Vitamin D and diabetes (original) (raw)
Abstract
Vitamin D deficiency predisposes individuals to type 1 and type 2 diabetes, and receptors for its activated form—1α,25-dihydroxyvitamin D3—have been identified in both beta cells and immune cells. Vitamin D deficiency has been shown to impair insulin synthesis and secretion in humans and in animal models of diabetes, suggesting a role in the development of type 2 diabetes. Furthermore, epidemiological studies suggest a link between vitamin D deficiency in early life and the later onset of type 1 diabetes. In some populations, type 1 diabetes is associated with certain polymorphisms within the vitamin D receptor gene. In studies in nonobese diabetic mice, pharmacological doses of 1α,25-dihydroxyvitamin D3, or its structural analogues, have been shown to delay the onset of diabetes, mainly through immune modulation. Vitamin D deficiency may, therefore, be involved in the pathogenesis of both forms of diabetes, and a better understanding of the mechanisms involved could lead to the development of preventive strategies.
Similar content being viewed by others
Explore related subjects
Discover the latest articles and news from researchers in related subjects, suggested using machine learning.
Introduction
The discovery of receptors for 1α,25-dihydroxyvitamin D3 (1,25(OH)2D3), the activated form of vitamin D, in tissues with no direct role in calcium and bone metabolism (e.g. pancreatic beta cells and cells of the immune system) has broadened our view of the physiological role of this molecule [1, 2]. An increased prevalence of type 2 diabetes has been described in vitamin D-deficient individuals [3–5], and insulin synthesis and secretion have been shown to be impaired in beta cells from vitamin D-deficient animals. Glucose tolerance is restored when vitamin D levels return to normal.
The identification of receptors for 1,25(OH)2D3 in cells of the immune system led to experiments in animal models of type 1 diabetes in which the administration of high doses of 1,25(OH)2D3 was shown to prevent type 1 diabetes [3, 6], mainly through immune regulation. It has been demonstrated that 1,25(OH)2D3 is one of the most powerful blockers of dendritic cell differentiation and that it directly blocks IL-12 secretion [7]. Lymphocyte proliferation is inhibited and regulator cell development is enhanced [8].
This review provides an overview of the data available on the role of vitamin D in type 1 and type 2 diabetes, and discusses possible applications of the molecule or its synthetic analogues [9, 10] in clinical disease. The terminology used in many papers to describe vitamin D and its metabolites is confusing, with misnomers leading to misunderstanding and over-interpretation of data. In this review the term vitamin D refers to the product that is in food (vitamins D2 and D3) and is synthesised in the skin under the influence of UVB radiation (vitamin D3), whereas the metabolically active molecule is referred to as 1,25(OH)2D3.
Vitamin D and its metabolism
In the 17th century, Whistler (in 1645) and Glisson (in 1650) described rickets as a bone disease of young children (for review see [11]), but it was only in 1923 that Goldblatt and Soames identified and characterised vitamin D [12]. Most vertebrates synthesise vitamin D in their skin under the influence of UV light [1, 13]. An ‘efficient’ sun exposure—exposure of the face and hands to the sun for 2 h/week—is probably sufficient to maintain normal levels. Food supplementation is required during pregnancy and lactation and for newborns and young children (especially in dark-skinned children living in northern countries). Vitamin D can be obtained from dietary sources of vegetable (vitamin D2, also known as ergocalciferol) or animal origin (vitamin D3, also known as cholecalciferol). The best food sources are fatty fish or their liver oils; however, small amounts are also found in butter, cream and egg yolk. Human and cows’ milk are poor sources of vitamin D. In many parts of the world, especially North America, fluid and dried milk, as well as some margarines, butter and cereals are supplemented with vitamin D. However, the real vitamin D content is frequently quite different from the labelling standard and often insufficient to reach the daily requirements for vitamin D (400–600 IU/day). Skimmed milk, in particular, frequently has no detectable vitamin D.
Vitamin D3 itself is biologically inert and requires two successive hydroxylations, one in the liver (on C25) and one in the kidney (on the α position of C1), to form its hormonally active metabolite, 1,25(OH)2D3 (Fig. 1). Liver 25-hydroxylases and kidney 1α-hydroxylase belong to the large family of cytochrome P450-dependent steroid hydroxylases [14]. The production of 1,25(OH)2D3 in the kidney is regulated by several factors, particularly by levels of parathyroid hormone, although kidney 1α-hydroxylase is also subject to direct negative feedback inhibition by 1,25(OH)2D3.
Fig. 1
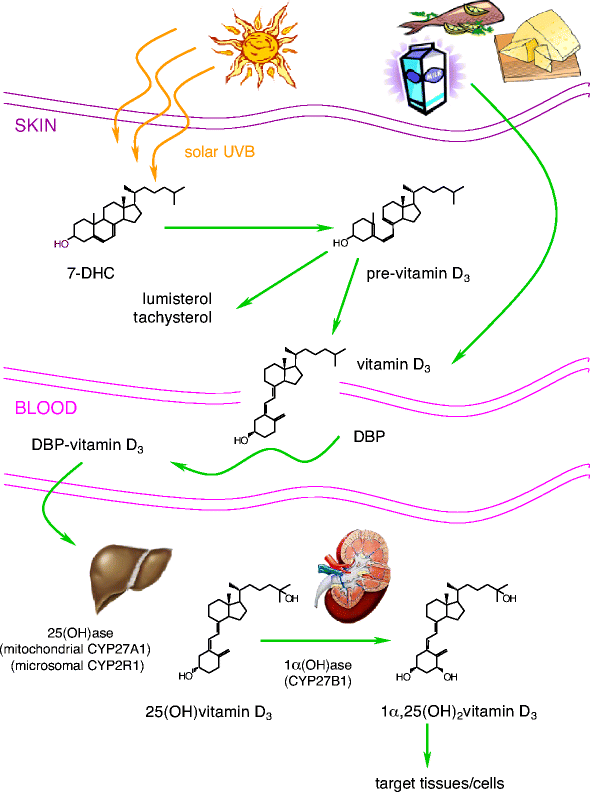
Synthesis and metabolism of 1α,25-dihydroxyvitamin D3. Vitamin D can be obtained from food (vitamin D2 and D3) or by photobiogenesis in the skin (vitamin D3). In the blood, all vitamin D metabolites are bound to vitamin D-binding protein (DBP). Vitamin D3 is converted by two successive hydroxylations in the liver (25-hydroxylases) and kidney (1α-hydroxylase) into its active hormonal form, 1,25(OH)2D3
The proximal renal tubule is the principal site of 1α-hydroxylation, although high levels of 1α-hydroxylase mRNA have also been found in human keratinocytes [15], dendritic cells [16] and macrophages [17]. This extra-renal production of 1,25(OH)2D3 is regulated in a completely different manner. For example, production in macrophages is resistant to stimulation by parathyroid hormone but may be directly stimulated by immune stimuli such as IFN-γ and lipopolysaccharide [17].
Another hydroxylation enzyme, 24-hydroxylase, initiates the catabolic cascade of 25-hydroxyvitamin D3 and 1,25(OH)2D3 [18]. In the circulation, all metabolites of vitamin D are bound to a carrier protein known as vitamin D-binding protein [19].
