A fibril-specific, conformation-dependent antibody recognizes a subset of Aβ plaques in Alzheimer disease, Down syndrome and Tg2576 transgenic mouse brain (original) (raw)
Abstract
Beta-amyloid (Aβ) is thought to be a key contributor to the pathogenesis of Alzheimer disease (AD) in the general population and in adults with Down syndrome (DS). Different assembly states of Aβ have been identified that may be neurotoxic. Aβ oligomers can assemble into soluble prefibrillar oligomers, soluble fibrillar oligomers and insoluble fibrils. Using a novel antibody, OC, recognizing fibrils and soluble fibrillar oligomers, we characterized fibrillar Aβ deposits in AD and DS cases. We further compared human specimens to those obtained from the Tg2576 mouse model of AD. Our results show that accumulation of fibrillar immunoreactivity is significantly increased in AD relative to nondemented aged subjects and those with select cognitive impairments (p < 0.0001). Further, there was a significant correlation between the extent of frontal cortex fibrillar deposit accumulation and dementia severity (MMSE r = −0.72). In DS, we observe an early age of onset and age-dependent accumulation of fibrillar OC immunoreactivity with little pathology in similarly aged non-DS individuals. Tg2576 mice show fibrillar accumulation that can be detected as young as 6 months. Interestingly, fibril-specific immunoreactivity was observed in diffuse, thioflavine S-negative Aβ deposits in addition to more mature neuritic plaques. These results suggest that fibrillar deposits are associated with disease in both AD and in adults with DS and their distribution within early Aβ pathology associated with diffuse plaques and correlation with MMSE suggest that these deposits may not be as benign as previously thought.
Similar content being viewed by others
Explore related subjects
Discover the latest articles and news from researchers in related subjects, suggested using machine learning.
Introduction
Alzheimer disease (AD) is associated with the progressive development of cognitive dysfunction and neuropathological accumulation of neurofibrillary tangles and senile plaques (SP) [37, 53, 55, 67]. A major constituent of SP is β-amyloid (Aβ), which is a 39–42 amino acid peptide cleaved from a longer amyloid precursor protein (APP) [[25](/article/10.1007/s00401-009-0530-3#ref-CR25 "Glenner GG, Wong CW (1984) Alzheimer’s disease and Down’s syndrome sharing of a unique cerebrovascular amyloid fibril protein. Biochem Biophys Res Commun 120:885–890. doi: 10.1016/S0006-291X(84)80190-4
"), [33](/article/10.1007/s00401-009-0530-3#ref-CR33 "Kang J, Lemaire HG, Unterbeck A, Salbaum JM, Masters CL, Grzeschik KH et al (1987) The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature 325(6106):733–736. doi:
10.1038/325733a0
")\]. Aβ can adopt a misfolded structure including prefibrillar oligomeric and fibrillar assembly states \[[9](/article/10.1007/s00401-009-0530-3#ref-CR9 "Caughey B, Lansbury PT (2003) Protofibrils, pores, fibrils, and neurodegeneration: separating the responsible protein aggregates from the innocent bystanders. Annu Rev Neurosci 26:267–298. doi:
10.1146/annurev.neuro.26.010302.081142
"), [24](/article/10.1007/s00401-009-0530-3#ref-CR24 "Glabe CG (2006) Common mechanisms of amyloid oligomer pathogenesis in degenerative disease. Neurobiol Aging 27(4):570–575. doi:
10.1016/j.neurobiolaging.2005.04.017
"), [28](/article/10.1007/s00401-009-0530-3#ref-CR28 "Harper JD, Wong SS, Lieber CM, Lansbury PT (1997) Observation of metastable Abeta amyloid protofibrils by atomic force microscopy. Chem Biol 4(2):119–125. doi:
10.1016/S1074-5521(97)90255-6
"), [42](/article/10.1007/s00401-009-0530-3#ref-CR42 "Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M et al (1998) Diffusible, nonfibrillar ligands derived from Aβ1–42 are potent central nervous system neurotoxins. Proc Natl Acad Sci USA 95:6448–6453. doi:
10.1073/pnas.95.11.6448
"), [70](/article/10.1007/s00401-009-0530-3#ref-CR70 "Walsh DM, Hartley DM, Kusumoto Y, Fezoui Y, Condron MM, Lomakin A et al (1999) Amyloid beta-protein fibrillogenesis. Structure and biological activity of protofibrillar intermediates. J Biol Chem 274(36):25945–25952. doi:
10.1074/jbc.274.36.25945
")\]. Some assembly states of Aβ may be more toxic than others \[[18](/article/10.1007/s00401-009-0530-3#ref-CR18 "Deshpande A, Mina E, Glabe C, Busciglio J (2006) Different conformations of amyloid beta induce neurotoxicity by distinct mechanisms in human cortical neurons. J Neurosci 26(22):6011–6018. doi:
10.1523/JNEUROSCI.1189-06.2006
"), [42](/article/10.1007/s00401-009-0530-3#ref-CR42 "Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M et al (1998) Diffusible, nonfibrillar ligands derived from Aβ1–42 are potent central nervous system neurotoxins. Proc Natl Acad Sci USA 95:6448–6453. doi:
10.1073/pnas.95.11.6448
"), [44](/article/10.1007/s00401-009-0530-3#ref-CR44 "Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A et al (2006) A specific amyloid-beta protein assembly in the brain impairs memory. Nature 440(7082):352–357. doi:
10.1038/nature04533
"), [71](/article/10.1007/s00401-009-0530-3#ref-CR71 "Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS et al (2002) Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416(6880):535–539. doi:
10.1038/416535a
")\]. For example, Aβ oligomers are toxic to neurons both in vitro \[[18](/article/10.1007/s00401-009-0530-3#ref-CR18 "Deshpande A, Mina E, Glabe C, Busciglio J (2006) Different conformations of amyloid beta induce neurotoxicity by distinct mechanisms in human cortical neurons. J Neurosci 26(22):6011–6018. doi:
10.1523/JNEUROSCI.1189-06.2006
"), [42](/article/10.1007/s00401-009-0530-3#ref-CR42 "Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M et al (1998) Diffusible, nonfibrillar ligands derived from Aβ1–42 are potent central nervous system neurotoxins. Proc Natl Acad Sci USA 95:6448–6453. doi:
10.1073/pnas.95.11.6448
")\] and in vivo \[[44](/article/10.1007/s00401-009-0530-3#ref-CR44 "Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A et al (2006) A specific amyloid-beta protein assembly in the brain impairs memory. Nature 440(7082):352–357. doi:
10.1038/nature04533
"), [71](/article/10.1007/s00401-009-0530-3#ref-CR71 "Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS et al (2002) Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416(6880):535–539. doi:
10.1038/416535a
")\]. Thus, measuring the extent of different and specific assembly states of Aβ may identify species that are more strongly linked to cognition in AD and can serve as a biomarker and possible therapeutic target \[[36](/article/10.1007/s00401-009-0530-3#ref-CR36 "Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW et al (2003) Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 300:486–489. doi:
10.1126/science.1079469
")\].Individuals with Down syndrome (DS) also develop AD pathology but in a progressive age-dependent manner [30, [49](/article/10.1007/s00401-009-0530-3#ref-CR49 "Mann DMA (1988) The pathological association between Down syndrome and Alzheimer disease. Mech Ageing Dev 43:99–136. doi: 10.1016/0047-6374(88)90041-3
"), [50](/article/10.1007/s00401-009-0530-3#ref-CR50 "Mann DMA, Esiri MM (1989) The pattern of acquisition of plaques and tangles in the brains of patients under 50 years of age with Down’s syndrome. J Neurol Sci 89:169–179. doi:
10.1016/0022-510X(89)90019-1
"), [77](/article/10.1007/s00401-009-0530-3#ref-CR77 "Wisniewski K, Wisniewski H, Wen G (1985) Occurrence of neuropathological changes and dementia of Alzheimer’s disease in Down’s syndrome. Ann Neurol 17:278–282. doi:
10.1002/ana.410170310
")\] and as such, are at high risk for the development of dementia \[[41](/article/10.1007/s00401-009-0530-3#ref-CR41 "Lai F, Williams MD (1989) A prospective study of Alzheimer disease in Down syndrome. Arch Neurol 46:849–853"), [46](/article/10.1007/s00401-009-0530-3#ref-CR46 "Lott IT, Head E (2001) Down syndrome and Alzheimer’s disease: a link between development and aging. Ment Retard Dev Disabil Res Rev 7(3):172–178. doi:
10.1002/mrdd.1025
")\]. Clinical signs of dementia are more commonly observed when individuals are over 50 years of age \[[8](/article/10.1007/s00401-009-0530-3#ref-CR8 "Bush A, Beail N (2004) Risk factors for dementia in people with Down syndrome: issues in assessment and diagnosis. Am J Ment Retard 109(2):83–97. doi:
10.1352/0895-8017(2004)109<83:RFFDIP>2.0.CO;2
"), [41](/article/10.1007/s00401-009-0530-3#ref-CR41 "Lai F, Williams MD (1989) A prospective study of Alzheimer disease in Down syndrome. Arch Neurol 46:849–853"), [63](/article/10.1007/s00401-009-0530-3#ref-CR63 "Prasher VP, Filer A (1995) Behavioural disturbance in people with Down’s syndrome and dementia. J Intellect Disabil Res 39(Pt. 5):432–436"), [69](/article/10.1007/s00401-009-0530-3#ref-CR69 "Tyrrell J, Cosgrave M, McCarron M, McPherson J, Calvert J, Kelly A et al (2001) Dementia in people with Down’s syndrome. Int J Geriatr Psychiatry 16(12):1168–1174. doi:
10.1002/gps.502
")\]. By 40 years of age, however, all individuals with DS have neuropathological changes including senile plaques and neurofibrillary tangles consistent with AD \[[50](/article/10.1007/s00401-009-0530-3#ref-CR50 "Mann DMA, Esiri MM (1989) The pattern of acquisition of plaques and tangles in the brains of patients under 50 years of age with Down’s syndrome. J Neurol Sci 89:169–179. doi:
10.1016/0022-510X(89)90019-1
"), [76](/article/10.1007/s00401-009-0530-3#ref-CR76 "Wisniewski K, Howe J, Williams G, Wisniewski HM (1978) Precocious aging and dementia in patients with Down’s syndrome. Biol Psychiatry 13(5):619–627"), [77](/article/10.1007/s00401-009-0530-3#ref-CR77 "Wisniewski K, Wisniewski H, Wen G (1985) Occurrence of neuropathological changes and dementia of Alzheimer’s disease in Down’s syndrome. Ann Neurol 17:278–282. doi:
10.1002/ana.410170310
")\]. By studying individuals with varying ages at death, the progressive accumulation of Aβ fibrils can be established as occurring early or late in AD pathogenesis.We hypothesized that one conformation of Aβ that may be linked to cognition in human brain is the accumulation of soluble and insoluble fibrils. We previously described a novel antibody, OC, that selectively recognized fibrils and fibrillar oligomers [[35](/article/10.1007/s00401-009-0530-3#ref-CR35 "Kayed R, Head E, Sarsoza F, Saing T, Cotman CW, Necula M et al (2007) Fibril specific, conformation dependent antibodies recognize a generic epitope common to amyloid fibrils and fibrillar oligomers that is absent in prefibrillar oligomers. Mol Neurodegener 2:18. doi: 10.1186/1750-1326-2-18
")\] that are distinct from prefibrillar oligomer assembly states. This new reagent was used to characterize and quantify the extent of fibrillar deposit accumulation in human brain by immunohistochemistry as a function of age (in DS) and dementia status (in AD). We further confirmed the age and disease-specific changes in Aβ fibrils by examining Tg2576 (overexpressing human APP with the Swedish mutation \[[31](/article/10.1007/s00401-009-0530-3#ref-CR31 "Hsaio K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G (1996) Correlative memory deficits, Ab elevation, and amyloid plaques in transgenic mice. Science 274:99–102. doi:
10.1126/science.274.5284.99
")\]) mice with a wide range of ages and quantifying fibril accumulation.Methods
Human autopsy cases
The first group of seven autopsy cases was used to characterize OC-associated pathology (neurofibrillary tangles, dystrophic neurites and glial activation). This study included two control cases (95 and 69 years) and five AD cases (65–89 years). A second group of 14 autopsy cases with MMSE scores ranging between 0 and 30 was used for OC load analyses and to measure associations between cognition and neuropathology (Table 1). Finally, tissues from a group of individuals with DS and similarly aged controls were compared (Table 1). Braak and Braak neurofibrillary tangle and senile plaque staging was provided as part of the standard neuropathology process [[5](/article/10.1007/s00401-009-0530-3#ref-CR5 "Braak H, Braak E (1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82(4):239–259. doi: 10.1007/BF00308809
")–[7](/article/10.1007/s00401-009-0530-3#ref-CR7 "Braak H, Braak E, Bohl J (1993) Staging of Alzheimer-related cortical destruction. Rev Clin Neurosci 33:403–408")\]. A final neuropathology diagnosis was made according to NIA/Reagan criteria \[[67](/article/10.1007/s00401-009-0530-3#ref-CR67 "The National Institute on Aging aRIWGoDCftNAoAsd (1997) Consensus recommendations for the post mortem diagnosis of Alzheimer’s disease. Neurobiol Aging 18(S4):1–2")\].Table 1 Case demographics
Brain tissue samples were obtained from the University of California Alzheimer Disease Research Center (UCI-ADRC) and from the NICHD Brain and Tissue Bank for Developmental Disorders (under contracts N01-4-3368 and N01-HD-4-3383). The hippocampus and midfrontal gyrus was dissected from 4% paraformaldehyde-fixed coronal slices and sectioned at 50 μm using a vibratome. Free-floating sections were stored in PBS with 0.02% sodium azide prior to use in immunohistochemical or histology experiments.
