Characterisation of glomerular haemodynamic responses to SGLT2 inhibition in patients with type 1 diabetes and renal hyperfiltration (original) (raw)
To the Editor: Renal hyperfiltration in experimental models of early diabetes is associated with increased intraglomerular pressure, and this condition may promote the development of diabetic nephropathy [1]. Despite the functional characterisation of renal hyperfiltration in animal models of diabetes indicating causal involvement of the afferent arteriole, confirmative data in humans are scarce. This is explained by the fact that direct measurements of human glomerular haemodynamic variables, such as afferent (RA) and efferent (RE) arteriolar resistances and glomerular hydrostatic pressure (PGLO), are technically not feasible. To elucidate mechanisms of renal disease in humans, Gomez et al published formulae derived from animal physiological studies in 1951 to estimate glomerular haemodynamic function by using measured mean arterial blood pressure (MAP), GFR, renal plasma flow, haematocrit and total protein concentrations [2].
More recently, selective sodium–glucose co-transporter 2 (SGLT2) inhibitors have been evaluated in studies involving patients with type 2 and type 1 diabetes. In this context, we recently demonstrated, in a stratified, open-label, single-arm 8 week trial, that SGLT2 inhibition with empagliflozin attenuates renal hyperfiltration in patients with type 1 diabetes, an effect likely due to modulation of tubuloglomerular feedback via increased distal tubular sodium delivery [3]. The objective of this exploratory, post hoc analysis was to apply the Gomez formulae to quantify glomerular haemodynamic variables in patients with type 1 diabetes analysed on the basis of filtration status.
Type 1 diabetic patients were treated with empagliflozin (25 mg daily) in the Adjunctive-To-Insulin and Renal MechAnistic pilot trial of empagliflozin in T1D (ATIRMA trial, ClinicalTrials.gov NCT01392560) [3]. Forty participants completed this study and the primary outcome of changes in renal function with empagliflozin have been published elsewhere [3]. None of the participants was taking medications that could affect blood pressure or other cardiovascular parameters. Other secondary endpoints related to glycaemia and arterial stiffness have also been reported [4, 5]. Studies were performed during two consecutive days at baseline and 8 weeks after empagliflozin treatment. A modified glucose clamp technique was used to maintain euglycaemic (approximately 4–6 mmol/l) conditions on the first day for approximately 6 h, followed by hyperglycaemic (approximately 9–11 mmol/l) conditions on the second day for approximately 6 h (electronic supplementary material [ESM] Fig. 1) [3]. All data are expressed as mean ± SD. For further details of the experimental design and statistical analysis, please see the ESM Methods. The study was approved by the Research Ethics Board at the University Health Network. All participants gave informed consent prior to the start of the study procedures.
Baseline characteristics, and serum and urine biochemical responses to empagliflozin in this cohort, have been reported elsewhere [3]. To assess renal sodium handling further, 24 h fractional excretion of sodium (FENa+) was calculated. FENa+ increased in type 1 diabetic patients with renal hyperfiltration (T1D-H; 0.54 ± 0.29 to 0.77 ± 0.48, p = 0.03) but not in type 1 diabetic patients with renal normofiltration (T1D-N; 0.63 ± 0.46 to 0.74 ± 0.61, p = 0.61). Using Gomez’s formulae at baseline during clamped euglycaemia, T1D-H patients exhibited lower RA, and increased PGLO and ΔPF (filtration pressure across the glomerular capillaries) than that of T1D-N patients (Fig. 1a–d, ESM Table 1; ESM Methods). There were no differences in RE or glomerular oncotic pressure (πG) between the two groups. In response to empagliflozin, RA increased in T1D-H patients (euglycaemia: 785 ± 442 vs 1,307 ± 615 dyn s cm−5, p < 0.0001; hyperglycaemia: 690 ± 637 vs 1,454 ± 814 dyn s cm−5, p < 0.0001; Fig. 1a). Furthermore, intraglomerular pressure (PGLO, Fig. 1c) decreased in response to empagliflozin in T1D-H patients during clamped euglycaemia (67.4 ± 5.4 vs 61.0 ± 5.2 mmHg, p < 0.0001) and hyperglycaemia (69.3 ± 6.5 vs 61.6 ± 6.3 mmHg, p < 0.0001). ΔPF (Fig. 1d) also decreased in response to empagliflozin in the T1D-H group during euglycaemia (33.1 ± 4.5 vs 26.7 ± 4.8 mmHg, p < 0.0001) and hyperglycaemia (35.8 ± 6.4 vs 27.5 ± 5.7 mmHg, p < 0.0001). In contrast, these variables remained unchanged in the T1D-N group (Fig. 1). There were no significant differences in RE in either group (Fig. 1b).