Molecular action of 1α,25-dihydroxyvitamin D3
Only the 1,25(OH)2D3 form of vitamin D is metabolically active, and this molecule exerts its effects by activating the nuclear vitamin D receptor (VDR). The VDR is a member of the nuclear receptor super family of ligand-activated transcription factors, which also includes the thyroid hormone receptor, the retinoic acid receptor and the peroxisome proliferator-activated receptor. The structure of the VDR has only very recently been described and the protein is yet to be fully characterised [20]. In humans the gene encoding the VDR is located on chromosome 12cen–q12 and shows extensive polymorphism, including a mononucleotide (A) n repeat polymorphism in the 3′ untranslated region and four RFLPs: _Fok_I within exon 2, _Bsm_I and _Apa_I within successive introns between exon 7 and exon 9, and _Taq_I within exon 9 (for review see [2]).
The binding of 1,25(OH)2D3 to the VDR leads to the transcription of genes regulated by 1,25(OH)2D3. The mechanism of this transcriptional regulation is very complex and is only just beginning to be unravelled (Fig. 2). The cognate vitamin D response element (VDRE) to which the VDR binds consists of a hexanucleotide direct repeat spaced by three nucleotides (DR3-type VDRE). The VDR usually binds as a heterodimer with the retinoic X receptor (RXR), and the classical effects of 1,25(OH)2D3 are the result of interactions with this nuclear receptor. With respect to its relevance in diabetes, the classical VDRE and other response sites are found within genes encoding proteins with important functions in beta cells and within genes encoding proteins with key roles throughout the immune system, such as cytokines and transcription factors [21–24].
Fig. 2
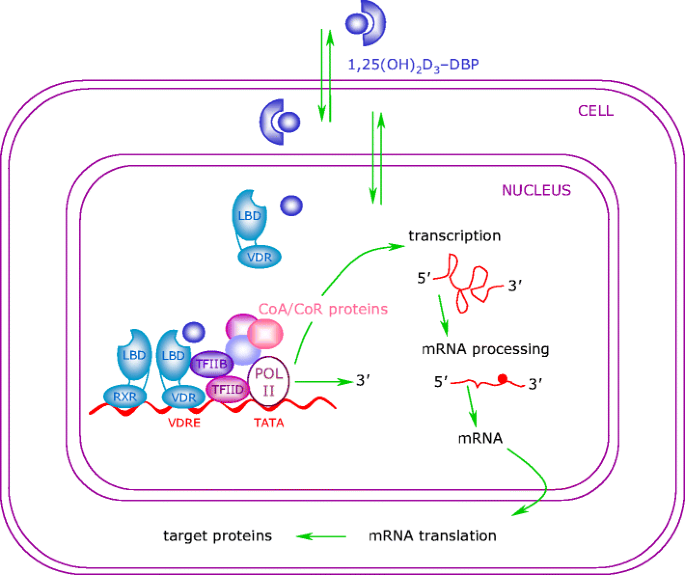
Genomic actions of 1α,25-dihydroxyvitamin D3. Molecules of 1,25(OH)2D3 easily penetrate the plasma membrane and exert their genomic effects by activating the VDR. Ligand binding to the VDR induces a conformational change in the receptor and subsequent heterodimerisation with RXR. The RXR–VDR complex binds to the VDRE, which is located within the 5′ flanking region of target genes. Thereafter, co-repressor (CoR) proteins are released from the surface of the VDR, allowing interaction with co-activator (CoA) proteins. These molecules modulate chromatin structure and allow the interaction of the receptor with the RNA polymerase II transcriptional complex (POL II), thus activating transcription of the target gene. LBD ligand-binding domain; TF transcription factor
The effects of 1,25(OH)2D3 on tissues other than calcium and bone can only be observed at concentrations (10−10 mol/l) that exceed the physiological levels needed for maintenance of calcium and bone homeostasis by a factor of 100–1,000. The fact that cells such as macrophages can secrete 1,25(OH)2D3 suggests that such high concentrations of 1,25(OH)2D3 may be attained locally within specific target areas (e.g. sites of inflammation).
The administration of 1,25(OH)2D3 in humans to exploit its ‘non-calcaemic’ effects is not realistic, since the doses needed to reach the high levels locally within target organs would lead to severe systemic hypercalcaemia and other (bone) side effects. Structural analogues containing side chain modifications or, more recently, CD-ring modifications have been synthesised that share the ‘non-classical’ effects of 1,25(OH)2D3 but have lesser effects on calcium and bone [25–27]. At present, more than 2,000 analogues have been patented, and some analogues have already reached the clinic (e.g. calcipotriol for psoriasis). A better understanding of the differential effects of these analogues on various tissues could permit targeted drug design and the development of analogues with an even greater dissociation of calcaemic and non-calcaemic effects.
Vitamin D and type 2 diabetes
Vitamin D deficiency was linked to IGT and type 2 diabetes in humans many years ago [5, 28]. These observations were confirmed in animal models, which demonstrated that pancreatic insulin secretion is inhibited by vitamin D deficiency [29]. Several reports have ascribed an active role to vitamin D in the functional regulation of the endocrine pancreas, particularly the beta cells. Not only are receptors for 1,25(OH)2D3 found in beta cells [30], but the effector part of the vitamin D pathway is also present in the form of vitamin D-dependent calcium-binding protein, also known as calbindin-D28k [31]. The expression of calbindin-D28K has been shown to protect beta cells from cytokine-mediated cell death [32].
Several studies have demonstrated a link between VDR gene polymorphisms and type 2 diabetes, although the findings differ from one population to another. A study in Bangladeshi Asians demonstrated that the _Apa_I RFLP influences insulin secretion in response to glucose [33], while associations between the VDR _Apa_I RFLP and higher fasting plasma glucose levels and glucose intolerance were observed in a community-based study of older adults without known diabetes [34]. More recently, genotyping for _Taq_I, _Apa_I, _Bsm_I and _Fok_I RFLPs revealed that the _Bsm_I RFLP is associated with high fasting glucose levels in young males with low physical activity [35].
Effects of vitamin D and its metabolites on insulin synthesis and secretion
Vitamin D deficiency leads to the impaired secretion of insulin—but not other islet hormones—in both animal models and humans, and induces glucose intolerance [29, 36, 37], while replenishment with vitamin D rectifies the abnormalities [38–40]. This impairment is primarily caused by the direct effect of vitamin D deficiency on the beta cell, but other effects of vitamin D deficiency, such as impaired food intake and hypocalcaemia, may also play a role. Data from VDR knock-out mice are conflicting, with some groups reporting IGT [41] and others reporting no impairment in glucose metabolism [42]. In these studies the genetic background of the mouse in which the VDR knock-out is introduced seems to be of critical importance.
More convincing support for a beneficial effect of 1,25(OH)2D3 on beta cell function has emerged from studies on islets isolated from normal animals in which insulin synthesis and release were provoked by glucose challenge in the presence of high doses of 1,25(OH)2D3 [43].
The mechanism by which 1,25(OH)2D3 might act on insulin secretion is suggested by the significant rise in cytosolic Ca2+ levels observed following 1,25(OH)2D3-stimulated secretion of insulin by islet cells. Controversy remains as to whether an influx of external Ca2+ via voltage-dependent Ca2+ channels is solely responsible for this rise, or whether the mobilisation of Ca2+ from intracellular organelles and the activation of release-potentiating systems via protein kinase C and protein kinase A pathways are also involved [44–46].