Transgenic mouse tissue
Fixed free-floating 4% paraformaldehyde-fixed sections that included the hippocampus from Tg2576 animals at 3, 6, 9, 12, 18 and 24 months (n = 5/group) were compared with aged wild type animals that were either 18 months (n = 5) or 24 months (n = 5) of age. Brain tissues were obtained from the UCI-ADRC.
Immunohistochemistry
For single-labeling experiments, all sections were washed with 0.1 M Tris-buffered saline (TBS), pH 7.5, and then pretreated with 3% hydrogen peroxide in 10% methanol to block endogenous peroxidase activity. Sections were subsequently washed in TBS with 0.1% Triton X-100 (TBS-A) and then blocked for 30 min in TBS-A with 3% bovine serum albumin (TBS-B). Sections were incubated overnight at room temperature in previously established optimal primary antibody dilutions as indicated in Table 2. Following two washes with TBS-A and a wash in TBS-B, sections were incubated in either goat anti-mouse or goat anti-rabbit biotinylated anti-IgG and then in avidin biotin complex (ABC) (Vector Laboratories, Burlingame, CA, USA). Antibodies were visualized using horseradish peroxidase/3,3′-diaminobenzidine (ABC Elite kit, Vector Laboratories, Burlingame, CA, USA). For quantification experiments, the frontal cortex from all cases used for quantification was immunostained in single experiments to reduce variability in the image analysis procedures and all immunohistochemical procedures were identical for the OC antibody. To determine the specificity of the OC antibody, IgG purified from serum at a concentration of 1:10,000 was preincubated in 10× and 100× concentration of fibrillar Aβ peptide for 2 h at RT. Subsequently, a section from the midfrontal cortex was incubated in the OC antibody/peptide solution and processed for immunohistochemistry.
Table 2 Antibodies used in the study
Immunofluorescence and confocal microscopy
A series of double label immunofluorescence experiments were conducted to determine the colocalization characteristics of OC with other markers of pathology. Primary antibody dilutions are indicated in Table 2. Sections were incubated in OC overnight and then detected using anti-rabbit IgG Alexa Fluor 568 (Molecular Probes, Eugene, OR 1:200). The appropriate second antibody was used to serve as a marker for tau pathology (PHF-1), Aβ (6E10), oligomers (A11), dystrophic neurites (ubiquitin), activated microglial cells (LN-3) and astrocytes (GFAP). In a subset of DS cases, additional double label immunofluorescence was completed using Aβ1-40 and Aβ1-42 antibodies after OC labeling. When possible, antibodies were selected that were raised in different hosts. When not possible, for double labeling experiments where both antibodies were raised in the same species an intervening formaldehyde treatment was used between the first and second immunolabel [[72](/article/10.1007/s00401-009-0530-3#ref-CR72 "Wang B, Larson L (1985) Simultaneous demonstration of multiple antigens by indirect immunofluorescence or immunogold staining. Histochemistry 83:47–56. doi: 10.1007/BF00495299
")\]. Alexa Fluoro 488 (Molecular Probes, Eugene, OR 1:200) was used to visualize the second label (PHF-1, ubiquitin, 6E10, LN-3, GFAP). Sections were allowed to dry on slides prior to rehydration and coverslipping with VectaShield (Vector Laboratories, Burlingame, CA, USA). Confocal images were collected on a BioRad Radiance 2000 Confocal Imaging System with an Olympus IX70 inverted microscope using a 40, 60 or 100× objective for image analysis. Each antibody label was excited and scanned separately using barrier filters at 510 and 480 nm. Channel 1 was used to acquire immunofluorescence with Alexa Fluor 488 and channel 2 was used to collect fluorescent labeling with Alexa Fluor 568\. A z-series scan at either 0.5 or 1 μm intervals was captured to determine the spatial colocalization characteristics of OC with other markers of pathology. A subset of sections was first immunostained for OC and visualization with Alexa Fluor 568 and then counterstained with thioflavin S.Quantification
OC loads
Immunostaining by the OC antibody was captured from a single section for each AD, DS and control case using a 20× objective, a Sony high-resolution CCD video camera (XC-77) and the built-in video capture capabilities of a Macintosh 8100/80AV. A random superficial cortical region containing OC immunostaining in samples that were positive was selected and captured. For control cases, OC immunostaining was scattered and thus the first sample was selected to include positive immunoreactivity if present, or a random area if absent. This sampling approach is slightly modified from that used by Cummings et al. [[13](/article/10.1007/s00401-009-0530-3#ref-CR13 "Cummings BJ, Cotman CW (1995) Image analysis of beta-amyloid “load” in Alzheimer’s disease and relation to dementia severity. Lancet 346:1524–1528. doi: 10.1016/S0140-6736(95)92053-6
")\], in that we did not begin with a sample that appeared to have the highest amount of OC immunoreactivity in AD cases. From that initial sample area, four adjacent superficial fields were captured that were non-overlapping. Subsequently, the tissue was shifted to capture five deep cortical fields that were non-overlapping but adjacent. Thus, a total of 10 fields (including 5 superficial and 5 fields containing deep layers of cortex) were quantified from the midfrontal gyrus for each human case. For the transgenic mouse study, OC loads were obtained from the frontoparietal cortex, the entorhinal cortex and from area CA1 in the hippocampus of individual animals. To do this, two samples from each cortical region was captured by starting above the cingulate cortex and moving laterally for the frontoparietal measures (fields captured included a superficial and deep layer sample), and from the most ventrolateral aspect of the entorhinal cortex and moving dorsally. Fewer samples were taken in mice as the cortical areas were smaller but to maintain consistency with the human study, the same magnification and capturing process was used. For area CA1 in the hippocampus, a single field was captured that contained CA1 neurons as a row of cells through the center of the field. All samples from a given region were captured sequentially during one session. Subsequently, public domain image analysis software (NIH Image 1.55) and a gray-scale threshold of 110 were used to separate positive staining from background and to calculate the percentage of area occupied by OC-positive labeling. The threshold cutoff point was selected to be conservative and resulted in a slight underestimation of the stained areas but minimized the impact of non-specific background reactivity. With this technique, individually captured fields measured 525 × 410 μm, which would allow us to quantify diffuse plaques and neuritic plaques, however, deposits that occupied less than 1% of any given area would not be included due to the thresholding.OC colocalization with neuritic and diffuse plaques
To obtain measures of co-localization between OC-positive immunolabeling and neuritic plaques or diffuse plaques, additional images were analyzed from confocal microscopy experiments. Confocal images captured from three Down syndrome cases and with three controls were used to obtain either neuritic plaque (thioflavin S-positive with OC) or diffuse plaque colocalization (using OC and 6E10 labeled sections in cases that were thioflavin S-negative). One image was captured (using a 20× objective such that 1 pixel = 0.18 μm) for each case for each double labeling experiment. Image J (Rasband, W. S., ImageJ, U. S. National Institutes of Health, Bethesda, MA, USA, http://rsb.info.nih.gov/ij/, 1997–2008) and the JACoP plug in [[4](/article/10.1007/s00401-009-0530-3#ref-CR4 "Bolte S, Cordelieres FP (2006) A guided tour into subcellular colocalization analysis in light microscopy. J Microsc 224(Pt 3):213–232. doi: 10.1111/j.1365-2818.2006.01706.x
