A novel IL2RG mutation associated with maternal T lymphocyte engraftment in a patient with severe combined immunodeficiency (original) (raw)
Introduction
Severe combined immunodeficiency (SCID) represents a genetically heterogeneous group of primary immunodeficiency disorders in which both T- and B-lymphocyte development and/or function is profoundly disturbed (Buckley 2004;Chan and Puck 2005). Patients with SCID may be engrafted with transplacentally derived maternal T-lymphocytes, which are not rejected by the immunodeficient infant (Muller et al. 2001). In a subset of such patients karyotype analysis may give perplexing results at first glance. We present a case in which genetic evaluation resulted in the identification of a novel mutation in the gene encoding the common γ chain (γc) of the IL-2 receptor (IL2RG) (MIM*308380) located on the X chromosome (Xq13.1) (for mutation reference see: http://www.research.nhgri.nih.gov/scid/).
Materials and methods
DNA was obtained from venous blood samples following standard procedures (Miller et al. 1988). IL2RG sequencing was performed as previously described (Niemela et al. 2000). For γc expression and functional studies, B cell lines from our patient, a healthy control and another patient with SCID due to a mutation of the initiating methionine codon (γc negative control) were established by transformation with Epstein Barr virus. Cells were stained with phycoerythrin-labeled anti-γc (TUGh4, Pharmingen, San Diego, CA) or control antibody and analyzed by flow cytometry. To measure STAT5 phosphorylation the cells were washed in RPMI medium (Gibco) acidified with CO2, exposed to medium with or without human recombinant IL-2 (1 μg/ml, Hoffman-LaRoche, Basel, Switzerland) for 15 min and fixed (Fix and Perm, Caltag, Burlingame, CA). The cells were then permeabilized and stained with anti-phospho-STAT5 (Zymed, San Francisco, CA). FITC-conjugated secondary antibody (Becton Dickinson, Franklin Lakes, NJ) was applied and the washed, resuspended cells were analyzed by flow cytometry.
Results
Case presentation
The 1-year-old boy was born weighing 3,500 g from an uncomplicated, non-consanguineous second pregnancy at 41 weeks gestation. The mother’s first pregnancy had been lost by spontaneous abortion, but there was no family history consistent with immunodeficiency. Perinatal adaptation was normal. He first presented at 9 months with a septic episode that resolved in a few days with broad spectrum antibiotic administration. However, a similar episode within a few weeks prompted workup for a possible immunodeficiency. A white blood cell count was 4 g/l (38% lymphocytes/CD5+ B cells: 28%; T- and NK cells: 7%). The low absolute number of T cells (0.24 g/l) suggested a T cell immunodeficiency. A fluorescence in situ hybridization (FISH) probe for DiGeorge syndrome showed no deletion of chromosome 22q11.2, but the concomitantly performed karyotype analysis [following phytohemagglutinin (PHA) stimulation] gave a perplexing 46,XX result. However, only 20 mitoses could be counted, showing that T cells responded extremely poorly to stimulation. Consecutive interphase FISH with X and Y chromosome α-satellite probes (Vysis, Downers Grove, IL) showed 46,XY(92%)/46,XX(8%) prior to and 46,XY(66%)/46,XX(33%) following PHA stimulation. The CD4/8 ratio increased from 0.5 to 2 after PHA culture, indicating a relative expansion of a 46,XX, CD4+ subset of T cells in the patient’s blood. Fibroblast and buccal mucosa karyotypes were 100% 46,XY, supporting the existence of the 46,XX T-cells as a chimeric lymphocyte population rather than a result of inherited mosaicism. Meanwhile, immunoglobulin levels were markedly low, with IgG of 1.3 g/l (normal for age 3.4–15.2), IgA, 0.28 g/l (0.2–1.5) and IgM, 0.31 g/l (0.43–2.42). Consequently, a diagnosis of SCID was made. Precise genetic diagnosis was pursued and the patient was referred for bone marrow transplantation (BMT).
We began with the sequencing of IL2RG, the most common genetic cause of SCID in males (Chan and Puck 2005). A novel missense mutation was detected within IL2RG exon 6: cDNA 816G>C; G268R (Fig. 1a, red arrow). The mother was a carrier of this defect. To determine whether this nonconservative amino acid change was sufficient to cause SCID, Epstein-Barr virus-transformed B cell lines were studied. Cell surface staining revealed no γc expression, comparable to cells from a known γc-deficient patient (Fig. 1b). Moreover, after exposure to IL-2, our patient’s cells failed to phosphorylate the downstream target STAT5 (Fig. 1c), a functional indication that signaling through the γc was absent.