Interventions with vitamin D and its metabolites in vivo: clinical implications for type 2 diabetes
The restoration of vitamin D reserves in vitamin D-deficient patients has been shown to improve glucose tolerance [47]. The amount of vitamin D administered to these patients with rickets was, however, subject to wide variation (from the daily oral administration of 2,000 IU [28] to a single intramuscular injection of 100,000 IU [3]), making it difficult to draw clear conclusions and establish guidelines. Nonetheless, it is clear that vitamin D deficiency should be treated, and that improved glucose tolerance is a desirable side effect. In contrast, repletion of 1,25(OH)2D3 in the relatively 1,25(OH)2D3-deficient state of uraemia only partially reverses glucose intolerance, probably because insulin resistance is also involved in this condition [48].
Studies on the administration of vitamin D supplements or even higher doses of 1,25(OH)2D3 to vitamin D-sufficient patients with IGT or type 2 diabetes have yielded conflicting results. Some have reported an improvement [49], others no effect [4]. One study even showed a worsening of type 2 diabetes: supplementation in three British Asians with vitamin D deficiency and type 2 diabetes led to increased insulin resistance and the deterioration of glycaemic control [50] (Table 1).
Table 1 Vitamin D in the aetiology of type 2 diabetes: published studies in humans
Vitamin D and type 1 diabetes
Several epidemiological studies have described an intriguing correlation between geographical latitude and the incidence of type 1 diabetes, and an inverse correlation between monthly hours of sunshine and the incidence of diabetes. A seasonal pattern of disease onset has also been described for type 1 diabetes [51], once again suggesting an inverse correlation between sunlight and the disease. Vitamin D is an obvious candidate as a mediator of this sunshine effect.
Dietary vitamin D supplementation is often recommended in pregnant women and in children to prevent vitamin D deficiency. Cod liver oil taken during the first year of life reportedly reduced the risk of childhood-onset type 1 diabetes [52], and a multicentre case-control study also showed an association between vitamin D supplementation in infancy and a decreased risk of type 1 diabetes [53]. A further study found that an intake of 2,000 IU of vitamin D during the first year of life diminished the risk of developing type 1 diabetes, and showed that the incidence of childhood diabetes was three times higher in subjects with suspected rickets [54]. More recently, the Diabetes Autoimmunity Study in the Young (DAISY) reported that the presence of islet auto-antibodies in offspring was inversely correlated with maternal dietary vitamin D intake during pregnancy [55] (Table 2).
Table 2 Vitamin D in the aetiology of type 1 diabetes: published studies in humans
It remains to be determined whether these observations are the result of supplementation of vitamin D to supraphysiological levels, or are simply the result of the prevention of vitamin D deficiency. Observations in animal models suggest the latter, since regular supplements of vitamin D in neonatal and early life offered no protection against type 1 diabetes in nonobese diabetic (NOD) mice or in BioBreeding (BB) rats [56, 57], whereas the prevalence of diabetes is doubled in NOD mice rendered vitamin D-deficient in early life [58, 59]. The results of genetic studies investigating a possible relationship between VDR polymorphisms and type 1 diabetes are highly confusing: a clear correlation exist in some populations [60–65], whereas no correlation can be found in others [66].
Immunomodulatory effects of 1α,25-dihydroxyvitamin D3 and its analogues
The identification of VDRs on almost all cells of the immune system, especially antigen-presenting cells (macrophages and dendritic cells) and activated T lymphocytes, prompted the investigation of 1,25(OH)2D3 as a potential immunomodulator (for reviews see [57, 67]). Immune cells—activated macrophages and dendritic cells in particular—contain the enzyme 1α-hydroxylase, which is necessary for the final activating step in the conversion of vitamin D3 to the metabolically active molecule, and are therefore able to synthesise and secrete 1,25(OH)2D3 [17, 68]. The 1α-hydroxylase present in immune cells is identical to the renal enzyme, but regulation of its expression and activity is different. Whereas the renal enzyme is principally under the control of calcaemic and bone signals (such as parathyroid hormone and 1,25(OH)2D3 itself), the macrophage enzyme is primarily regulated by immune signals, with IFN-γ and Toll-like receptor agonists being powerful stimulators. It is of interest that a defect in the upregulation of 1α-hydroxylase in response to immune stimuli has been reported in NOD mice [17].
A unique feature of 1,25(OH)2D3 as an immunomodulator is that not only does it interact with T cells, but it also—more importantly—targets the central cell in the immune cascade, the antigen-presenting cell (for reviews see [69, 70]) (Fig. 3). In vitro, 1,25(OH)2D3 stimulates the phagocytosis and killing of bacteria by macrophages but suppresses the antigen-presenting capacity of these cells and dendritic cells [71]. It is noteworthy that the expression of MHC-II molecules and adhesion molecules necessary for full T cell stimulation, such as B7.2, is suppressed [72]. Cytokines secreted by antigen-presenting cells for the recruitment and activation of T cells are also directly influenced by 1,25(OH)2D3, and several authors have described the inhibition of a key cytokine in the immune system, IL-12, by 1,25(OH)2D3 and its analogues [7, 73, 74]. This protein, which is produced by macrophages and dendritic cells, is the major determinant of the direction in which the immune system will be activated, since it stimulates the development of CD4 T-helper type 1 (Th-1) cells and inhibits the development of CD4 Th-2 lymphocytes. This inhibition is observed in vitro, but IL-12 suppression and a shift from Th1 to Th2 predominance can also be observed after in vivo administration of 1,25(OH)2D3 or its analogues [75–77]. Inhibition of IL-12 is achieved through a direct interaction between 1,25(OH)2D3 bound to the VDR (as a heterodimer with RXR) and nuclear factor κB (NF-κB), which interferes with the NF-κB-induced transcription of IL-12 [7]. The secretion of other cytokines by macrophages/dendritic cells is also influenced by 1,25(OH)2D3. Prostaglandin E2, a suppressive cytokine, is stimulated, while the monocyte recruiter granulocyte-macrophage-colony-stimulating factor (GM-CSF) is suppressed. Suppression of GM-CSF is achieved via binding of ligand-bound monomers of the VDR (in the absence of RXR) to a DNA element in the promotor region of the gene encoding GM-CSF [78].
Fig. 3
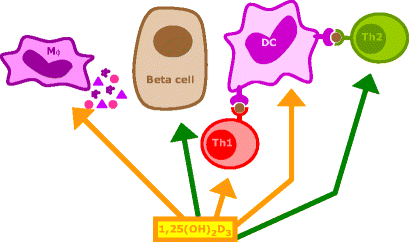
Mechanisms of action of 1α,25-dihydroxyvitamin D3 and its analogues in type 1 diabetes (with courtesy to E. van Etten). 1,25(OH)2D3 and its analogues can modulate the immune responses via several mechanisms. They are necessary for normal insulin secretion and biosynthesis and are reported to protect beta cells against cytokines. They downregulate antigen presentation and co-stimulatory molecule expression by antigen-presenting cells, especially dendritic cells (DCs), thus indirectly inhibiting the development of Th lymphocytes along the Th-1 pathway and promoting the induction of Th-2 lymphocytes and/or regulatory T lymphocytes. 1,25(OH)2D3 and its analogues also exert direct effects on T lymphocytes: IL-2 and IFN-γ production by Th-1 lymphocytes is inhibited, whereas the production of the Th2 cytokine IL-4 is directly induced, both shifting the Th balance towards the Th-2 phenotype. Mφ macrophage
Several T cell cytokines are direct targets of 1,25(OH)2D3 and its analogues. The secretion of IL-2 is suppressed through the inhibition of nuclear factor of activated T cells (NFAT) complex formation by ligand-bound VDR–RXR heterodimers through binding to the distal NFAT binding site in the human IL-2 promoter [79]. Another key T cell cytokine, IFN-γ, is downregulated by 1,25(OH)2D3 [80]. The promoter region of the gene encoding this protein contains a classical VDRE (DR3 type), and binding of the ligand-bound VDR–RXR heterodimers causes direct suppression of transcription [81].