Fig. 1
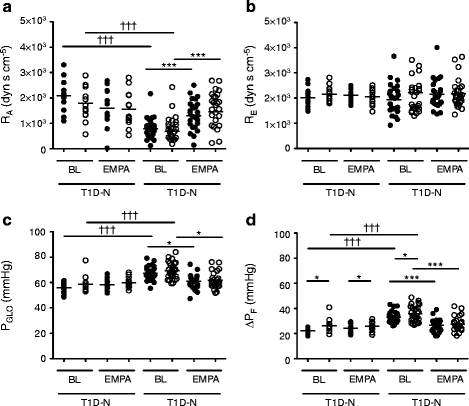
Glomerular haemodynamics estimated by Gomez’s equations (KFG = 0.0867) in participants with normofiltration (T1D-N) or hyperfiltration (T1D-H) at baseline (BL) and after empagliflozin (EMPA, 25 mg daily) treatment during clamped euglycaemic (black circles) and hyperglycaemic (open circles) conditions. *p < 0.05 and ***p < 0.001 for within-group differences using a paired Student’s t test. ††† p < 0.001 for between-group differences (T1D-N [n = 13] vs T1D-H [n = 27]) using Tukey’s post hoc test after ANOVA analysis. Horizontal lines represent means of haemodynamic variables
Owing to higher GFR values in T1D-H patients (mean GFR 172 ± 23 ml min−1 [1.73 m2]−1) compared with the cohort used to derive the ultrafiltration coefficient (KFG) in previous literature (mean GFR < 130 ml min−1 [1.73 m2]−1), we estimated a KFG of 0.115 for T1D-H and repeated the Gomez formulae. Using this adjusted KFG, similar results were obtained (ESM Fig. 2). To assess the appropriateness of the serum albumin concentration to be used, instead of total protein to calculate RA and PGLO variables, albumin concentration was multiplied by a calculated conversation factor of 1.695. Haemodynamic variables were strongly correlated (r = 0.91 to 0.99, p < 0.0001) using either serum albumin or total protein (ESM Fig. 3).
Our first main observation was that in a cohort of normotensive, normoalbuminuric patients, T1D-H participants exhibited evidence of afferent vasodilation characterised by lower RA compared with T1D-N participants. This is consistent with previous animal models of diabetes demonstrating a dominant state of afferent vasodilatation relative to effects at the efferent arteriole [6]. Our second main observation was that treatment with empagliflozin for 8 weeks decreased RA and also reduced PGLO in T1D-H patients, with no effect on RE. On the basis of these haemodynamic responses, empagliflozin significantly improved intraglomerular hypertension and achieved a mean intraglomerular pressure approximating that in T1D-N participants. These first-in-human observations suggested that SGLT2 inhibition exerts a clinically meaningful impact on early renal haemodynamic abnormalities characteristic of type 1 diabetes. Future randomised, appropriately controlled studies should determine whether these haemodynamic effects also occur in patients with renal impairment, and whether SGLT2 inhibition may, as a consequence, be renal protective.