Fig. 1
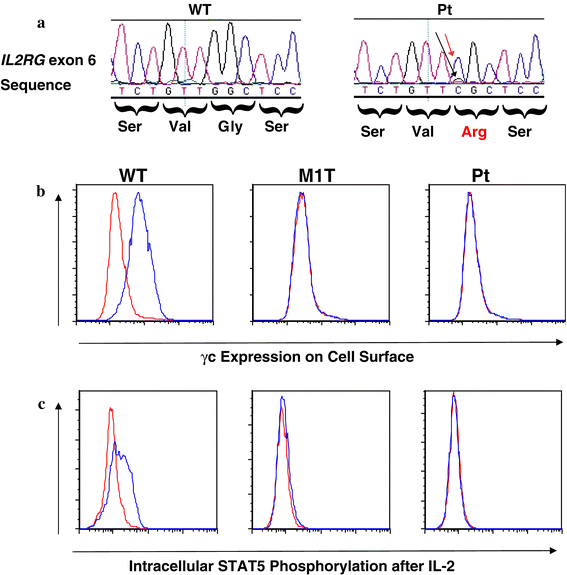
a Exon 6 IL2RG sequence tracing of genomic DNA from blood cells of a wild type control (WT) and the patient (Pt), who has a predominant mutant nucleotide C (red arrow) at position 818 (transcription start site =1) with a trace of wild type G (black arrow). The resulting missense mutation of glycine 268 (initiating methionine =1) to arginine introduces a charged residue in the membrane spanning portion of the protein. b B cell lines stained with nonspecific control (red) or antibody to γc (blue) from a wild type control (WT), a known SCID patient with an IL2RG mutation of the first methionine residue and no γc expression (M1T), and the current G268R patient (Pt). c STAT5 phosphorylation after treatment with medium only (red) or IL-2 (blue) in the same cells lines as in (b)
Upon HLA typing, the presence of the maternal genotype could be detected above background. Similarly, the presence of the wild type (WT) guanine at cDNA 816 could be observed on the fluorescent tracing of the patient’s IL2RG sequence (Fig. 1a, black arrow). The patient is currently receiving antibiotic prophylaxis against Pneumocystis and monthly IV Ig therapy and is doing well clinically except for a mild-to-moderate recurrent eczematous rash, consistent with spontaneous graft versus host disease from recognition of maternally derived cells (Muller et al. 2001). He is scheduled for BMT.
Discussion
Twelve pathogenic genes have already been identified in SCID. Among these, defective IL2RG can be detected in 40–50% of patients. This ratio is even higher if we take the presence of B cells into consideration (Chan and Puck 2005). Therefore, in the case of a male, B-cell-positive SCID patient, genetic evaluation of IL2RG is recommended first. The missense mutation introducing a charged amino acid into the transmembrane domain of γc would be predicted to interfere with localization at the cell surface, and indeed cell surface staining and assay of IL-2 response proved this mutation to be pathogenic. Once a precise molecular diagnosis is made in SCID, carrier testing and prenatal diagnosis can be offered to promote optimal family planning. Irrespective of the genetic defect, substantial numbers of transplacentally acquired maternal T lymphocytes are present in 40% of SCID cases. Among these patients, a subgroup carries mostly CD4+ T cells responsive to PHA stimulation (Muller et al. 2001). Consequently, the detection of a 46,XX karyotype in a male SCID patient may be observed due to the persistent engraftment of such maternal lymphocytes. It has been proposed that these cells may promote near-term survival in SCID, leading to a rather benign clinical picture in the first few years of life (Tezcan et al. 2005). However, despite of the presence of maternal T cells, untreated cases have been fatal. Consequently, definitive therapy should be promoted emergently for such patients.
References
- Buckley RH (2004) Molecular defects in human severe combined immunodeficiency and approaches to immune reconstitution. Annu Rev Immunol 22:625–655
Article CAS Google Scholar - Chan K, Puck JM (2005) Development of population-based newborn screening for severe combined immunodeficiency. J Allergy Clin Immunol 115:391–398
Article Google Scholar - Miller SA, Dykes DD, Polesky HF (1988) A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 16:1215
Article CAS Google Scholar - Muller SM, Ege M, Pottharst A, Schulz AS, Schwarz K, Friedrich W (2001) Transplacentally acquired maternal T lymphocytes in severe combined immunodeficiency: a study of 121 patients. Blood 98:1847–1851
Article CAS Google Scholar - Niemela JE, Puck JM, Fischer RE, Fleisher TA, Hsu AP (2000) Efficient detection of thirty-seven new IL2RG mutations in human X-linked severe combined immunodeficiency. Clin Immunol 95:33–38
Article CAS Google Scholar - Tezcan I, Ersoy F, Sanal O, Turul T, Uckan D, Balci S, Hicsonmez G, Prieur M, Caillat-Zucmann S, Le Deist F, De Saint BG (2005) Long-term survival in severe combined immune deficiency: the role of persistent maternal engraftment. J Pediatr 146:137–140
Article Google Scholar
Acknowledgements
This research was supported in part by the Intramural Research Program of the National Human Genome Research Institute, National Institutes of Health, USA (A.P.H., J.M.P.) and OTKA T49017 (L.M.).
Author information
Authors and Affiliations
- Department of Medical Genetics and Child Development, University of Pécs, József A. u. 7, 7623, Pécs, Hungary
Richard Kellermayer, Kinga Hadzsiev & György Kosztolányi - Genetics and Molecular Biology Branch, National Human Genome Research Institute, NIH, DHHS, Bethesda, MD, USA
Amy P. Hsu & Jennifer M. Puck - Department of Pediatrics, University of Pécs, Pécs, Hungary
József Stankovics, Ágnes Vojcek & Pál Kajtár - Department of Immunology and Biotechnology, University of Pécs, Pécs, Hungary
Péter Balogh - Department of Infectious Diseases and Pediatric Immunology, University of Debrecen, Debrecen, Hungary
László Maródi - MTA-PTE Clinical Genetics Research Group, Pécs, Hungary
György Kosztolányi
Authors
- Richard Kellermayer
- Amy P. Hsu
- József Stankovics
- Péter Balogh
- Kinga Hadzsiev
- Ágnes Vojcek
- László Maródi
- Pál Kajtár
- György Kosztolányi
- Jennifer M. Puck
Corresponding author
Correspondence toRichard Kellermayer.
Rights and permissions
About this article
Cite this article
Kellermayer, R., Hsu, A.P., Stankovics, J. et al. A novel IL2RG mutation associated with maternal T lymphocyte engraftment in a patient with severe combined immunodeficiency.J Hum Genet 51, 495–497 (2006). https://doi.org/10.1007/s10038-006-0386-5
- Received: 07 December 2005
- Accepted: 24 January 2006
- Published: 01 May 2006
- Issue Date: May 2006
- DOI: https://doi.org/10.1007/s10038-006-0386-5