Taken together, these observations suggest a physiological role for 1,25(OH)2D3 in the immune system, with a tightly regulated secretion of 1,25(OH)2D3 by macrophages and dendritic cells upon immune stimulation on the one hand and a direct inhibitory effect of the molecule on antigen presentation and T cell proliferation and cytokine secretion on the other hand. These immune effects are typically mediated through the VDR. Therefore, the fact that VDR knock-out mice have no apparent immune abnormalities suggests the redundancy of 1,25(OH)2D3 as a signal in the immune system [42].
Beta cell protection by 1α,25-dihydroxyvitamin D3 and its analogues
Direct beta cell damage by cytokines and other inflammatory agents is an important step in the pathogenesis of type 1 diabetes. The inhibition of beta cell function (insulin synthesis and insulin secretion) induced by IL-1β or IFN-γ in vitro is prevented by 1,25(OH)2D3 and its analogues, MC903 and KH1060 [82, 83]. In contrast, Mauricio et al observed no effect of 1,25(OH)2D3 on IL-1β-induced beta cell dysfunction [84]. This discrepancy may be due to the fact that an incubation time of 24 h was used in the latter study, whereas incubation periods ranging from 48 to 72 h were used in the studies describing protection. In addition to the alteration of the effects of cytokines on beta cell function, 1,25(OH)2D3 blocks the induction of surface markers by these cytokines [83, 85, 86]. The upregulation of expression of MHC-II molecules and intercellular adhesion molecule-1 on rat islets after exposure to IFN-γ was markedly decreased by co-incubation with 1,25(OH)2D3 or an analogue (ZXY1106) [83]. More recently, Riachy et al demonstrated that 1,25(OH)2D3 might preserve the insulin content of human islets and prevent MHC-I expression, IL-6 production and NO release [85]. Moreover, it was reported that 1,25(OH)2D3 induced and maintained high levels of A20, an anti-apoptotic protein, in rat RINm5F cells and human islets after exposure to inflammatory cytokines [86]. We recently performed a comparative study of different cell systems (whole rat islets, FACS-purified beta cells and INS-1E cells) and did not observe direct protection by 1,25(OH)2D3 against cytokine-induced beta cell death, but demonstrated decreased expression of chemokines by beta cells treated with 1,25(OH)2D3 [87].
In summary, 1,25(OH)2D3 protects beta cells from cytokine-induced beta cell dysfunction but cannot protect them from direct cytokine-induced cell death (Fig. 3).
Effects of 1α,25-dihydroxyvitamin D3 and its analogues in animal models of type 1 diabetes
The chronic administration of pharmacological doses of 1,25(OH)2D3 reduces the incidence of both insulitis and diabetes in NOD mice [6, 88]. When 1,25(OH)2D3 was administered lifelong from weaning in NOD mice, the incidence of insulitis was reduced from 80% in control mice to 58% in 1,25(OH)2D3-treated mice, and the incidence of diabetes was reduced from 56 to 8%. These findings have been confirmed in other studies [77]. Extensive immunological screening, including FACS analysis of major lymphocyte subsets, evaluation of T cell proliferation and natural killer cell function, did not reveal any major changes in the treated mice compared with the control mice [6]. The reported restoration of deficient suppressor cell function of NOD mice in response to treatment with 1,25(OH)2D3 represents a major finding [89]. Adorini et al demonstrated that the regulatory T cell induced by 1,25(OH)2D3 or its analogues is most likely a CD4CD25 cell [77]. However, it remains to be determined whether this restoration of suppressor cells is the main mechanism involved in the protection afforded by 1,25(OH)2D3 against diabetes. Protection against insulitis was also observed, suggesting interference with the induction of autoimmunity itself. This is consistent with the observation that 1,25(OH)2D3 protects NOD mice against cyclophosphamide-induced diabetes [90].
The molecular basis of protection by 1,25(OH)2D3 seems to involve the reshaping of the immune repertoire, with elimination of effector cells, although the direct protective effects of 1,25(OH)2D3 on the beta cell might also play a role in disease prevention. It has been shown that there is a shift in the production of T cell cytokines from predominantly Th1 (IL-2, IFN-γ) in control mice to Th2 (IL-4, IL-10) in mice treated with 1,25(OH)2D3 or an analogue of the molecule [75]. The shift appeared to be antigen specific and is probably caused by the direct interference of 1,25(OH)2D3 or its analogues by the antigen-presenting dendritic cells. Indeed, 1,25(OH)2D3 induces a reshaping of dendritic cells towards tolerogenic cells [69, 70, 77]. We have also demonstrated that dendritic cells generated in the presence of 1,25(OH)2D3 or an analogue can re-direct already committed T cell clones derived from a type 1 diabetic patient towards non-proliferation [8, 91]. In the NOD mouse, the reshaping of the immune system happens centrally, in the thymus, where treatment with 1,25(OH)2D3 restores the sensitivity of T lymphocytes towards apoptosis-inducing signals, thereby enhancing the elimination of autoimmune effector cells [92].
In marked contrast to the NOD mouse, no significant difference in the incidence of diabetes was observed between control BB rats (33%) and BB rats treated with 1,25(OH)2D3 from weaning up to 120 days (24%) [93]. This finding confirms the basic differences in disease pathogenesis in the two available animal models for type 1 diabetes, and indicates that caution is warranted when transferring findings from either of these models to the human situation.
A major obstacle to the administration of 1,25(OH)2D3 in humans is its important effects on calcium and bone metabolism. However, in view of the therapeutic potential, structural analogues have been designed and synthesised with the aim of achieving a dissociation between calcaemic and immune effects. Several analogues developed in different chemical laboratories have been successfully tested in the NOD mouse [89]. The mechanism of protection against insulitis and diabetes appears to be similar to that of 1,25(OH)2D3. Effects of the analogues on dendritic cell phenotype, regulator cell induction and beta cell protection have been described [8, 77, 83].
The optimal analogue should provide a combination of beta cell protection, immune modulation and low calcaemic effects. Although several analogues have provided promising results [93], long-term safety data will have to be collated before long-term interventions in individuals at high risk of type 1 diabetes are initiated. A critical question as regards the applicability of analogues in the human situation is whether these 1,25(OH)2D3 analogues are able to arrest progression to clinically overt diabetes if administered when active beta cell destruction is already present—the situation in pre-diabetic subjects in whom immune intervention is considered [94]. Casteels et al demonstrated that, when combined with a short induction course of a classical immunosuppressant such as cyclosporine A, some of these analogues can arrest the progression of the disease when administered after autoimmunity has started [95]. This approach of combining a short induction course of a classical immunosuppressant with non-hypercalcaemic analogues of 1,25(OH)2D3 is promising and might open new perspectives for the prevention of autoimmune diabetes in humans.