The preferential effect of SGLT2 inhibition on RA, rather than RE, is important in light of how these agents could potentially be used in patients taking renin–angiotensin–aldosterone system (RAAS) inhibitors [7]. The renal effects of SGLT2 inhibition could be complementary to the well described changes in RE achieved with RAAS blockade, an intriguing concept for the design of future renal protection studies using the combination of dual SGLT2–RAAS inhibition (ACE inhibition or angiotensin II receptor blockade) to normalise glomerular hyperfiltration (ESM Fig. 4). Although existing studies suggest that SGLT2 inhibitors have an acceptable renal safety profile, further work is required to determine whether combined SGLT2–RAAS inhibition could fully normalise the intraglomerular hypertension and hyperfiltration associated with the early stages of diabetes, and whether this effect would lead to the long-term preservation of renal function. On the other hand, whether such a combination may reduce intraglomerular pressure to an extent that could increase the risk for acute kidney injury events needs to be elucidated. In light of findings from the Ongoing Telmisartan Alone and in Combination With Ramipril Global Endpoint Trial (ONTARGET), Aliskiren Trial in Type 2 Diabetes Using Cardiovascular and Renal Disease Endpoints (ALTITUDE) and Veterans Affairs Nephropathy in Diabetes trial (VA-NEPHRON-D), which use strategies of dual RAAS blockade [8–10], this is an important consideration that requires further human physiological investigation and surveillance in ongoing clinical trials. Finally, our observations may have particular implications for primary renal preventive strategies in patients with type 1 diabetes, since RAAS blockade monotherapy is ineffective in normotensive, normoalbuminuric patients with preserved renal function. Future studies of SGLT2–RAAS blockade in patients with uncomplicated diabetes are therefore warranted, since simultaneous constriction of the afferent arteriole (SGLT2 inhibition) with efferent vasodilatation (RAAS blockade) has the potential to reverse early renal haemodynamic abnormalities that contribute to hyperfiltration.
Abbreviations
∆PF :
Filtration pressure across the glomerular capillaries
πG :
Glomerular oncotic pressure
FENa+ :
Fractional excretion of sodium
KFG :
Ultrafiltration coefficient
PGLO :
Glomerular hydrostatic pressure
RA :
Afferent renal arteriolar resistances
RAAS:
Renin–angiotensin–aldosterone system
RE :
Efferent renal arteriolar resistances
SGLT2:
Sodium–glucose co-transporter 2
T1D-N:
Type 1 diabetic patients with renal normofiltration
T1D-H:
Type 1 diabetic patients with renal hyperfiltration
References
- Hostetter TH, Rennke HG, Brenner BM (1982) The case for intrarenal hypertension in the initiation and progression of diabetic and other glomerulopathies. Am J Med 72:375–380
Article CAS PubMed Google Scholar - Gomez DM (1951) Evaluation of renal resistances, with special reference to changes in essential hypertension. J Clin Invest 30:1143–1155
Article CAS PubMed PubMed Central Google Scholar - Cherney DZI, Perkins BA, Soleymanlou N et al (2014) Renal hemodynamic effect of sodium-glucose cotransporter 2 inhibition in patients with type 1 diabetes mellitus. Circulation 129:587–597
Article CAS PubMed Google Scholar - Cherney DZ, Perkins BA, Soleymanlou N et al (2014) The effect of empagliflozin on arterial stiffness and heart rate variability in subjects with uncomplicated type 1 diabetes mellitus. Cardiovasc Diabetol 13:28
Article PubMed PubMed Central Google Scholar - Perkins BA, Cherney DZ, Partridge H et al (2014) Sodium-glucose cotransporter 2 inhibition and glycemic control in type 1 diabetes: results of an 8-week open-label proof-of-concept trial. Diabetes Care 37:1480–1483
Article PubMed Google Scholar - Vallon V, Richter K, Blantz RC, Thomson S, Osswald H (1999) Glomerular hyperfiltration in experimental diabetes mellitus: potential role of tubular reabsorption. J Am Soc Nephrol 10:2569–2576
CAS PubMed Google Scholar - Luik PT, Hoogenberg K, van Der Kleij FG et al (2002) Short-term moderate sodium restriction induces relative hyperfiltration in normotensive normoalbuminuric type I diabetes mellitus. Diabetologia 45:535–541
Article CAS PubMed Google Scholar - Investigators O, Yusuf S, Teo KK et al (2008) Telmisartan, ramipril, or both in patients at high risk for vascular events. N Engl J Med 358:1547–1559
Article Google Scholar - Parving H-H, Brenner BM, McMurray JJV et al (2012) Cardiorenal end points in a trial of aliskiren for type 2 diabetes. N Engl J Med 367:2204–2213
Article CAS PubMed Google Scholar - Fried LF, Emanuele N, Zhang JH et al (2013) Combined angiotensin inhibition for the treatment of diabetic nephropathy. N Engl J Med 369:1892–1903
Article CAS PubMed Google Scholar
Acknowledgements
The authors would like to thank P. Yip and J. Cheung-Hum (University Health Network, Toronto, ON, Canada) for their invaluable assistance with biochemical assays included in this work. Finally, the authors are grateful to the study participants, whose time and effort are critical to the success of our research programme.