")\] was used to obtain Manders’ overlap coefficients, which provides a proportion of the “green signal” coincident with the “red signal”. Images were thresholded prior to quantification by first determining a cut off for the marker (OC or thioflavin or 6E10) that occupied the most area in the image with the second marker automatically set to the same threshold.Statistical analysis
Linear regression analyses using the Huber–White robust standard error estimator [[74](/article/10.1007/s00401-009-0530-3#ref-CR74 "White H (1980) A heteroskedasticity-consistent covariance matrix estimator and a direct test for heteroskedasticity. Econometrica 48:817–830. doi: 10.2307/1912934
")\] were used to evaluate group differences while co-varying for post mortem interval and age at death.Results
OC antibody characteristics
To determine if OC labeling was sensitive to formic acid pretreatment, a procedure used to improve immunostaining for fibrils [39], serial sections from an AD case were pretreated with 0, 10, 50, 70 and 90% formic acid for 4 min prior to incubation in OC antibody. After visualization with anti-rabbit conjugated to Alex Fluor 568, sections were washed and then incubated in 6E10, which shows an increase in immunolabeling with increasing formic acid. Figure 1 shows confocal images from a similar region in each serial section illustrating that increasing formic acid concentration leads to increasingly extensive 6E10 labeling but there is no change in OC labeling. We next examined whether OC immunoreactivity co-localizes with thioflavine-S, a marker for beta-pleated sheet fibrils but that there would also be distinct deposits. Figure 2 shows that some OC-positive deposits are also positive for thioflavin S suggesting an association with mature plaques (63.4% of thioflavin S was positive for OC). However, additional thioflavin-negative deposits were detected by the OC antibody, particularly at the periphery of mature plaques. It is possible that the OC antibody can detect other types of amyloid such as, for example, fibrillar oligomers that are thioflavine S-negative or alpha synuclein [[35](/article/10.1007/s00401-009-0530-3#ref-CR35 "Kayed R, Head E, Sarsoza F, Saing T, Cotman CW, Necula M et al (2007) Fibril specific, conformation dependent antibodies recognize a generic epitope common to amyloid fibrils and fibrillar oligomers that is absent in prefibrillar oligomers. Mol Neurodegener 2:18. doi: 10.1186/1750-1326-2-18
")\]. Further, OC-positive deposits had a distinct morphology from thioflavine S-positive fibrils (Fig. [2](/article/10.1007/s00401-009-0530-3#Fig2)a–c). Using tissue from a nondemented elderly subject (Fig. [2](/article/10.1007/s00401-009-0530-3#Fig2)d–f) and a younger individual with DS (Fig. [2](/article/10.1007/s00401-009-0530-3#Fig2)g–i), both having primarily diffuse plaques (thioflavine S-negative), we observed that diffuse plaques are also OC-positive (9.3% of OC-positive plaques were thioflavin S-positive).Fig. 1
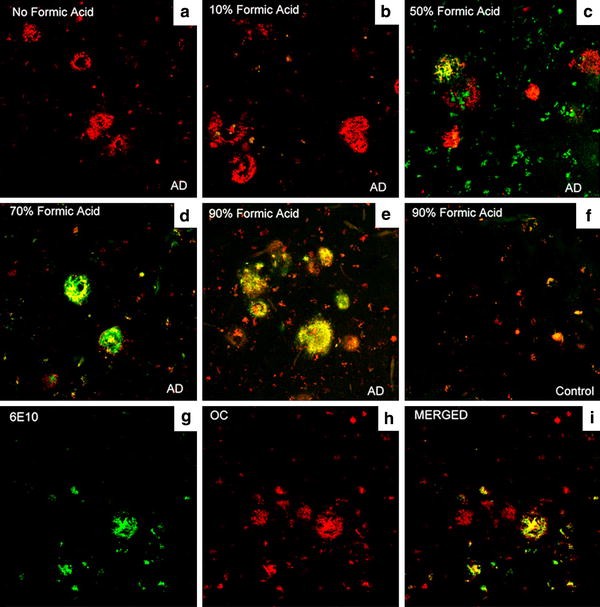
Formic acid pretreatment has little effect on OC labeling intensity. Increasing concentrations of formic acid pretreatment (a none, b 10%, c 50%, d 70%, e 90%) were used in an AD case (frontal cortex) to illustrate no change in OC (red fluorescence) immunolabeling but an increase in 6E10 (green fluorescence) labeling. Note that with increasing formic acid and corresponding increase in 6E10 labeling, the extent of co-localization of the two markers also increases (orange fluorescence). In comparison, a control case shows neither significant OC nor 6E10 immunolabeling (f). However, a subset of OC-positive deposits (g and i) was negative for 6E10 (h and i)
Fig. 2
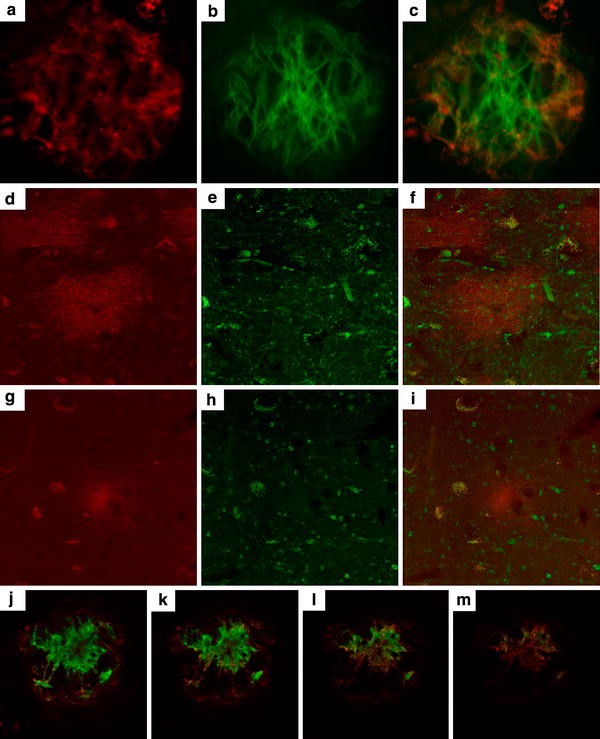
OC immunolabeling is distinct from thioflavine S-positive fibrils within plaques and also identifies diffuse plaque deposits. OC-positive deposits (a) had a distinct morphology that appears as shorter aggregates (red) as compared to thioflavine S-positive fibrils (green) (b). Further, when OC and thioflavine-S was colocalized within plaques (orange), OC labeling was more intense in the periphery (c). Using tissue from a nondemented elderly subject (d–f) and a younger individual with DS (g–i), both having primarily diffuse plaques (thioflavine S-negative—green), we observed that diffuse plaques are also OC-positive (red). A z-scan shows that OC-positive deposits (green) could also be distinguished from A11-positive oligomers (red) in mature plaques observed in DS frontal cortex
Additional experiments in DS and control cases were used to measure the overlap in immunostaining between OC and Aβ40 or Aβ42. When both OC and Aβ40 were present, we observed a range of colocalization from 17.6 to 62.5% of OC-positive deposits containing Aβ40. However, we observed individual plaques that were Aβ40-positive and OC-negative. Similarly for OC and Aβ42, when both were present, OC occupied 38.5% of Aβ42-positive plaques. However, we observed Aβ42-positive plaques that were also negative for OC. Thus, in this small set of samples, we observed both Aβ40 and Aβ42-positive plaques that were negative for OC but when both are present, there was a higher association with OC to Aβ40. OC-positive deposits could also be distinguished from A11-positive oligomers in mature plaques observed in DS (Fig. 2j–m).
OC-positive deposits were characterized further to determine if they were associated with dystrophic neurites and glial cell responses (Fig. 3). OC-positive deposits contain PHF-1 (Fig. 3a) and ubiquitin (Fig. 3c) positive dystrophic neurites but a subset were negative (Fig. 3b). Microglial activation (Fig. 3d, e) was associated with a subset of OC-positive fibrils and a subset of microglia appear to have phagocytosed OC-positive material within vacuoles (Fig. 3f). OC-positive deposits were also surrounded by hypertrophic astrocytes (Fig. 3g, h).
Fig. 3
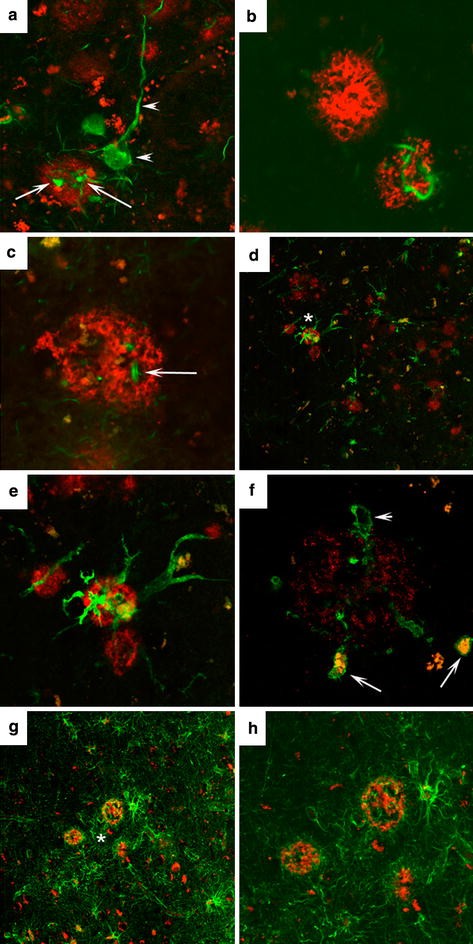
Characteristics of OC-positive deposits. A subset of OC deposits (red) contained dystrophic neurites labeled by PHF (green arrows) that could be distinguished from tangle bearing neurons (arrowheads) (a). Not all OC (red) deposits contained PHF positive dystrophic neurites (green) (b). Dystrophic neurites labeled with ubiquitin (green) were also found in association with a subset of OC deposits (red) (c). A subset of OC (red) deposits was associated with activated microglial (LN-3 green) (d) Area in d indicated by asterisk at higher magnification illustrating two activated microglial cells (green) surrounding an OC-positive deposit (red) (e). A subset of activated microglial cells (green) appear to have engulfed OC-positive material (arrows red/orange) whereas other microglial cells appear OC-negative (arrowhead) (f). OC-positive deposits (red) are surrounded by astrocytes (green) (g). A higher magnification of the region in g indicated by the asterisk shows that some astrocytes in proximity to OC appear hypertrophied (h)
OC immunostaining in AD and controls
To test the hypothesis that the extent of OC-positive fibrils was higher in AD cases relative to controls, and intermediate within those cases with select cognitive impairments (cognitively impaired not demented—CIND or MCI), OC labeled frontal cortex sections were quantified. Robust linear regression was used to test for group differences, adjusting for PMI and age at death. After adjusting for PMI and age, there was a statistically significantly higher OC percentage load in AD patients compared to controls (p value = 0.002, 95% CI for difference = 12.09, 37.56) (Fig. 4a). Significant differences in OC load were observed as a function of Braak and Braak tangle staging primarily due to higher levels in Braak stage V/VI (p = 0.002)(data not shown). Figure 4b shows the results of a linear regression that included the interval between last clinic visit and death, that as dementia severity increases (low MMSE score) there was a corresponding increase in OC load (p = 0.0007).