The capacity of some of the analogues of 1,25(OH)2D3 to prevent disease recurrence after islet transplantation has been evaluated in spontaneously diabetic NOD mice. The most convincing results were obtained using a combination of KH1060 and cyclosporine A [96]. A similar synergism has been reported between 1,25(OH)2D3 or an analogue and mycophenolate mofetil [97]. One novel approach involved the combination of analogues of 1,25(OH)2D3 with other physiological immune modulators, such as recombinant IFN-β [98]. In this study, the combination of recombinant IFN-β and ZXY1106 markedly delayed the recurrence of autoimmune diabetes following islet transplantation [98].
Conclusion
Solid evidence exists that vitamin D deficiency is detrimental to beta cell function, leads to glucose intolerance in animal models and humans, and predisposes to type 2 diabetes. Vitamin D deficiency in early life predisposes NOD mice and humans to the later development of autoimmune diabetes. A major practical conclusion that can be drawn from the studies conducted on vitamin D and diabetes to date is that vitamin D deficiency is undesirable, not only for calcium and bone, but also for glucose metabolism.
Administration of high doses of the activated form of vitamin D, 1,25(OH)2D3, has immunomodulatory effects that lead to diabetes prevention in animal models of type 1 diabetes. The availability of structural analogues of 1,25(OH)2D3 opens new perspectives for the exploitation of these interesting properties in humans. Carefully designed intervention studies using these analogues, either alone or in combination with other immunomodulators, will yield the answer to the question of their clinical potential in diabetes prevention.
| Clinical perspectives |
|---|
| The metabolically active form of vitamin D, 1,25(OH)2D3, and its analogues have been shown to have effects on the major players involved in the pathogenesis of type 1 and type 2 diabetes. Beta cell function has been shown to be improved by 1,25(OH)2D3 in vitro and in vivo, and the avoidance of vitamin D deficiency is essential for normal beta cell function. In NOD mice, 1,25(OH)2D3 protects against insulitis, diabetes and disease recurrence after islet transplantation, primarily through immunomodulatory effects. |
| The use of 1,25(OH)2D3 for the prevention or cure of diabetes is limited by its hypercalcaemic and bone remodelling effects, as its protective effects are only observed in response to supra-physiological doses. Future applications of this therapy in human diabetes are conceivable, since the calcaemic and immunomodulatory effects of 1,25(OH)2D3 can be dissociated by structural analogues of the molecule. |
| It is likely that these analogues will find a use as beta-cell-protective and -stimulating agents as adjuncts to the current treatment for type 2 diabetes. These agents could play a major role in preventative strategies for type 1 diabetes in humans, because of their profile as both beta cell-protective and immuno-active drugs. More information is required concerning their mechanism of action and long-term safety before they can be routinely administered in humans. |
Abbreviations
1,25(OH)2D3 :
1α,25-dihydroxyvitamin D3
BB:
BioBreeding
GM-CSF:
granulocyte-macrophage-colony-stimulating factor
NF-κB:
nuclear factor-κB
NFAT:
nuclear factor of activated T cell
NOD:
nonobese diabetic
RXR:
retinoid X receptor
Th:
T helper
VDR:
vitamin D receptor
VDRE:
vitamin D response element
References
- Holick MF (2003) Vitamin D: a millenium perspective. J Cell Biochem 88:296–307
Article CAS PubMed Google Scholar - Haussler MR, Whitfield GK, Haussler CA et al (1998) The nuclear vitamin D receptor: biological and molecular regulatory properties revealed. J Bone Miner Res 13:325–349
CAS PubMed Google Scholar - Boucher BJ, Mannan N, Noonan K, Hales CN, Evans SJ (1995) Glucose intolerance and impairment of insulin secretion in relation to vitamin D deficiency in east London Asians. Diabetologia 38:1239–1245
Article CAS PubMed Google Scholar - Isaia G, Giorgino R, Adami S (2001) High prevalence of hypovitaminosis D in female type 2 diabetic population. Diabetes Care 24:1496
CAS Google Scholar - Chiu KC, Chu A, Go VL, Saad MF (2004) Hypovitaminosis D is associated with insulin resistance and beta cell dysfunction. Am J Clin Nutr 79:820–825
CAS PubMed Google Scholar - Mathieu C, Waer M, Laureys J, Rutgeerts O, Bouillon R (1994) Prevention of autoimmune diabetes in NOD mice by 1,25 dihydroxyvitamin D3. Diabetologia 37:552–558
CAS PubMed Google Scholar - D’Ambrosio D, Cippitelli M, Cocciolo MG et al (1998) Inhibition of IL-12 production by 1,25-dihydroxyvitamin D3. Involvement of NF-kappaB downregulation in transcriptional repression of the p40 gene. J Clin Invest 101:252–262
CAS PubMed Google Scholar - van Halteren AG, Van Etten E, de Jong EC, Bouillon R, Roep BO, Mathieu C (2002) Redirection of human autoreactive T-cells upon interaction with dendritic cells modulated by TX527, an analog of 1,25 dihydroxyvitamin D3. Diabetes 51:2119–2125
PubMed Google Scholar - Bouillon R, Verstuyf A, Verlinden L, Eelen G, Mathieu C (2003) Prospects for vitamin D receptor modulators as candidate drugs for cancer and (auto)immune diseases. Recent Results Cancer Res 164:353–356
CAS PubMed Google Scholar - Bouillon R, Garmyn M, Verstuyf A, Segaert S, Casteels K, Mathieu C (1995) Paracrine role for calcitriol in the immune system and skin creates new therapeutic possibilities for vitamin D analogs. Eur J Endocrinol 133:7–16
CAS PubMed Google Scholar - Rajakumar K (2003) Vitamin D, cod-liver oil, sunlight, and rickets: a historical perspective. Pediatrics 112:e132–e135
Article PubMed Google Scholar - Windaus A, Linsert O, Luttringhaus A, Weidlinch G (1932) Uber das krystallistierte Vitamin D2. Justus Liebigs Ann Chem 492:226–231
CAS Google Scholar - Holick MF, Clark MB (1978) The photobiogenesis and metabolism of vitamin D. Fed Proc 37:2567–2574
CAS PubMed Google Scholar - Inouye K, Sakaki T (2001) Enzymatic studies on the key enzymes of vitamin D metabolism: 1 alpha-hydroxylase (CYP27B1) and 24-hydroxylase (CYP24). Biotechnol Annu Rev 7:179–194
Article CAS PubMed Google Scholar - Fu GK, Lin D, Zhang MY et al (1997) Cloning of human 25-hydroxyvitamin D-1 alpha-hydroxylase and mutations causing vitamin D-dependent rickets type 1. Mol Endocrinol 11:1961–1970
