tree to examine fungal community composition using our ITS1 data.
objs$taxmap_its %>%
filter_taxa(! grepl(x = taxon_names, "NA", ignore.case = TRUE)) %>%
metacoder::heat_tree(node_label = taxon_names,
node_size = n_obs,
node_color = n_obs,
node_color_axis_label = "ASV count",
node_size_axis_label = "Total Abundance of Taxa",
layout = "da", initial_layout = "re")
Now we make a heat tree to showcase oomycete community composition using our rps10 data
objs$taxmap_rps10 %>%
filter_taxa(! grepl(x = taxon_names, "NA", ignore.case = TRUE)) %>%
metacoder::heat_tree(node_label = taxon_names,
node_size = n_obs,
node_color = n_obs,
node_color_axis_label = "ASV count",
node_size_axis_label = "Total Abundance of Taxa",
layout = "da", initial_layout = "re")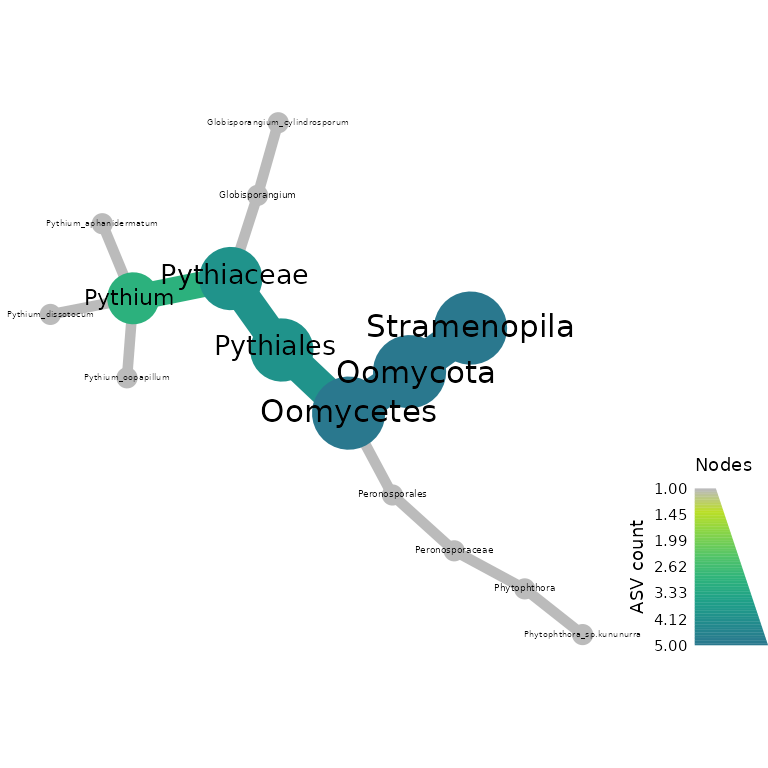
We can also do a variety of analyses, if we convert to**phyloseq** object
Here we demonstrate how to make a stacked bar plot of the relative abundance of taxa by sample for the ITS1-barcoded samples
data <- objs$phyloseq_its %>%
phyloseq::transform_sample_counts(function(x) {x/sum(x)} ) %>%
phyloseq::psmelt() %>%
dplyr::filter(Abundance > 0.02) %>%
dplyr::arrange(Genus)
abund_plot <- ggplot2::ggplot(data, ggplot2::aes(x = Sample, y = Abundance, fill = Genus)) +
ggplot2::geom_bar(stat = "identity", position = "stack", color = "black", size = 0.2) +
ggplot2::scale_fill_viridis_d() +
ggplot2::theme_minimal() +
ggplot2::labs(
y = "Relative Abundance",
title = "Relative abundance of taxa by sample",
fill = "Genus"
) +
ggplot2::theme(
axis.text.x = ggplot2::element_text(angle = 90, hjust = 1, vjust = 0.5, size = 14),
panel.grid.major = ggplot2::element_blank(),
panel.grid.minor = ggplot2::element_blank(),
legend.position = "top",
legend.text = ggplot2::element_text(size = 14),