Fig. 4
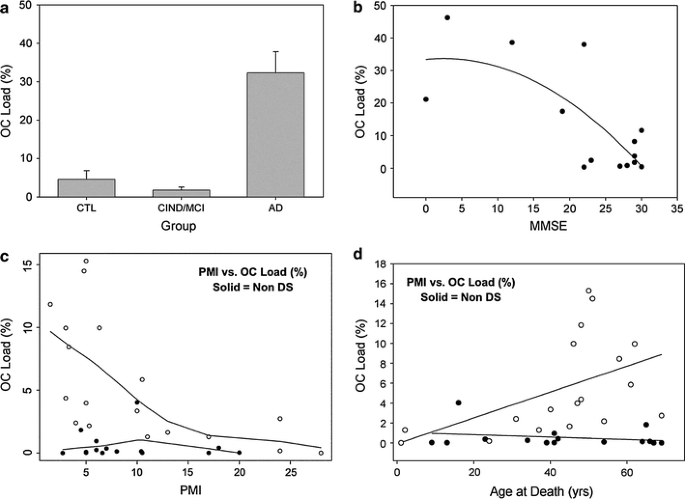
The extent of OC labeling in AD and DS brain. More extensive OC immunoreactivity was observed in the AD cases relative to both the control and MCI/CIND cases (a). Higher MMSE scores were associated with less extensive OC immunolabeling (r = −0.72, p < 0.001) (b). In DS cases used in experiment 2, OC labeling appears to be significantly reduced when the PMI is over 10 h (c). A significant increase in OC was observed in adults with DS over the age of 40 years (open circles d) but not in similarly aged non-DS controls (closed circles). When OC loads are plotted as a function of individual age, there is an exponential rise in Aβ fibril accumulation after the age of 40 years in DS (open circles) but not in non-DS controls (closed circles) (d). Bars represent means and error bars, standard errors of the mean. In b–d, the lines represent a best fit function
OC in Down syndrome
We next asked the question whether OC immunoreactivity accumulates as a function of age in DS cases, where typically, virtually all cases over the age of 40 years have sufficient pathology for a diagnosis of AD [[50](/article/10.1007/s00401-009-0530-3#ref-CR50 "Mann DMA, Esiri MM (1989) The pattern of acquisition of plaques and tangles in the brains of patients under 50 years of age with Down’s syndrome. J Neurol Sci 89:169–179. doi: 10.1016/0022-510X(89)90019-1
")\]. We first tested the hypothesis that PMI was not a significant contributor to OC load. However, we observed a significant association of PMI with OC load (_p_ \= 0.001) with longer PMI’s associated with a loss of OC immunoreactivity and a 2.5% decrease in OC labeling with each 10-h block of PMI (Fig. [4](/article/10.1007/s00401-009-0530-3#Fig4)c). We further found a significant interaction between age and genotype group (DS vs. non-DS) with aged DS individuals having higher overall levels of OC labeling (_p_ \= 0.0004) (Fig. [4](/article/10.1007/s00401-009-0530-3#Fig4)d).OC in Tg2576 mice
Age-associated increases in OC labeling in individuals with DS may be due to overexpression of the human APP gene. To confirm and extend this hypothesis, we used tissue from Tg2576 mice, which overexpress mutant human APP [[31](/article/10.1007/s00401-009-0530-3#ref-CR31 "Hsaio K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G (1996) Correlative memory deficits, Ab elevation, and amyloid plaques in transgenic mice. Science 274:99–102. doi: 10.1126/science.274.5284.99
")\]. In a study of the brains of 30 Tg2576 animals, we find a significant main effect of age on OC loads in the frontoparietal cortex (_F_(5,29) = 5.90, _p_ \= 0.001), in the entorhinal cortex (_F_(5,29) = 9.91, _p_ < 0.0005) and in area CA1 of the hippocampus (_F_(5,29) = 9.94, _p_ < 0.0005). Figure [5](/article/10.1007/s00401-009-0530-3#Fig5) shows that as with DS cases, there was a sudden rise in the extent of OC labeling after the age of 12 months in Tg2576 animals. Initial deposits were observed at 6 months in one animal and in 4/5 animals at 9 months of age. When 18- and 24-month old Tg2576 mice were directly compared to wild type animals of the same ages, the latter group had undetectable amounts of OC-positive deposits (data not shown).Fig. 5
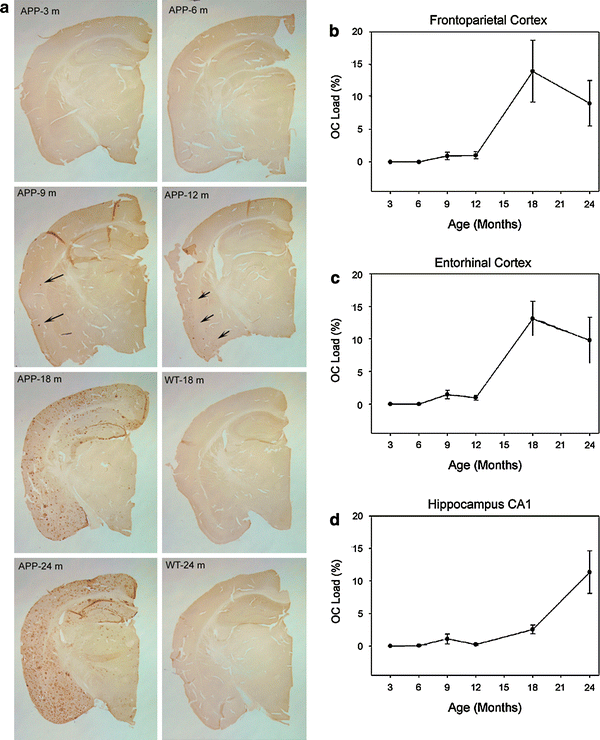
OC fibril labeling in Tg2576 animals. a Tg2576 animals show increasing OC accumulation with age while no OC was observed in comparably aged wild type animals. OC loads were obtained from the b frontoparietal cortex, c entorhinal cortex and d from area CA1 in the hippocampus of individual animals. Mean loads are plotted as a function of age for each brain region and show a dramatic increase after 12 months of age in cortical regions but with a slower more progressive rise in the hippocampus. Error bars represent standard errors of the mean (n = 5 animals/age group)
Discussion
Using a newly developed conformation-dependent, fibril-specific, polyclonal antibody [[35](/article/10.1007/s00401-009-0530-3#ref-CR35 "Kayed R, Head E, Sarsoza F, Saing T, Cotman CW, Necula M et al (2007) Fibril specific, conformation dependent antibodies recognize a generic epitope common to amyloid fibrils and fibrillar oligomers that is absent in prefibrillar oligomers. Mol Neurodegener 2:18. doi: 10.1186/1750-1326-2-18
")\] that recognizes a generic epitope associated with fibrils and soluble fibrillar oligomers we have characterized AD and age-associated changes in Aβ fibril accumulation. Several technical aspects to the use of the OC antibody were identified in the current study. First, formic acid pretreatment, typically used to enhance Aβ immunostaining \[[39](/article/10.1007/s00401-009-0530-3#ref-CR39 "Kitamoto T, Ogomori K, Tateishi J, Prusiner SB (1987) Formic acid pretreatment enhances immunostaining of cerebral and systemic amyloids. Lab Invest 57(2):230–236")\] does not significantly improve OC labeling. This suggests that the epitope/conformation recognized by the OC antibody is not obscured when fibrils form and have not adopted a full beta-pleated sheet assembly state. Second, based on our studies in DS brain, post mortem interval may be a key factor to consider when using this antibody. Longer postmortem intervals (>10 h) may be associated with a degradation of the signal, possibly related to protease activity. Whether this represents a loss of tissue morphology, a spontaneous disassembly of fibrils or oligomeric protofibrils or the opposite, a conversion into beta-pleated sheet fibrils post mortem, is difficult to determine. Further, a PMI effect on OC immunolabeling may potentially distinguish sporadic AD deposits from DS with AD deposits and there is some evidence that the properties of Aβ in these two groups may behave differently \[[1](/article/10.1007/s00401-009-0530-3#ref-CR1 "Armstrong RA (1999) Do beta-amyloid (Abeta) deposits in patients with Alzheimer’s disease and Down’s syndrome grow according to the log-normal model? Neurosci Lett 261(1–2):97–100. doi:
10.1016/S0304-3940(99)00009-9
")\].OC-positive fibrillar deposits accumulate in human brain as overlapping deposits with thioflavine-S plaques but are also found in deposits that are diffuse, i.e. thioflavine S-negative. By examining the brains of individuals with and without dementia, we show increased fibrillar accumulation in AD but when compared to controls. However, CIND/MCI cases showed similar OC immunolabeling as controls. Further, the extent of fibril accumulation in AD, CIND/MCI and control brain correlates with the severity of cognitive decline measured by MMSE scores. Similarly, in individuals with DS, we observe fibril accumulation that is both age- and AD neuropathology-associated but with a rapid rise in levels after the age of 40 years. Finally, in one of the most commonly used animal models of Aβ pathogenesis, we find a rapid age-dependent accumulation of fibrils that is first detectable at 6 months of age but dramatically increases after 12 months of age after overexpression of mutant human APP in the Tg2576 model system. Thus, OC-positive deposits may be visible histologically at an earlier age than Aβ-positive deposits reported previously [34] and may represent an early neuropathological feature more consistent with the onset of behavioral dysfunction [[31](/article/10.1007/s00401-009-0530-3#ref-CR31 "Hsaio K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G (1996) Correlative memory deficits, Ab elevation, and amyloid plaques in transgenic mice. Science 274:99–102. doi: 10.1126/science.274.5284.99
")\].OC-positive deposits that are thioflavine S-negative may reflect fibrillar oligomers. This is consistent with reports that protofibrillar forms of Aβ have low thioflavine-T response [[75](/article/10.1007/s00401-009-0530-3#ref-CR75 "Williams AD, Sega M, Chen M, Kheterpal I, Geva M, Berthelier V et al (2005) Structural properties of A beta protofibrils stabilized by a small molecule. Proc Natl Acad Sci USA 102(20):7115–7120. doi: 10.1073/pnas.0408582102
")\]. The morphology of the OC-positive, thioflavine S-negative deposits are distinct and appear to be shorter and wider than thioflavine S-positive fibrils. In addition, we frequently find OC-positive deposits at the periphery of thioflavine S-positive plaques providing further evidence that soluble fibrillar oligomers may represent a reservoir for fibril formation \[[35](/article/10.1007/s00401-009-0530-3#ref-CR35 "Kayed R, Head E, Sarsoza F, Saing T, Cotman CW, Necula M et al (2007) Fibril specific, conformation dependent antibodies recognize a generic epitope common to amyloid fibrils and fibrillar oligomers that is absent in prefibrillar oligomers. Mol Neurodegener 2:18. doi:
10.1186/1750-1326-2-18
")\]. OC, interestingly, was also able to detect diffuse plaques characterized as being thioflavine S-negative but also containing intact neurons. This may suggest that the pathogenesis of diffuse plaques involves the accumulation of thioflavine S-negative fibrils and soluble fibrillar oligomers. Interestingly, a similar conclusion was made in an earlier study by Davies and Mann \[[16](/article/10.1007/s00401-009-0530-3#ref-CR16 "Davies CA, Mann DMA (1993) Is the “Preamyloid” of diffuse plaques in Alzheimer’s disease really nonfibrillar? Am J Pathol 143(6):1594–1605")\] based on electron microscopy in biopsy samples from AD cases showing diffuse plaques contained fibrillar material. Parallel studies in the canine model, that only develop diffuse Aβ plaques also suggests that these early deposits are fibrillar in nature \[[68](/article/10.1007/s00401-009-0530-3#ref-CR68 "Torp R, Head E, Milgram NW, Hahn F, Ottersen OP, Cotman CW (2000) Ultrastructural evidence of fibrillar β-amyloid associated with neuronal membranes in behaviorally characterized aged dog brains. Neuroscience 93(3):495–506. doi:
10.1016/S0306-4522(99)00568-0
")\]. Indeed, diffuse plaques, though to be one of the earlier plaque subtypes accumulating Aβ pathology \[[27](/article/10.1007/s00401-009-0530-3#ref-CR27 "Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297(5580):353–356. doi:
10.1126/science.1072994
")\], contain primarily the longer toxic Aβ1-42 peptide \[[15](/article/10.1007/s00401-009-0530-3#ref-CR15 "Cummings BJ, Satou T, Head E, Milgram NW, Cole GM, Savage MJ et al (1996) Diffuse plaques contain C-terminal A beta 42 and not A beta 40: evidence from cats and dogs. Neurobiol Aging 17(4):653–659"), [26](/article/10.1007/s00401-009-0530-3#ref-CR26 "Gravina SA, Ho L, Eckman CB, Long KE, Otvos L, Younkin LH, Suzuki N, Younkin SG (1995) Amyloid β-protein (Aβ) in Alzheimer’s disease brain. Biochemical and immunocytochemical analysis with antibodies specific for forms of Aβ(40) and Aβ42(43). J Biol Chem 270:7013–7016. doi:
10.1074/jbc.270.13.7013
"), [32](/article/10.1007/s00401-009-0530-3#ref-CR32 "Iwatsubo T, Mann DM, Odaka A, Suzuki N, Ihara Y (1995) Amyloid beta protein (A beta) deposition: A beta 42(43) precedes A beta 40 in Down syndrome. Ann Neurol 37(3):294–299. doi:
10.1002/ana.410370305
")\], are observed at the threshold of detectable dementia \[[56](/article/10.1007/s00401-009-0530-3#ref-CR56 "Morris JC, Storandt M, McKeel DW Jr, Rubin EH, Price JL, Grant EA et al (1996) Cerebral amyloid deposition and diffuse plaques in “normal” aging: evidence for presymptomatic and very mild Alzheimer’s disease. Neurology 46(3):707–719")\], can occur after traumatic brain injury leaving affected patients at higher risk for developing AD \[[17](/article/10.1007/s00401-009-0530-3#ref-CR17 "DeKosky ST, Abrahamson EE, Ciallella JR, Paljug WR, Wisniewski SR, Clark RS et al (2007) Association of increased cortical soluble abeta42 levels with diffuse plaques after severe brain injury in humans. Arch Neurol 64(4):541–544. doi:
10.1001/archneur.64.4.541
")\] and may reflect synaptic activity induced Aβ production and release into the interstitial space \[[10](/article/10.1007/s00401-009-0530-3#ref-CR10 "Cirrito JR, Kang JE, Lee J, Stewart FR, Verges DK, Silverio LM et al (2008) Endocytosis is required for synaptic activity-dependent release of amyloid-beta in vivo. Neuron 58(1):42–51. doi:
10.1016/j.neuron.2008.02.003
")\]. Further, individuals with early onset AD caused by a missense mutation in APP (T714I), develop primarily diffuse Aβ deposits and an aggressive form of dementia \[[40](/article/10.1007/s00401-009-0530-3#ref-CR40 "Kumar-Singh S, De Jonghe C, Cruts M, Kleinert R, Wang R, Mercken M et al (2000) Nonfibrillar diffuse amyloid deposition due to a gamma(42)-secretase site mutation points to an essential role for N-truncated A beta(42) in Alzheimer’s disease. Hum Mol Genet 9(18):2589–2598. doi:
10.1093/hmg/9.18.2589
")\]. In combination, diffuse plaques may not be entirely benign as has been suggested previously \[[51](/article/10.1007/s00401-009-0530-3#ref-CR51 "Masliah E, Terry RD, Mallory M, Alford M, Hansen LA (1990) Diffuse plaques do not accentuate synapse loss in Alzheimer’s disease. Am J Pathol 137(6):1293–1297")\] although they may be “clinically silent”.When OC is detected in more compact plaques, we observe dystrophic neurites, microglial and astrocyte involvement similar to previous reports of associated neurodegeneration in these more mature plaque subtypes [[19](/article/10.1007/s00401-009-0530-3#ref-CR19 "Dickson DW (1997) The pathogenesis of senile plaques. J Neuropathol Exp Neurol 56(4):321–339. doi: 10.1097/00005072-199704000-00001
")\]. Further, microglial cells may engulf OC-positive deposits for degradation as we observe OC-positive material within phagocytic vacuoles within HLA-DR expressing microglial cells. We have observed a similar phenomenon with oxidatively modified Aβ \[[29](/article/10.1007/s00401-009-0530-3#ref-CR29 "Head E, Garzon-Rodriguez W, Johnson JK, Lott IT, Cotman CW, Glabe C (2001) Oxidation of Aβ and plaque biogenesis in Alzheimer’s disease and Down syndrome. Neurobiol Dis 8:792–806. doi:
10.1006/nbdi.2001.0431
")\] and others have reported Aβ within microglial cells in vitro and within AD brain previously \[[21](/article/10.1007/s00401-009-0530-3#ref-CR21 "El Khoury J, Hickman SE, Thomas CA, Cao L, Silverstein SC, Loike JD (1996) Scavenger receptor mediated adhesion of microglia to beta-amyloid fibrils. Nature 382:716–719. doi:
10.1038/382716a0
"), [22](/article/10.1007/s00401-009-0530-3#ref-CR22 "El Khoury J, Hickman SE, Thomas CA, Loike JD, Sliverstein SC (1998) Microglia, scavenger receptors and the pathogenesis of Alzheimer’s disease. Neurobiol Aging 19:S81. doi:
10.1016/S0197-4580(98)00036-0
"), [60](/article/10.1007/s00401-009-0530-3#ref-CR60 "Paresce DM, Chung H, Maxfield FR (1997) Slow degradation of aggregates of the Alzheimer’s disease amyloid β-protein by microglial cells. J Biol Chem 272(46):29390–29397. doi:
10.1074/jbc.272.46.29390
"), [61](/article/10.1007/s00401-009-0530-3#ref-CR61 "Paresce DM, Gosh R, Maxfield FR (1996) Microglial cells internalize aggregates of the Alzheimer’s disease amyloid β-protein via a scavenger receptor. Neuron 17:553–565. doi:
10.1016/S0896-6273(00)80187-7
"), [65](/article/10.1007/s00401-009-0530-3#ref-CR65 "Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Liao Z, Lieberburg I, Motter R, Mutter L, Soriano F, Shopp G, Vasquez N, Vandervert C, Walker S, Wogulis M, Yednock T, Games D, Seubert P (1999) Immunization with amyloid-β attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature 400:173–177. doi:
10.1038/22124
")\]. A critical role for microglial cells in clearing Aβ from the brain has been established in transgenic mice \[[3](/article/10.1007/s00401-009-0530-3#ref-CR3 "Bolmont T, Haiss F, Eicke D, Radde R, Mathis CA, Klunk WE et al (2008) Dynamics of the microglial/amyloid interaction indicate a role in plaque maintenance. J Neurosci 28(16):4283–4292. doi:
10.1523/JNEUROSCI.4814-07.2008
")\] and the results from this study also suggest efforts by microglial cells to clear soluble and insoluble fibrils.OC fibrils in AD
We next used the OC antibody in studies to quantify the extent of fibril formation in individuals with and without AD. OC-positive deposits were ~7-fold higher in AD brain relative to controls. However, based on previous studies in the brains of individuals with MCI [[57](/article/10.1007/s00401-009-0530-3#ref-CR57 "Morris JC, Storandt M, Miller JP, McKeel DW, Price JL, Rubin EH et al (2001) Mild cognitive impairment represents early-stage Alzheimer disease. Arch Neurol 58(3):397–405. doi: 10.1001/archneur.58.3.397
")\], we also predicted that subjects with MCI or with a cognitive impairment although not demented, may be either equivalent or intermediate with respect to extent of OC-positive deposits. However, we found that OC fibrils were similar in MCI/CIND as compared to nondemented aged controls. There are two possible interpretations of these data. The first is that given our sample consisted primarily of CIND subjects; there is a less clear relationship between CIND and progression to dementia as in MCI \[[62](/article/10.1007/s00401-009-0530-3#ref-CR62 "Petersen RC, Smith GE, Waring SC, Ivnik RJ, Tangalos EG, Kokmen E (1999) Mild cognitive impairment: clinical characterization and outcome. Arch Neurol 56(3):303–308. doi:
10.1001/archneur.56.3.303
")\]. Second, fibrils may be more closely associated with full-blown dementia rather than a possible pro-dromal selective clinical impairment. By immunohistochemistry, we may also be detecting primarily fibrils as opposed to soluble oligomeric fibrils, the latter may be optimally detected using biochemical methods \[[35](/article/10.1007/s00401-009-0530-3#ref-CR35 "Kayed R, Head E, Sarsoza F, Saing T, Cotman CW, Necula M et al (2007) Fibril specific, conformation dependent antibodies recognize a generic epitope common to amyloid fibrils and fibrillar oligomers that is absent in prefibrillar oligomers. Mol Neurodegener 2:18. doi:
10.1186/1750-1326-2-18
")\]. Measures of soluble fibrillar oligomers from protein extracts of frozen tissue in these cases may be more sensitive and reveal higher levels of OC in MCI/CIND cases relative to controls because they have the potential of distinguishing insoluble fibrils and soluble fibrillar oligomers; both of which react with OC. Another important consideration is that the current study focused on plaque pathology in the frontal cortex and had we sampled regions in other vulnerable areas, particularly the entorhinal cortex (which was not available for all cases), we may have observed OC levels that are more consistent between MCI/CIND cases and AD cases.In AD brain, there are various reports of a correlation between the extent of Aβ deposition in plaques and the severity of cognitive decline. For example, several studies show a significant inverse correlation with MMSE scores, suggesting that lower scores associated with dementia are linked to more extensive plaque formation [[2](/article/10.1007/s00401-009-0530-3#ref-CR2 "Blessed G, Tomlinson BE, Roth M (1968) The association between quantitative measures of dementia and of senile change in the cerebral grey matter of elderly subjects. Br J Psychiatry 114:797–811. doi: 10.1192/bjp.114.512.797
"), [13](/article/10.1007/s00401-009-0530-3#ref-CR13 "Cummings BJ, Cotman CW (1995) Image analysis of beta-amyloid “load” in Alzheimer’s disease and relation to dementia severity. Lancet 346:1524–1528. doi:
10.1016/S0140-6736(95)92053-6
"), [20](/article/10.1007/s00401-009-0530-3#ref-CR20 "Dickson DW, Crystal HA, Bevona C, Honer W, Vincent I, Davies P (1995) Correlations of synaptic and pathological markers with cognition of the elderly. Neurobiol Aging 16(3):285–304. doi:
10.1016/0197-4580(95)00013-5
"), [58](/article/10.1007/s00401-009-0530-3#ref-CR58 "Nelson PT, Jicha GA, Schmitt FA, Liu H, Davis DG, Mendiondo MS et al (2007) Clinicopathologic correlations in a large Alzheimer disease center autopsy cohort: neuritic plaques and neurofibrillary tangles “do count” when staging disease severity. J Neuropathol Exp Neurol 66(12):1136–1146. doi:
10.1097/nen.0b013e31815c5efb
")\]. In contrast, additional studies do not show this relationship and/or find that neurofibrillary tangles or synapse protein loss, other key pathological features of AD, are also tightly linked to cognition \[[12](/article/10.1007/s00401-009-0530-3#ref-CR12 "Cummings BJ (1997) Plaques and tangles: searching for primary events in a forest of data. Neurobiol Aging 18(4):358–362. doi:
10.1016/S0197-4580(97)00049-3
"), [14](/article/10.1007/s00401-009-0530-3#ref-CR14 "Cummings BJ, Pike CJ, Shankle R, Cotman CW (1996) Beta-amyloid deposition and other measures of neuropathology predict cognitive status in Alzheimer’s disease. Neurobiol Aging 17(6):921–933. doi:
10.1016/S0197-4580(96)00170-4