Article CAS PubMed Google Scholar - Hewison M, Freeman L, Hughes SV et al (2003) Differential regulation of vitamin D receptor and its ligand in human monocyte-derived dendritic cells. J Immunol 170:5382–5390
CAS PubMed Google Scholar - Overbergh L, Decallonne B, Valckx D et al (2000) Identification and immune regulation of 25-hydroxyvitamin D-1-alpha-hydroxylase in murine macrophages. Clin Exp Immunol 120:139–146
Article CAS PubMed Google Scholar - Tanaka Y, Castillo L, Deluca HF (1977) The 24-hydroxylation of 1,25-dihydroxyvitamin D3. J Biol Chem 252:1421–1424
Google Scholar - Haddad JG, Matsuoka LY, Hollis BW, Hu YZ, Wortsman J (1993) Human plasma transport of vitamin D after its endogenous synthesis. J Clin Invest 91:2552–2555
CAS PubMed Google Scholar - Rochel N, Wurtz JM, Mitschler A, Klaholz B, Moras D (2000) The crystal structure of the nuclear receptor for vitamin D bound to its natural ligand. Mol Cell 5:173–179
Article CAS PubMed Google Scholar - Vidal M, Ramana CV, Dusso AS (2002) Stat1–vitamin D receptor interactions antagonize 1,25-dihydroxyvitamin D transcriptional activity and enhance stat1-mediated transcription. Mol Cell Biol 22:2777–2787
Article CAS PubMed Google Scholar - Christakos S, Barletta F, Huening M et al (2003) Vitamin D target proteins: function and regulation. J Cell Biochem 88:238–244
Article CAS PubMed Google Scholar - Maestro B, Davila N, Carranza MC, Calle C (2003) Identification of a vitamin D response element in the human insulin receptor gene promoter. J Steroid Biochem Mol Biol 84:223–230
Article CAS PubMed Google Scholar - Eerligh P, Koeleman BP, Dudbridge F, Jan BG, Roep BO, Giphart MJ (2004) Functional genetic polymorphisms in cytokines and metabolic genes as additional genetic markers for susceptibility to develop type 1 diabetes. Genes Immun 5:36–40
Article CAS PubMed Google Scholar - Verstuyf A, Verlinden L, van Baelen H et al (1998) The biological activity of nonsteroidal vitamin D hormone analogs lacking both the C- and D-rings. J Bone Miner Res 13:549–558
CAS PubMed Google Scholar - Verstuyf A, Verlinden L, Van Etten E et al (2000) Biological activity of CD-ring modified 1alpha,25-dihydroxyvitamin D analogues: C-ring and five-membered D-ring analogues. J Bone Miner Res 15:237–252
CAS PubMed Google Scholar - De Clercq PJ, Murad I, Gao LJ et al (2004) Biological activity and conformational analysis of C20 and C14 epimers of CD-ring modified _trans_-decalin 1alpha,25-dihydroxyvitamin D analogs. J Steroid Biochem Mol Biol 89–90:61–66
Article PubMed Google Scholar - Gedik O, Akalin S (1986) Effects of vitamin D deficiency and repletion on insulin and glucagon secretion in man. Diabetologia 29:142–145
CAS PubMed Google Scholar - Norman AW, Frankel JB, Heldt AM, Grodsky GM (1980) Vitamin D deficiency inhibits pancreatic secretion of insulin. Science 209:823–825
CAS PubMed Google Scholar - Lee S, Clark SA, Gill RK, Christakos S (1994) 1,25-Dihydroxyvitamin D3 and pancreatic beta-cell function: vitamin D receptors, gene expression, and insulin secretion. Endocrinology 134:1602–1610
Article CAS PubMed Google Scholar - Sooy K, Schermerhorn T, Noda M et al (1999) Calbindin-D(28k) controls [Ca(2+)](i) and insulin release. Evidence obtained from calbindin-D(28k) knockout mice and beta cell lines. J Biol Chem 274:34343–34349
Google Scholar - Rabinovitch A, Suarez-Pinzon WL, Sooy K, Strynadka K, Christakos S (2001) Expression of calbindin-D(28k) in a pancreatic islet beta-cell line protects against cytokine-induced apoptosis and necrosis. Endocrinology 142:3649–3655
Article CAS PubMed Google Scholar - Hitman GA, Mannan N, McDermott MF et al (1998) Vitamin D receptor gene polymorphisms influence insulin secretion in Bangladeshi Asians. Diabetes 47:688–690
CAS PubMed Google Scholar - Oh JY, Barrett-Connor E (2002) Association between vitamin D receptor polymorphism and type 2 diabetes or metabolic syndrome in community-dwelling older adults: the Rancho Bernardo Study. Metabolism 51:356–359
Article CAS PubMed Google Scholar - Ortlepp JR, Metrikat J, Albrecht M, von Korff A, Hanrath P, Hoffmann R (2003) The vitamin D receptor gene variant and physical activity predicts fasting glucose levels in healthy young men. Diabet Med 20:451–454
Article CAS PubMed Google Scholar - Chertow BS, Sivitz WI, Baranetsky NG, Clark SA, Waite A, Deluca HF (1983) Cellular mechanisms of insulin release: the effects of vitamin D deficiency and repletion on rat insulin secretion. Endocrinology 113:1511–1518
CAS PubMed Google Scholar - Cade C, Norman AW (1986) Vitamin D3 improves impaired glucose tolerance and insulin secretion in the vitamin D-deficient rat in vivo. Endocrinology 119:84–90
CAS PubMed Google Scholar - Kadowaki S, Norman AW (1984) Dietary vitamin D is essential for normal insulin secretion from the perfused rat pancreas. J Clin Invest 73:759–766
CAS PubMed Google Scholar - Nyomba BL, Bouillon R, De Moor P (1984) Influence of vitamin D status on insulin secretion and glucose tolerance in the rabbit. Endocrinology 115:191–197
CAS PubMed Google Scholar - Tanaka Y, Seino Y, Ishida M et al (1984) Effect of vitamin D3 on the pancreatic secretion of insulin and somatostatin. Acta Endocrinol (Copenh) 105:528–533
CAS Google Scholar - Zeitz U, Weber K, Soegiarto DW, Wolf E, Balling R, Erben RG (2003) Impaired insulin secretory capacity in mice lacking a functional vitamin D receptor. FASEB J 17:509–511
CAS PubMed Google Scholar - Mathieu C, Van Etten E, Gysemans C et al (2001) In vitro and in vivo analysis of the immune system of vitamin D receptor knockout mice. J Bone Miner Res 16:2057–2065
CAS PubMed Google Scholar - d’Emden MC, Dunlop M, Larkins RG, Wark JD (1989) The in vitro effect of 1 alpha,25-dihydroxyvitamin D3 on insulin production by neonatal rat islets. Biochem Biophys Res Commun 164:413–418
CAS PubMed Google Scholar - de Boland AR, Norman AW (1990) Influx of extracellular calcium mediates 1,25-dihydroxyvitamin D3-dependent transcaltachia (the rapid stimulation of duodenal Ca2+ transport). Endocrinology 127:2475–2480
PubMed Google Scholar - Billaudel BJ, Faure AG, Sutter BC (1990) Effect of 1,25 dihydroxyvitamin D3 on isolated islets from vitamin D3-deprived rats. Am J Physiol 258:E643–E648
CAS PubMed Google Scholar - Bourlon PM, Faure-Dussert A, Billaudel B (1997) Modulatory role of 1,25 dihydroxyvitamin D3 on pancreatic islet insulin release via the cyclic AMP pathway in the rat. Br J Pharmacol 121:751–758