The results presented in this paper have not been published previously in whole or in part, except in abstract form at the 74th Scientific Sessions of the American Diabetes Association, 2014.
Funding
This work was supported by Boehringer Ingelheim and Eli Lilly (to DZIC and BAP). DZIC was supported by a Kidney Foundation of Canada Scholarship and a Canadian Diabetes Association-KRESCENT Program Joint New Investigator Award and receives operating support from the Canadian Institutes of Health Research and the Kidney Foundation of Canada. BAP and DZIC hold joint funding from the JDRF.
Duality of interest
DZIC has received speaker honoraria from Boehringer Ingelheim and Eli Lilly, and BAP received operational funding with DZIC for this work. HJW, MvE, NS, OEJ, TH and UCB are employees of Boehringer Ingelheim.
Contributions
DZIC, BAP, MŠ, GKY, MSil, YL, HJW, NS, OEJ, UCB, MvE and TH made substantial contributions to the conception and design, acquisition of data, or analysis and interpretation of data. All authors either drafted or revised the manuscript for important intellectual content. All authors have approved the final version of this manuscript. DZIC and MŠ are guarantors of the work.
Author information
Authors and Affiliations
- Department of Medicine, Division of Nephrology, Toronto General Hospital, University of Toronto, 585 University Ave, 8N-845, Toronto, ON, Canada, M5G 2N2
Marko Škrtić, Gary K. Yang, Yuliya Lytvyn, Melvin Silverman & David Z. I. Cherney - Department of Medicine, Division of Endocrinology, Mount Sinai Hospital, University of Toronto, Toronto, ON, Canada
Bruce A. Perkins - Boehringer Ingelheim Canada, Burlington, ON, Canada
Nima Soleymanlou - Boehringer Ingelheim Pharmaceuticals, Ridgefield, CT, USA
Maximilian von Eynatten - Boehringer Ingelheim Pharma GmbH & Co.KG, Ingelheim, Germany
Hans J. Woerle, Uli C. Broedl & Thomas Hach - Boehringer Ingelheim Norway KS, Asker, Norway
Odd Erik Johansen
Authors
- Marko Škrtić
You can also search for this author inPubMed Google Scholar - Gary K. Yang
You can also search for this author inPubMed Google Scholar - Bruce A. Perkins
You can also search for this author inPubMed Google Scholar - Nima Soleymanlou
You can also search for this author inPubMed Google Scholar - Yuliya Lytvyn
You can also search for this author inPubMed Google Scholar - Maximilian von Eynatten
You can also search for this author inPubMed Google Scholar - Hans J. Woerle
You can also search for this author inPubMed Google Scholar - Odd Erik Johansen
You can also search for this author inPubMed Google Scholar - Uli C. Broedl
You can also search for this author inPubMed Google Scholar - Thomas Hach
You can also search for this author inPubMed Google Scholar - Melvin Silverman
You can also search for this author inPubMed Google Scholar - David Z. I. Cherney
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toDavid Z. I. Cherney.
Additional information
An erratum to this article is available at http://dx.doi.org/10.1007/s00125-017-4241-3.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Škrtić, M., Yang, G.K., Perkins, B.A. et al. Characterisation of glomerular haemodynamic responses to SGLT2 inhibition in patients with type 1 diabetes and renal hyperfiltration.Diabetologia 57, 2599–2602 (2014). https://doi.org/10.1007/s00125-014-3396-4
- Received: 23 August 2014
- Accepted: 10 September 2014
- Published: 04 October 2014
- Issue Date: December 2014
- DOI: https://doi.org/10.1007/s00125-014-3396-4