"), [23](/article/10.1007/s00401-009-0530-3#ref-CR23 "Giannakopoulos P, Herrmann FR, Bussiere T, Bouras C, Kovari E, Perl DP et al (2003) Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer’s disease. Neurology 60(9):1495–1500"), [52](/article/10.1007/s00401-009-0530-3#ref-CR52 "McKee AC, Kosik KS, Kowall NW (1991) Neuritic pathology and dementia in Alzheimer’s disease. Ann Neurol 30:156–165. doi:
10.1002/ana.410300206
"), [58](/article/10.1007/s00401-009-0530-3#ref-CR58 "Nelson PT, Jicha GA, Schmitt FA, Liu H, Davis DG, Mendiondo MS et al (2007) Clinicopathologic correlations in a large Alzheimer disease center autopsy cohort: neuritic plaques and neurofibrillary tangles “do count” when staging disease severity. J Neuropathol Exp Neurol 66(12):1136–1146. doi:
10.1097/nen.0b013e31815c5efb
"), [66](/article/10.1007/s00401-009-0530-3#ref-CR66 "Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R et al (1991) Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol 30(4):572–580. doi:
10.1002/ana.410300410
")\]. Various interpretations of these results have been suggested but one hypothesis is that assembly state of Aβ may be a critical factor in determining its toxicity and thus the link to impaired cognition. The current study suggests a significant correlation between OC-positive fibrillar deposits and MMSE scores with higher scores associated with intact cognition linked to lower levels of OC. Interestingly, we may be observing a threshold effect as OC loads increase significantly when MMSE scores drop below 20 but this should be confirmed in a larger cohort of cases.OC fibrils in Down syndrome
In the most common form of DS, trisomy 21, chromosome 21 is present in triplicate and leads to lifelong overexpression of the APP gene (21q21.2) [64]. Despite this life-long overexpression of APP in brain and in peripheral lymphocytes [[59](/article/10.1007/s00401-009-0530-3#ref-CR59 "Pallister C, Jung SS, Shaw I, Nalbantoglu J, Gauthier S, Cashman NR (1997) Lymphocyte content of amyloid precursor protein is increased in Down’s syndrome and aging. Neurobiol Aging 18(1):97–103. doi: 10.1016/S0197-4580(96)00207-2
"), [64](/article/10.1007/s00401-009-0530-3#ref-CR64 "Rumble B, Retallack R, Hilbich C, Simms G, Multhaup G, Martins R et al (1989) Amyloid A4 and its precursor in Down’s syndrome and Alzheimer’s disease. N Engl J Med 320:1446–1462")\], Aβ accumulation in plaques does not typically begin until after the age of 30 years \[[50](/article/10.1007/s00401-009-0530-3#ref-CR50 "Mann DMA, Esiri MM (1989) The pattern of acquisition of plaques and tangles in the brains of patients under 50 years of age with Down’s syndrome. J Neurol Sci 89:169–179. doi:
10.1016/0022-510X(89)90019-1
")\], although there have been reports of plaques in younger individuals \[[43](/article/10.1007/s00401-009-0530-3#ref-CR43 "Lemere CA, Blusztajn JK, Yamaguchi H, Wisniewski T, Saido TC, Selkoe DJ (1996) Sequence of deposition of heterogeneous amyloid beta-peptides and APOE in Down syndrome: implications for initial events in amyloid plaque formation. Neurobiol Dis 3:16–32. doi:
10.1006/nbdi.1996.0003
"), [45](/article/10.1007/s00401-009-0530-3#ref-CR45 "Leverenz JB, Raskind MA (1998) Early amyloid deposition in the medial temporal lobe of young Down syndrome patients: a regional quantitative analysis. Exp Neurol 150:296–304. doi:
10.1006/exnr.1997.6777
")\]. Interestingly, the incidence of dementia typically does not increase until adults with DS are over the age of 50 years \[[41](/article/10.1007/s00401-009-0530-3#ref-CR41 "Lai F, Williams MD (1989) A prospective study of Alzheimer disease in Down syndrome. Arch Neurol 46:849–853")\], suggesting the possibility that either the brain can compensate for significant Aβ accumulation or that Aβ must adopt a specific conformation to become neurotoxic.To test this hypothesis, we measured OC loads in individuals with DS with a range of ages. In the second experiment, we observed an age-associated increase in OC in DS that was not observed in similarly aged non-DS subjects without dementia. In DS, it is possible to differentiate initiation from acceleration phases of AD pathogenesis. Between the ages of 30–40 years, the first signs of Aβ plaques appear as diffuse deposits [47, [48](/article/10.1007/s00401-009-0530-3#ref-CR48 "Mann DM, Marcyniuk B, Yates PO, Neary D, Snowden JS (1988) The progression of the pathological changes of Alzheimer’s disease in frontal and temporal neocortex examined both at biopsy and at autopsy. Neuropathol Appl Neurobiol 14(3):177–195. doi: 10.1111/j.1365-2990.1988.tb00880.x
")\]. Between 40 and 50 years, neurofibrillary tangles also form and neuritic plaques appear suggesting an acceleration phase. We may also consider from 50 years and older as when functional declines may occur \[[41](/article/10.1007/s00401-009-0530-3#ref-CR41 "Lai F, Williams MD (1989) A prospective study of Alzheimer disease in Down syndrome. Arch Neurol 46:849–853")\]. If we classify adults with DS into these groups, OC-positive fibrils are found as early as the initiation phase suggesting a key role in Aβ pathogenesis in DS. Despite life-long overexpression of APP, the brains of individuals with DS under the age of 30 years were negative for fibrils, however, a larger sample of individuals between 20 and 30 years would extend these findings. Thus, OC-positive deposits appear prior to the age at which dementia prevalence rises in DS and may play a role in AD pathogenesis in DS.OC fibrils in Tg2576
Similar to individuals with DS, transgenic mouse models that overexpress mutant human APP develop Aβ pathology in a progressive age-dependent manner [[31](/article/10.1007/s00401-009-0530-3#ref-CR31 "Hsaio K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G (1996) Correlative memory deficits, Ab elevation, and amyloid plaques in transgenic mice. Science 274:99–102. doi: 10.1126/science.274.5284.99
"), [34](/article/10.1007/s00401-009-0530-3#ref-CR34 "Kawarabayashi T, Younkin LH, Saido TC, Shoji M, Ashe KH, Younkin SG (2001) Age-dependent changes in brain, CSF, and plasma amyloid β protein in the Tg2576 transgenic mouse model of Alzheimer’s disease. J Neurosci 21(2):372–381")\]. In a systematic study of the different types and assembly states of Aβ that accumulate in Tg2576 mice, Kawarabayashi and colleagues \[[34](/article/10.1007/s00401-009-0530-3#ref-CR34 "Kawarabayashi T, Younkin LH, Saido TC, Shoji M, Ashe KH, Younkin SG (2001) Age-dependent changes in brain, CSF, and plasma amyloid β protein in the Tg2576 transgenic mouse model of Alzheimer’s disease. J Neurosci 21(2):372–381")\] showed that Aβ in the form of plaques appear as early as 7–8 months whereas diffuse plaques appear between 12 and 15 months. However, Aβ extracted with formic acid can be detected using biochemical approaches younger ages between 6 and 8 months. Further, Aβ oligomers are detected in older animals \~12 months of age as variably being increased \[[34](/article/10.1007/s00401-009-0530-3#ref-CR34 "Kawarabayashi T, Younkin LH, Saido TC, Shoji M, Ashe KH, Younkin SG (2001) Age-dependent changes in brain, CSF, and plasma amyloid β protein in the Tg2576 transgenic mouse model of Alzheimer’s disease. J Neurosci 21(2):372–381")\] or decreased \[[44](/article/10.1007/s00401-009-0530-3#ref-CR44 "Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A et al (2006) A specific amyloid-beta protein assembly in the brain impairs memory. Nature 440(7082):352–357. doi:
10.1038/nature04533
")\]. However, behavioral impairments may appear prior to significant Aβ plaque pathology as early as 3 months \[[38](/article/10.1007/s00401-009-0530-3#ref-CR38 "King DL, Arendash GW, Crawford F, Sterk T, Menendez J, Mullan MJ (1999) Progressive and gender-dependent cognitive impairment in the APP(SW) transgenic mouse model for Alzheimer’s disease. Behav Brain Res 103(2):145–162. doi:
10.1016/S0166-4328(99)00037-6
")\], 6 months of age \[[73](/article/10.1007/s00401-009-0530-3#ref-CR73 "Westerman MA, Cooper-Blacketer D, Mariash A, Kotilinek L, Kawarabayashi T, Younkin LH, Carlson GA, Younkin SG, Ashe KH (2002) The relationship between Aβ and memory in the Tg2576 mouse model of Alzheimer’s disease. J Neurosci 22(5):1858–1867")\] or older \[[11](/article/10.1007/s00401-009-0530-3#ref-CR11 "Corcoran KA, Lu Y, Turner RS, Maren S (2002) Overexpression of hAPPswe impairs rewarded alternation and contextual fear conditioning in a transgenic mouse model of Alzheimer’s disease. Learn Mem 9(5):243–252. doi:
10.1101/lm.51002
"), [31](/article/10.1007/s00401-009-0530-3#ref-CR31 "Hsaio K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G (1996) Correlative memory deficits, Ab elevation, and amyloid plaques in transgenic mice. Science 274:99–102. doi:
10.1126/science.274.5284.99
"), [54](/article/10.1007/s00401-009-0530-3#ref-CR54 "Morgan D, Diamond DM, Gottschall PE, Ugen KE, Dickey C, Hardy J et al (2000) A beta peptide vaccination prevents memory loss in an animal model of Alzheimer’s disease. Nature 408(6815):982–985. doi:
10.1038/35050116
")\]. The discrepancy between the onset of behavioral dysfunction in Tg2576 mice and the age at which Aβ plaques first appear suggests a critical role for different assembly states of Aβ on neuronal function. The earliest age at which we consistently observed OC-positive deposits in the Tg2576 mice in the current study was 9 months, with one animal at 6 months of age showing pathology, but this does not rule out soluble fibrillar oligomers, which may not be detected by immunohistochemistry, from being a contributor to neuronal dysfunction. Indeed, Aβ\*56 oligomers that appear as early as 7 months are A11-positive, indicating that they are prefibrillar oligomers that would not be expected to react with OC \[[44](/article/10.1007/s00401-009-0530-3#ref-CR44 "Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A et al (2006) A specific amyloid-beta protein assembly in the brain impairs memory. Nature 440(7082):352–357. doi:
10.1038/nature04533
")\].In summary, using a novel antibody that detects amyloid fibrils and soluble oligomeric protofibrils we show a differential distribution from beta-pleated sheet fibrils and association with AD in both the general population and in adults with DS. Further, OC-positive fibrils also accumulate in Tg2576 mice suggesting that several of the dynamics involved with production and accumulation of fibrils may be modeled in this animal. It would be interesting to selectively reduce OC-positive fibrils through a vaccination approach to determine their functional impact.