Google Scholar - Kumar S, Davies M, Zakaria Y et al (1994) Improvement in glucose tolerance and beta-cell function in a patient with vitamin D deficiency during treatment with vitamin D. Postgrad Med J 70:440–443
CAS PubMed Google Scholar - Allegra V, Luisetto G, Mengozzi G, Martimbianco L, Vasile A (1994) Glucose-induced insulin secretion in uremia: role of 1 alpha,25(OH)2-vitamin D3. Nephron 68:41–47
CAS PubMed Google Scholar - Borissova AM, Tankova T, Kirilov G, Dakovska L, Kovacheva R (2003) The effect of vitamin D3 on insulin secretion and peripheral insulin sensitivity in type 2 diabetic patients. Int J Clin Pract 57:258–261
Google Scholar - Taylor AV, Wise PH (1998) Vitamin D replacement in Asians with diabetes may increase insulin resistance. Postgrad Med J 74:365–366
CAS PubMed Google Scholar - Karvonen M, Jantti V, Muntoni S et al (1998) Comparison of the seasonal pattern in the clinical onset of IDDM in Finland and Sardinia. Diabetes Care 21:1101–1109
CAS PubMed Google Scholar - Stene LC, Joner G (2003) Use of cod liver oil during the first year of life is associated with lower risk of childhood-onset type 1 diabetes: a large, population-based, case-control study. Am J Clin Nutr 78:1128–1134
CAS PubMed Google Scholar - The EURODIAB Substudy 2 Study Group (1999) Vitamin D supplement in early childhood and risk for type I (insulin-dependent) diabetes mellitus. Diabetologia 42:51–54
Google Scholar - Hypponen E, Laara E, Reunanen A, Jarvelin MR, Virtanen SM (2001) Intake of vitamin D and risk of type 1 diabetes: a birth-cohort study. Lancet 358:1500–1503
CAS PubMed Google Scholar - Fronczak CM, Baron AE, Chase HP et al (2003) In utero dietary exposures and risk of islet autoimmunity in children. Diabetes Care 26:3237–3242
PubMed Google Scholar - Mathieu C, Van Etten E, Gysemans C, Decallonne B, Bouillon R (2002) Seasonality of birth in patients with type 1 diabetes. Lancet 359:1248
Article Google Scholar - Mathieu C, Van Etten E, Decallonne B et al (2004) Vitamin D and 1,25-dihydroxyvitamin D(3) as modulators in the immune system. J Steroid Biochem Mol Biol 89–90:449–452
PubMed Google Scholar - Giulietti A, Gysemans C, Stoffels K et al (2004) Vitamin D deficiency in early life accelerates type 1 diabetes in non-obese diabetic mice. Diabetologia 47:451–462
CAS PubMed Google Scholar - Zella JB, McCary LC, Deluca HF (2003) Oral administration of 1,25-dihydroxyvitamin D3 completely protects NOD mice from insulin-dependent diabetes mellitus. Arch Biochem Biophys 417:77–80
CAS PubMed Google Scholar - McDermott MF, Ramachandran A, Ogunkolade BW et al (1997) Allelic variation in the vitamin D receptor influences susceptibility to IDDM in Indian Asians. Diabetologia 40:971–975
CAS PubMed Google Scholar - Pani MA, Knapp M, Donner H et al (2000) Vitamin D receptor allele combinations influence genetic susceptibility to type 1 diabetes in Germans. Diabetes 49:504–507
CAS PubMed Google Scholar - Ban Y, Taniyama M, Yanagawa T et al (2001) Vitamin D receptor initiation codon polymorphism influences genetic susceptibility to type 1 diabetes mellitus in the Japanese population. BMC Med Genet 2:7
CAS PubMed Google Scholar - Ogunkolade BW, Boucher BJ, Prahl JM et al (2002) Vitamin D receptor (VDR) mRNA and VDR protein levels in relation to vitamin D status, insulin secretory capacity, and VDR genotype in Bangladeshi Asians. Diabetes 51:2294–2300
CAS PubMed Google Scholar - Yokota I, Satomura S, Kitamura S et al (2002) Association between vitamin D receptor genotype and age of onset in juvenile Japanese patients with type 1 diabetes. Diabetes Care 25:1244
Google Scholar - Motohashi Y, Yamada S, Yanagawa T et al (2003) Vitamin D receptor gene polymorphism affects onset pattern of type 1 diabetes. J Clin Endocrinol Metab 88:3137–3140
CAS PubMed Google Scholar - Nejentsev S, Cooper JD, Godfrey L et al (2004) Analysis of the vitamin D receptor gene sequence variants in type 1 diabetes. Diabetes 53:2709–2712
CAS PubMed Google Scholar - Mathieu C, Adorini L (2002) The coming of age of 1,25-dihydroxyvitamin D(3) analogs as immunomodulatory agents. Trends Mol Med 8:174–179
CAS PubMed Google Scholar - Adams JS, Sharma OP, Gacad MA, Singer FR (1983) Metabolism of 25-hydroxyvitamin D3 by cultured pulmonary alveolar macrophages in sarcoidosis. J Clin Invest 72:1856–1860
CAS PubMed Google Scholar - Adorini L (2003) Tolerogenic dendritic cells induced by vitamin D receptor ligands enhance regulatory T cells inhibiting autoimmune diabetes. Ann N Y Acad Sci 987:258–261
CAS PubMed Google Scholar - Adorini L, Penna G, Giarratana N et al (2004) Dendritic cells as key targets for immunomodulation by vitamin D receptor ligands. J Steroid Biochem Mol Biol 89–90:437–441
PubMed Google Scholar - Van Etten E, Decallonne B, Bouillon R, Mathieu C (2004) NOD bone marrow-derived dendritic cells are modulated by analogs of 1,25-dihydroxyvitamin D(3). J Steroid Biochem Mol Biol 89–90:457–459
PubMed Google Scholar - Griffin MD, Lutz WH, Phan VA, Bachman LA, McKean DJ, Kumar R (2000) Potent inhibition of dendritic cell differentiation and maturation by vitamin D analogs. Biochem Biophys Res Commun 270:701–708
Google Scholar - Penna G, Adorini L (2000) 1 Alpha,25-dihydroxyvitamin D3 inhibits differentiation, maturation, activation, and survival of dendritic cells leading to impaired alloreactive T cell activation. J Immunol 164:2405–2411
CAS PubMed Google Scholar - Griffin MD, Lutz W, Phan VA, Bachman LA, McKean DJ, Kumar R (2001) Dendritic cell modulation by 1alpha,25 dihydroxyvitamin D3 and its analogs: a vitamin D receptor-dependent pathway that promotes a persistent state of immaturity in vitro and in vivo. Proc Natl Acad Sci U S A 98:6800–6805
CAS PubMed Google Scholar - Overbergh L, Decallonne B, Waer M et al (2000) 1alpha,25-dihydroxyvitamin D3 induces an autoantigen-specific T-helper 1/T-helper 2 immune shift in NOD mice immunized with GAD65 (p524-543). Diabetes 49:1301–1307
CAS PubMed Google Scholar - Boonstra A, Barrat FJ, Crain C, Health VL, Savelkoul HF, O’Garra A (2001) 1alpha,25-dihydroxyvitamin D3 has a direct effect on naive CD4(+) T cells to enhance the development of Th2 cells. J Immunol 167:4974–4980
CAS PubMed Google Scholar - Gregori S, Giarratana N, Smiroldo S, Uskokovic M, Adorini L (2002) A 1alpha,25-dihydroxyvitamin D(3) analog enhances regulatory T-cells and arrests autoimmune diabetes in NOD mice. Diabetes 51:1367–1374