legend.title = ggplot2::element_text(size = 14),
strip.text = ggplot2::element_text(size = 14),
strip.background = ggplot2::element_blank()
) +
ggplot2::guides(
fill = ggplot2::guide_legend(
reverse = TRUE,
keywidth = 1,
keyheight = 1,
title.position = "top",
title.hjust = 0.5, # Center the legend title
label.theme = ggplot2::element_text(size = 10)
)
)
print(abund_plot)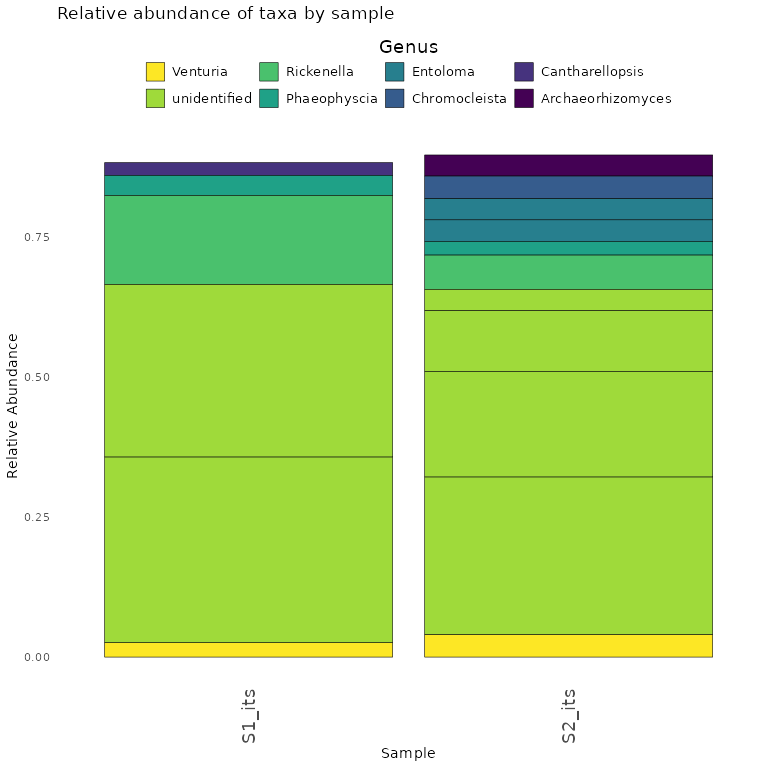
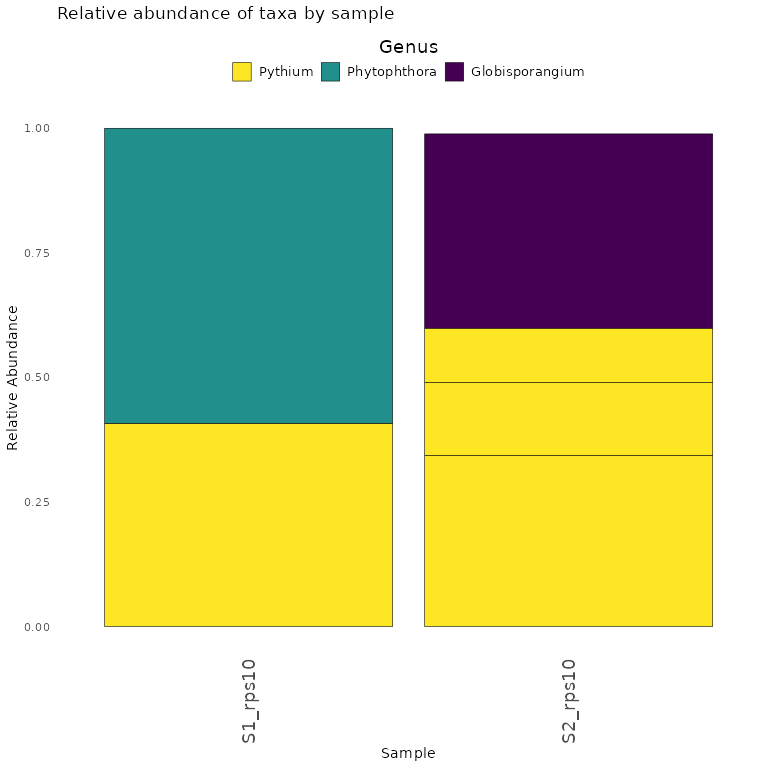
Finally, we demonstrate how to make a stacked bar plot of the relative abundance of taxa by sample for the rps10-barcoded samples
data <- objs$phyloseq_rps10 %>%
phyloseq::transform_sample_counts(function(x) {x/sum(x)} ) %>%
phyloseq::psmelt() %>%
dplyr::filter(Abundance > 0.02) %>%
dplyr::arrange(Genus)
abund_plot <- ggplot2::ggplot(data, ggplot2::aes(x = Sample, y = Abundance, fill = Genus)) +
ggplot2::geom_bar(stat = "identity", position = "stack", color = "black", size = 0.2) +
ggplot2::scale_fill_viridis_d() +
ggplot2::theme_minimal() +
ggplot2::labs(
y = "Relative Abundance",
title = "Relative abundance of taxa by sample",
fill = "Genus"
) +
ggplot2::theme(
axis.text.x = ggplot2::element_text(angle = 90, hjust = 1, vjust = 0.5, size = 14),
panel.grid.major = ggplot2::element_blank(),
panel.grid.minor = ggplot2::element_blank(),
legend.position = "top",
legend.text = ggplot2::element_text(size = 14),
legend.title = ggplot2::element_text(size = 14), # Adjust legend title size
strip.text = ggplot2::element_text(size = 14),
strip.background = ggplot2::element_blank()
) +
ggplot2::guides(
fill = ggplot2::guide_legend(
reverse = TRUE,
keywidth = 1,
keyheight = 1,
title.position = "top",
title.hjust = 0.5, # Center the legend title
label.theme = ggplot2::element_text(size = 10) # Adjust the size of the legend labels
)
)
print(abund_plot)