References
- Armstrong RA (1999) Do beta-amyloid (Abeta) deposits in patients with Alzheimer’s disease and Down’s syndrome grow according to the log-normal model? Neurosci Lett 261(1–2):97–100. doi:10.1016/S0304-3940(99)00009-9
Article PubMed CAS Google Scholar - Blessed G, Tomlinson BE, Roth M (1968) The association between quantitative measures of dementia and of senile change in the cerebral grey matter of elderly subjects. Br J Psychiatry 114:797–811. doi:10.1192/bjp.114.512.797
Article PubMed CAS Google Scholar - Bolmont T, Haiss F, Eicke D, Radde R, Mathis CA, Klunk WE et al (2008) Dynamics of the microglial/amyloid interaction indicate a role in plaque maintenance. J Neurosci 28(16):4283–4292. doi:10.1523/JNEUROSCI.4814-07.2008
Article PubMed CAS Google Scholar - Bolte S, Cordelieres FP (2006) A guided tour into subcellular colocalization analysis in light microscopy. J Microsc 224(Pt 3):213–232. doi:10.1111/j.1365-2818.2006.01706.x
Article PubMed CAS Google Scholar - Braak H, Braak E (1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82(4):239–259. doi:10.1007/BF00308809
Article PubMed CAS Google Scholar - Braak H, Braak E (1995) Staging of Alzheimer’s disease-related neurofibrillary changes. Neurobiol Aging 16(3):271–284. doi:10.1016/0197-4580(95)00021-6
Article PubMed CAS Google Scholar - Braak H, Braak E, Bohl J (1993) Staging of Alzheimer-related cortical destruction. Rev Clin Neurosci 33:403–408
CAS Google Scholar - Bush A, Beail N (2004) Risk factors for dementia in people with Down syndrome: issues in assessment and diagnosis. Am J Ment Retard 109(2):83–97. doi:10.1352/0895-8017(2004)109<83:RFFDIP>2.0.CO;2
Article PubMed Google Scholar - Caughey B, Lansbury PT (2003) Protofibrils, pores, fibrils, and neurodegeneration: separating the responsible protein aggregates from the innocent bystanders. Annu Rev Neurosci 26:267–298. doi:10.1146/annurev.neuro.26.010302.081142
Article PubMed CAS Google Scholar - Cirrito JR, Kang JE, Lee J, Stewart FR, Verges DK, Silverio LM et al (2008) Endocytosis is required for synaptic activity-dependent release of amyloid-beta in vivo. Neuron 58(1):42–51. doi:10.1016/j.neuron.2008.02.003
Article PubMed CAS Google Scholar - Corcoran KA, Lu Y, Turner RS, Maren S (2002) Overexpression of hAPPswe impairs rewarded alternation and contextual fear conditioning in a transgenic mouse model of Alzheimer’s disease. Learn Mem 9(5):243–252. doi:10.1101/lm.51002
Article PubMed Google Scholar - Cummings BJ (1997) Plaques and tangles: searching for primary events in a forest of data. Neurobiol Aging 18(4):358–362. doi:10.1016/S0197-4580(97)00049-3
Article PubMed CAS Google Scholar - Cummings BJ, Cotman CW (1995) Image analysis of beta-amyloid “load” in Alzheimer’s disease and relation to dementia severity. Lancet 346:1524–1528. doi:10.1016/S0140-6736(95)92053-6
Article PubMed CAS Google Scholar - Cummings BJ, Pike CJ, Shankle R, Cotman CW (1996) Beta-amyloid deposition and other measures of neuropathology predict cognitive status in Alzheimer’s disease. Neurobiol Aging 17(6):921–933. doi:10.1016/S0197-4580(96)00170-4
Article PubMed CAS Google Scholar - Cummings BJ, Satou T, Head E, Milgram NW, Cole GM, Savage MJ et al (1996) Diffuse plaques contain C-terminal A beta 42 and not A beta 40: evidence from cats and dogs. Neurobiol Aging 17(4):653–659
PubMed CAS Google Scholar - Davies CA, Mann DMA (1993) Is the “Preamyloid” of diffuse plaques in Alzheimer’s disease really nonfibrillar? Am J Pathol 143(6):1594–1605
PubMed CAS Google Scholar - DeKosky ST, Abrahamson EE, Ciallella JR, Paljug WR, Wisniewski SR, Clark RS et al (2007) Association of increased cortical soluble abeta42 levels with diffuse plaques after severe brain injury in humans. Arch Neurol 64(4):541–544. doi:10.1001/archneur.64.4.541
Article PubMed Google Scholar - Deshpande A, Mina E, Glabe C, Busciglio J (2006) Different conformations of amyloid beta induce neurotoxicity by distinct mechanisms in human cortical neurons. J Neurosci 26(22):6011–6018. doi:10.1523/JNEUROSCI.1189-06.2006
Article PubMed CAS Google Scholar - Dickson DW (1997) The pathogenesis of senile plaques. J Neuropathol Exp Neurol 56(4):321–339. doi:10.1097/00005072-199704000-00001
Article PubMed CAS Google Scholar - Dickson DW, Crystal HA, Bevona C, Honer W, Vincent I, Davies P (1995) Correlations of synaptic and pathological markers with cognition of the elderly. Neurobiol Aging 16(3):285–304. doi:10.1016/0197-4580(95)00013-5
Article PubMed CAS Google Scholar - El Khoury J, Hickman SE, Thomas CA, Cao L, Silverstein SC, Loike JD (1996) Scavenger receptor mediated adhesion of microglia to beta-amyloid fibrils. Nature 382:716–719. doi:10.1038/382716a0
Article PubMed CAS Google Scholar - El Khoury J, Hickman SE, Thomas CA, Loike JD, Sliverstein SC (1998) Microglia, scavenger receptors and the pathogenesis of Alzheimer’s disease. Neurobiol Aging 19:S81. doi:10.1016/S0197-4580(98)00036-0
Article PubMed CAS Google Scholar - Giannakopoulos P, Herrmann FR, Bussiere T, Bouras C, Kovari E, Perl DP et al (2003) Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer’s disease. Neurology 60(9):1495–1500
PubMed CAS Google Scholar - Glabe CG (2006) Common mechanisms of amyloid oligomer pathogenesis in degenerative disease. Neurobiol Aging 27(4):570–575. doi:10.1016/j.neurobiolaging.2005.04.017
Article PubMed CAS Google Scholar - Glenner GG, Wong CW (1984) Alzheimer’s disease and Down’s syndrome sharing of a unique cerebrovascular amyloid fibril protein. Biochem Biophys Res Commun 120:885–890. doi:10.1016/S0006-291X(84)80190-4
Article PubMed CAS Google Scholar - Gravina SA, Ho L, Eckman CB, Long KE, Otvos L, Younkin LH, Suzuki N, Younkin SG (1995) Amyloid β-protein (Aβ) in Alzheimer’s disease brain. Biochemical and immunocytochemical analysis with antibodies specific for forms of Aβ(40) and Aβ42(43). J Biol Chem 270:7013–7016. doi:10.1074/jbc.270.13.7013
Article PubMed CAS Google Scholar - Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297(5580):353–356. doi:10.1126/science.1072994
Article PubMed CAS Google Scholar - Harper JD, Wong SS, Lieber CM, Lansbury PT (1997) Observation of metastable Abeta amyloid protofibrils by atomic force microscopy. Chem Biol 4(2):119–125. doi:10.1016/S1074-5521(97)90255-6
Article PubMed CAS Google Scholar - Head E, Garzon-Rodriguez W, Johnson JK, Lott IT, Cotman CW, Glabe C (2001) Oxidation of Aβ and plaque biogenesis in Alzheimer’s disease and Down syndrome. Neurobiol Dis 8:792–806. doi:10.1006/nbdi.2001.0431
Article PubMed CAS Google Scholar - Hof PR, Bouras C, Perl DP, Sparks DL, Mehta N, Morrison JH (1995) Age-related distribution of neuropathologic changes in the cerebral cortex of patients with Down’s syndrome. Arch Neurol 52:379–391
PubMed CAS Google Scholar - Hsaio K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G (1996) Correlative memory deficits, Ab elevation, and amyloid plaques in transgenic mice. Science 274:99–102. doi:10.1126/science.274.5284.99
Article Google Scholar - Iwatsubo T, Mann DM, Odaka A, Suzuki N, Ihara Y (1995) Amyloid beta protein (A beta) deposition: A beta 42(43) precedes A beta 40 in Down syndrome. Ann Neurol 37(3):294–299. doi:10.1002/ana.410370305
Article PubMed CAS Google Scholar - Kang J, Lemaire HG, Unterbeck A, Salbaum JM, Masters CL, Grzeschik KH et al (1987) The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature 325(6106):733–736. doi:10.1038/325733a0
Article PubMed CAS Google Scholar - Kawarabayashi T, Younkin LH, Saido TC, Shoji M, Ashe KH, Younkin SG (2001) Age-dependent changes in brain, CSF, and plasma amyloid β protein in the Tg2576 transgenic mouse model of Alzheimer’s disease. J Neurosci 21(2):372–381
PubMed CAS Google Scholar - Kayed R, Head E, Sarsoza F, Saing T, Cotman CW, Necula M et al (2007) Fibril specific, conformation dependent antibodies recognize a generic epitope common to amyloid fibrils and fibrillar oligomers that is absent in prefibrillar oligomers. Mol Neurodegener 2:18. doi:10.1186/1750-1326-2-18
Article PubMed CAS Google Scholar - Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW et al (2003) Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 300:486–489. doi:10.1126/science.1079469
Article PubMed CAS Google Scholar - Khachaturian ZS (1985) Diagnosis of Alzheimer’s disease. Arch Neurol 42:1097–1106
PubMed CAS Google Scholar - King DL, Arendash GW, Crawford F, Sterk T, Menendez J, Mullan MJ (1999) Progressive and gender-dependent cognitive impairment in the APP(SW) transgenic mouse model for Alzheimer’s disease. Behav Brain Res 103(2):145–162. doi:10.1016/S0166-4328(99)00037-6
Article PubMed CAS Google Scholar - Kitamoto T, Ogomori K, Tateishi J, Prusiner SB (1987) Formic acid pretreatment enhances immunostaining of cerebral and systemic amyloids. Lab Invest 57(2):230–236
PubMed CAS Google Scholar - Kumar-Singh S, De Jonghe C, Cruts M, Kleinert R, Wang R, Mercken M et al (2000) Nonfibrillar diffuse amyloid deposition due to a gamma(42)-secretase site mutation points to an essential role for N-truncated A beta(42) in Alzheimer’s disease. Hum Mol Genet 9(18):2589–2598. doi:10.1093/hmg/9.18.2589
Article PubMed CAS Google Scholar - Lai F, Williams MD (1989) A prospective study of Alzheimer disease in Down syndrome. Arch Neurol 46:849–853
PubMed CAS Google Scholar - Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M et al (1998) Diffusible, nonfibrillar ligands derived from Aβ1–42 are potent central nervous system neurotoxins. Proc Natl Acad Sci USA 95:6448–6453. doi:10.1073/pnas.95.11.6448
Article PubMed CAS Google Scholar - Lemere CA, Blusztajn JK, Yamaguchi H, Wisniewski T, Saido TC, Selkoe DJ (1996) Sequence of deposition of heterogeneous amyloid beta-peptides and APOE in Down syndrome: implications for initial events in amyloid plaque formation. Neurobiol Dis 3:16–32. doi:10.1006/nbdi.1996.0003
Article PubMed CAS Google Scholar - Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A et al (2006) A specific amyloid-beta protein assembly in the brain impairs memory. Nature 440(7082):352–357. doi:10.1038/nature04533