CAS PubMed Google Scholar - Towers TL, Staeva TP, Freedman LP (1999) A two-hit mechanism for vitamin D3-mediated transcriptional repression of the granulocyte-macrophage colony-stimulating factor gene: vitamin D receptor competes for DNA binding with NFAT1 and stabilizes c-Jun. Mol Cell Biol 19:4191–4199
CAS PubMed Google Scholar - Takeuchi A, Reddy GS, Kobayashi T, Okano T, Park J, Sharma S (1998) Nuclear factor of activated T cells (NFAT) as a molecular target for 1alpha,25-dihydroxyvitamin D3-mediated effects. J Immunol 160:209–218
CAS PubMed Google Scholar - Reichel H, Koeffler HP, Tobler A, Norman AW (1987) 1 alpha,25-dihydroxyvitamin D3 inhibits gamma-interferon synthesis by normal human peripheral blood lymphocytes. Proc Natl Acad Sci U S A 84:3385–3389
CAS PubMed Google Scholar - Cippitelli M, Santoni A (1998) Vitamin D3: a transcriptional modulator of the interferon-gamma gene. Eur J Immunol 28:3017–3030
CAS PubMed Google Scholar - Sandler S, Buschard K, Bendtzen K (1994) Effects of 1,25-dihydroxyvitamin D3 and the analogues MC903 and KH1060 on interleukin-1 beta-induced inhibition of rat pancreatic islet beta-cell function in vitro. Immunol Lett 41:73–77
CAS PubMed Google Scholar - Hahn HJ, Kuttler B, Mathieu C, Bouillon R (1997) 1,25-Dihydroxyvitamin D3 reduces MHC antigen expression on pancreatic beta-cells in vitro. Transplant Proc 29:2156–2157
CAS PubMed Google Scholar - Mauricio D, Andersen H, Karlsen A, Mandrup-Poulsen T, Nerup J (1995) Effect of steroidal hormones on interleukin-1beta action on rat islets. Diabetologia 38S:A80
Google Scholar - Riachy R, Vandewalle B, Belaich S et al (2001) Beneficial effect of 1,25 dihydroxyvitamin D3 on cytokine-treated human pancreatic islets. J Endocrinol 169:161–168
CAS PubMed Google Scholar - Riachy R, Vandewalle B, Kerr CJ et al (2002) 1,25-dihydroxyvitamin D3 protects RINm5F and human islet cells against cytokine-induced apoptosis: implication of the antiapoptotic protein A20. Endocrinology 143:4809–4819
CAS PubMed Google Scholar - Gysemans CA, Cardozo AK, Callewaert H et al (2005) 1,25-Dihydroxyvitamin D3 modulates expression of chemokines and cytokines in pancreatic islets: implications for prevention of diabetes in nonobese diabetic mice. Endocrinology 146:1956–1964
CAS PubMed Google Scholar - Mathieu C, Laureys J, Sobis H, Vandeputte M, Waer M, Bouillon R (1992) 1,25-Dihydroxyvitamin D3 prevents insulitis in NOD mice. Diabetes 41:1491–1495
CAS PubMed Google Scholar - Mathieu C, Waer M, Casteels K, Laureys J, Bouillon R (1995) Prevention of type I diabetes in NOD mice by nonhypercalcemic doses of a new structural analog of 1,25-dihydroxyvitamin D3, KH1060. Endocrinology 136:866–872
CAS PubMed Google Scholar - Casteels K, Waer M, Bouillon R et al (1998) 1,25-Dihydroxyvitamin D3 restores sensitivity to cyclophosphamide-induced apoptosis in non-obese diabetic (NOD) mice and protects against diabetes. Clin Exp Immunol 112:181–187
CAS PubMed Google Scholar - van Halteren AG, Tysma OM, van Etten E, Mathieu C, Roep BO (2004) 1alpha,25-dihydroxyvitamin D3 or analogue treated dendritic cells modulate human autoreactive T cells via the selective induction of apoptosis. J Autoimmun 23:233–239
PubMed Google Scholar - Casteels KM, Gysemans CA, Waer M et al (1998) Sex difference in resistance to dexamethasone-induced apoptosis in NOD mice: treatment with 1,25(OH)2D3 restores defect. Diabetes 47:1033–1037
CAS PubMed Google Scholar - Mathieu C, Casteels K, Bouillon R (1997) Vitamin D and diabetes. In: Feldman D, Glorieux F, Pike J (eds) Vitamin D. Academic, San Diego, pp 1183–1196
Google Scholar - Bach JF (1994) Predictive medicine in autoimmune diseases: from the identification of genetic predisposition and environmental influence to precocious immunotherapy. Clin Immunol Immunopathol 72:156–161
CAS PubMed Google Scholar - Casteels K, Waer M, Bouillon R, Allewaert K, Laureys J, Mathieu C (1996) Prevention of type I diabetes by late intervention with nonhypercalcemic analogues of vitamin D3 in combination with cyclosporin A. Transplant Proc 28:3095
CAS PubMed Google Scholar - Mathieu C, Laureys J, Waer M, Bouillon R (1994) Prevention of autoimmune destruction of transplanted islets in spontaneously diabetic NOD mice by KH1060, a 20-epi analog of vitamin D: synergy with cyclosporine. Transplant Proc 26:3128–3129
CAS PubMed Google Scholar - Gregori S, Casorati M, Amuchastegui S, Smiroldo S, Davalli AM, Adorini L (2001) Regulatory T cells induced by 1 alpha,25-dihydroxyvitamin D3 and mycophenolate mofetil treatment mediate transplantation tolerance. J Immunol 167:1945–1953
CAS PubMed Google Scholar - Gysemans C, Van Etten E, Overbergh L et al (2002) Treatment of autoimmune diabetes recurrence in non-obese diabetic mice by mouse interferon-beta in combination with an analogue of 1alpha,25-dihydroxyvitamin-D3. Clin Exp Immunol 128:213–220
CAS PubMed Google Scholar - Orwoll E, Riddle M, Prince M (1994) Effects of vitamin D on insulin and glucagon secretion in non-insulin-dependent diabetes mellitus. Am J Clin Nutr 59:1083–1087
CAS PubMed Google Scholar
Acknowledgements
The authors would like to thank E. van Etten for drawing Fig. 3. They would also like to thank the Belgium Program on Interuniversity Poles of Attraction initiated by the Belgian State (IUAP P5/17), the Catholic University of Leuven (Geconcerteerde Onderzoeksacties [GOA] 2004/10), the Flemish Research Foundation (FWO G.0084.02 and G.0233.04), the Juvenile Diabetes Research Foundation Centre for Prevention of Beta Cell Destruction in Europe (grant number 4-2002-457), and the European Community concerted Sixth Framework Program (grant number TONECA 503245) for support. C. Mathieu has a clinical research fellowship (FWO), C. Gysemans a postdoctoral fellowship (FWO), and A. Giulietti a doctoral scholarship from the Catholic University of Leuven (Interfaculty Council for Development Co-operation).
Author information
Authors and Affiliations
- Laboratory of Experimental Medicine and Endocrinology (LEGENDO), Catholic University of Leuven, Herestraat 49, 3000, Leuven, Belgium
C. Mathieu, C. Gysemans, A. Giulietti & R. Bouillon
Authors
- C. Mathieu
- C. Gysemans
- A. Giulietti
- R. Bouillon
Corresponding author
Correspondence toC. Mathieu.
Rights and permissions
About this article
Cite this article
Mathieu, C., Gysemans, C., Giulietti, A. et al. Vitamin D and diabetes.Diabetologia 48, 1247–1257 (2005). https://doi.org/10.1007/s00125-005-1802-7
- Received: 30 June 2004
- Accepted: 21 April 2005
- Published: 22 June 2005
- Issue Date: July 2005
- DOI: https://doi.org/10.1007/s00125-005-1802-7