Article PubMed CAS Google Scholar - Leverenz JB, Raskind MA (1998) Early amyloid deposition in the medial temporal lobe of young Down syndrome patients: a regional quantitative analysis. Exp Neurol 150:296–304. doi:10.1006/exnr.1997.6777
Article PubMed CAS Google Scholar - Lott IT, Head E (2001) Down syndrome and Alzheimer’s disease: a link between development and aging. Ment Retard Dev Disabil Res Rev 7(3):172–178. doi:10.1002/mrdd.1025
Article PubMed CAS Google Scholar - Mann DM (1992) Down syndrome and Alzheimer’s disease: towards an understanding of pathogenesis. In: Hunter AJ, Clarke S (eds) Neurodegeneration. Academic Press, San Diego, pp 21–31
Google Scholar - Mann DM, Marcyniuk B, Yates PO, Neary D, Snowden JS (1988) The progression of the pathological changes of Alzheimer’s disease in frontal and temporal neocortex examined both at biopsy and at autopsy. Neuropathol Appl Neurobiol 14(3):177–195. doi:10.1111/j.1365-2990.1988.tb00880.x
Article PubMed CAS Google Scholar - Mann DMA (1988) The pathological association between Down syndrome and Alzheimer disease. Mech Ageing Dev 43:99–136. doi:10.1016/0047-6374(88)90041-3
Article PubMed CAS Google Scholar - Mann DMA, Esiri MM (1989) The pattern of acquisition of plaques and tangles in the brains of patients under 50 years of age with Down’s syndrome. J Neurol Sci 89:169–179. doi:10.1016/0022-510X(89)90019-1
Article PubMed CAS Google Scholar - Masliah E, Terry RD, Mallory M, Alford M, Hansen LA (1990) Diffuse plaques do not accentuate synapse loss in Alzheimer’s disease. Am J Pathol 137(6):1293–1297
PubMed CAS Google Scholar - McKee AC, Kosik KS, Kowall NW (1991) Neuritic pathology and dementia in Alzheimer’s disease. Ann Neurol 30:156–165. doi:10.1002/ana.410300206
Article PubMed CAS Google Scholar - Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM et al (1991) The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology 41(4):479–486
PubMed CAS Google Scholar - Morgan D, Diamond DM, Gottschall PE, Ugen KE, Dickey C, Hardy J et al (2000) A beta peptide vaccination prevents memory loss in an animal model of Alzheimer’s disease. Nature 408(6815):982–985. doi:10.1038/35050116
Article PubMed CAS Google Scholar - Morris JC, Mohs RC, Rogers H, Fillenbaum G, Heyman A (1988) Consortium to establish a registry for Alzheimer’s disease (CERAD) clinical and neuropsychological assessment of Alzheimer’s disease. Psychopharmacol Bull 24(4):641–652
PubMed CAS Google Scholar - Morris JC, Storandt M, McKeel DW Jr, Rubin EH, Price JL, Grant EA et al (1996) Cerebral amyloid deposition and diffuse plaques in “normal” aging: evidence for presymptomatic and very mild Alzheimer’s disease. Neurology 46(3):707–719
PubMed CAS Google Scholar - Morris JC, Storandt M, Miller JP, McKeel DW, Price JL, Rubin EH et al (2001) Mild cognitive impairment represents early-stage Alzheimer disease. Arch Neurol 58(3):397–405. doi:10.1001/archneur.58.3.397
Article PubMed CAS Google Scholar - Nelson PT, Jicha GA, Schmitt FA, Liu H, Davis DG, Mendiondo MS et al (2007) Clinicopathologic correlations in a large Alzheimer disease center autopsy cohort: neuritic plaques and neurofibrillary tangles “do count” when staging disease severity. J Neuropathol Exp Neurol 66(12):1136–1146. doi:10.1097/nen.0b013e31815c5efb
Article PubMed Google Scholar - Pallister C, Jung SS, Shaw I, Nalbantoglu J, Gauthier S, Cashman NR (1997) Lymphocyte content of amyloid precursor protein is increased in Down’s syndrome and aging. Neurobiol Aging 18(1):97–103. doi:10.1016/S0197-4580(96)00207-2
Article PubMed CAS Google Scholar - Paresce DM, Chung H, Maxfield FR (1997) Slow degradation of aggregates of the Alzheimer’s disease amyloid β-protein by microglial cells. J Biol Chem 272(46):29390–29397. doi:10.1074/jbc.272.46.29390
Article PubMed CAS Google Scholar - Paresce DM, Gosh R, Maxfield FR (1996) Microglial cells internalize aggregates of the Alzheimer’s disease amyloid β-protein via a scavenger receptor. Neuron 17:553–565. doi:10.1016/S0896-6273(00)80187-7
Article PubMed CAS Google Scholar - Petersen RC, Smith GE, Waring SC, Ivnik RJ, Tangalos EG, Kokmen E (1999) Mild cognitive impairment: clinical characterization and outcome. Arch Neurol 56(3):303–308. doi:10.1001/archneur.56.3.303
Article PubMed CAS Google Scholar - Prasher VP, Filer A (1995) Behavioural disturbance in people with Down’s syndrome and dementia. J Intellect Disabil Res 39(Pt. 5):432–436
Article PubMed Google Scholar - Rumble B, Retallack R, Hilbich C, Simms G, Multhaup G, Martins R et al (1989) Amyloid A4 and its precursor in Down’s syndrome and Alzheimer’s disease. N Engl J Med 320:1446–1462
PubMed CAS Google Scholar - Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Liao Z, Lieberburg I, Motter R, Mutter L, Soriano F, Shopp G, Vasquez N, Vandervert C, Walker S, Wogulis M, Yednock T, Games D, Seubert P (1999) Immunization with amyloid-β attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature 400:173–177. doi:10.1038/22124
Article PubMed CAS Google Scholar - Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R et al (1991) Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol 30(4):572–580. doi:10.1002/ana.410300410
Article PubMed CAS Google Scholar - The National Institute on Aging aRIWGoDCftNAoAsd (1997) Consensus recommendations for the post mortem diagnosis of Alzheimer’s disease. Neurobiol Aging 18(S4):1–2
Google Scholar - Torp R, Head E, Milgram NW, Hahn F, Ottersen OP, Cotman CW (2000) Ultrastructural evidence of fibrillar β-amyloid associated with neuronal membranes in behaviorally characterized aged dog brains. Neuroscience 93(3):495–506. doi:10.1016/S0306-4522(99)00568-0
Article Google Scholar - Tyrrell J, Cosgrave M, McCarron M, McPherson J, Calvert J, Kelly A et al (2001) Dementia in people with Down’s syndrome. Int J Geriatr Psychiatry 16(12):1168–1174. doi:10.1002/gps.502
Article PubMed CAS Google Scholar - Walsh DM, Hartley DM, Kusumoto Y, Fezoui Y, Condron MM, Lomakin A et al (1999) Amyloid beta-protein fibrillogenesis. Structure and biological activity of protofibrillar intermediates. J Biol Chem 274(36):25945–25952. doi:10.1074/jbc.274.36.25945
Article PubMed CAS Google Scholar - Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS et al (2002) Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416(6880):535–539. doi:10.1038/416535a
Article PubMed CAS Google Scholar - Wang B, Larson L (1985) Simultaneous demonstration of multiple antigens by indirect immunofluorescence or immunogold staining. Histochemistry 83:47–56. doi:10.1007/BF00495299
Article PubMed CAS Google Scholar - Westerman MA, Cooper-Blacketer D, Mariash A, Kotilinek L, Kawarabayashi T, Younkin LH, Carlson GA, Younkin SG, Ashe KH (2002) The relationship between Aβ and memory in the Tg2576 mouse model of Alzheimer’s disease. J Neurosci 22(5):1858–1867
PubMed CAS Google Scholar - White H (1980) A heteroskedasticity-consistent covariance matrix estimator and a direct test for heteroskedasticity. Econometrica 48:817–830. doi:10.2307/1912934
Article Google Scholar - Williams AD, Sega M, Chen M, Kheterpal I, Geva M, Berthelier V et al (2005) Structural properties of A beta protofibrils stabilized by a small molecule. Proc Natl Acad Sci USA 102(20):7115–7120. doi:10.1073/pnas.0408582102
Article PubMed CAS Google Scholar - Wisniewski K, Howe J, Williams G, Wisniewski HM (1978) Precocious aging and dementia in patients with Down’s syndrome. Biol Psychiatry 13(5):619–627
PubMed CAS Google Scholar - Wisniewski K, Wisniewski H, Wen G (1985) Occurrence of neuropathological changes and dementia of Alzheimer’s disease in Down’s syndrome. Ann Neurol 17:278–282. doi:10.1002/ana.410170310
Article PubMed CAS Google Scholar
Acknowledgments
Funding provided by the UCI-ADRC National Institutes on Health/National Institute on Aging Grant number P50 AG16573, National Institutes on Health/National Institute on Aging P01 AG000538. NICHD Brain and Tissue Bank for Developmental Disorders is supported under contracts N01-4-3368 and N01-HD-4-3383.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Author notes
- Elizabeth Head
Present address: Department of Molecular and Biomedical Pharmacology, Sanders-Brown Center on Aging, University of Kentucky, 203 Sanders-Brown Building, 800 South Limestone Street, Lexington, KY, 40536, USA
Authors and Affiliations
- Institute for Brain Aging and Dementia, University of California, Irvine, CA, 92697, USA
Floyd Sarsoza, Tommy Saing, Robert Dahlin, Malcolm Dick, Camille Broadwater-Hollifield, Scott Mobley, Ira Lott, Eric Doran, Daniel Gillen, Clifford Anderson-Bergman, David H. Cribbs, Charles Glabe & Elizabeth Head - Department of Molecular Biology and Biochemistry, University of California, Irvine, CA, 92697, USA
Rakez Kayed & Charles Glabe - Department of Neurology, University of California, Irvine, CA, 92697, USA
Ira Lott, David H. Cribbs & Elizabeth Head - Department of Pediatrics, University of California, Irvine, CA, 92697, USA
Ira Lott & Eric Doran - Department of Statistics, University of California, Irvine, CA, 92697, USA
Daniel Gillen & Clifford Anderson-Bergman
Authors
- Floyd Sarsoza
- Tommy Saing
- Rakez Kayed
- Robert Dahlin
- Malcolm Dick
- Camille Broadwater-Hollifield
- Scott Mobley
- Ira Lott
- Eric Doran
- Daniel Gillen
- Clifford Anderson-Bergman
- David H. Cribbs
- Charles Glabe
- Elizabeth Head
Corresponding author
Correspondence toElizabeth Head.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Sarsoza, F., Saing, T., Kayed, R. et al. A fibril-specific, conformation-dependent antibody recognizes a subset of Aβ plaques in Alzheimer disease, Down syndrome and Tg2576 transgenic mouse brain.Acta Neuropathol 118, 505–517 (2009). https://doi.org/10.1007/s00401-009-0530-3
- Received: 04 December 2008
- Revised: 11 March 2009
- Accepted: 29 March 2009
- Published: 10 April 2009
- Issue Date: October 2009
- DOI: https://doi.org/10.1007/s00401-009-0530-3