Contrasting patterns of the 5S and 45S rDNA evolutions in the Byblis liniflora complex (Byblidaceae) (original) (raw)
Introduction
The 5S and 45S ribosomal RNA genes (rDNAs) reside as independent tandem arrays in a plant genome. Their characteristic positions on chromosomes provide useful landmarks in chromosome study (Heslop-Harrison 2000). Physical mapping of the 5S and 45S rDNA provides universally applicable markers for chromosome and genome characterization in many plants. On the other hand, nucleotide sequences of the nontranscribed spacer of 5S rDNA (5S-NTS) and the internal transcribed spacer of 45S rDNA (ITS) are used globally as molecular markers for resolving species-level phylogenetic relationships (Persson 2000; Becerra 2003). Fluorescence in situ hybridization (FISH) detection and phylogenetic analysis allow us to learn the chromosomal-locus-level and nucleotide-sequence-level dynamics of these tandem arrays during diversification of the plant taxa. Chromosomal location of rDNA arrays has been suggested to affect the degree and speed of homogenization through concerted evolution (Wendel et al. 1995; Zhang and Sang 1999). Therefore, the combination of FISH and phylogenetic analyses may lead to a more sophisticated interpretation of rDNA evolution. However, chromosomal and molecular studies have often been carried out separately. This situation has masked evolutionary dynamics of rDNAs in plant taxa.
To trace the dynamics of 5S and 45S rDNA arrays in plant genomes, we investigated the chromosomal location and phylogenetic relationship of rDNAs in the Byblis liniflora complex. Byblis Salisb. (Byblidaceae) comprises seven species essentially native to Australia (Lowrie and Conran 2008). All members of the genus prey on small insects by aerial parts covered with secretory glands, which bear sticky droplets containing digestive enzymes (Bruce 1905; Hartmeyer and Hartmeyer 2005; Płachno et al. 2006). Their glistening appearance lends them the common name “the rainbow plants” (McPherson 2008). Currently recognized species fall into two complexes, the B. gigantea complex and the B. liniflora complex (Conran et al. 2002a). The former consists of temperate, perennial, shrubby plants distributed in South Western Australia, whereas the latter consists of tropical, annual, herbaceous plants mainly native to Northern Australia. Additionally, these complexes have been cytogenetically characterized by basic chromosome number: x = 9 for the B. gigantea complex and x = 8 for the B. liniflora complex (Conran et al. 2002a). The B. liniflora complex consists of three diploid species (B. aquatica Lowrie & Conran, B. filifolia Planch., and B. rorida Lowrie & Conran) and two tetraploid species (B. liniflora and B. guehoi Lowrie & Conran), whereas the B. gigantea complex comprises two diploids (B. gigantea Lindl. and B. lamellata Conran & Lowrie). Byblis liniflora, the representative member of the B. liniflora complex, is the most widespread species, being distributed up to New Guinea as well as to a wide area of northern Australia (Steenis 1971; Lowrie and Conran 1998). A seed fossil, which shows morphological resemblance to the B. liniflora complex, was discovered in clay lens from Eocene South Australia (Conran and Christophel 2004). Molecular phylogenetic studies have supported the placement of the family Byblidaceae in Lamiales (Albert et al. 1992; Müller et al. 2004). Despite the progress in phylogenetic investigations at higher taxonomic levels, the intrageneric relationships of Byblis have been less studied. The lack of a phylogenetic framework has made it difficult to understand the evolutionary history of this genus.
In this study, we investigated the 5S and 45S rDNA arrays in the B. liniflora complex through (1) chromosomal physical mapping by FISH and (2) phylogenetic analyses using 5S-NTS and ITS sequences. Furthermore, phylogenetic analysis using plastid sequences (rbcL and trnK intron) was used to gain further insight into the evolutionary history of the complex.
Materials and methods
Plant materials
Six species were used in this study: one species from the B. gigantea complex (B. gigantea) and five species from all taxa in the B. liniflora complex (B. aquatica, B. filifolia, B. guehoi, B. liniflora, and B. rorida). Seeds and seedlings of Byblis species were purchased from commercial growers. Taxon names, cytotypes, voucher information, and GenBank accession numbers of DNA sequences presented in this study are given in Table 1. Voucher specimens were deposited in our laboratory of Tokai University. To test intraspecific variation, 3 to 5 accessions were analyzed for B. aquatica, B. filifolia, B. liniflora and B. rorida. Byblis gigantea and B. guehoi were sampled for single accession each. We sampled a total of 17 accessions from 6 species.
Table 1 Voucher accessions of Byblis species sampled in this study and GenBank accessions of rbcL, trnK intron, ITS and 5S-NTS sequences
Seeds and shoot tips were surface sterilized with 1% benzalkonium chlorite and with 1% sodium hypochlorite for 5 min each, repeatedly rinsed with sterilized water, and then sown on half-strength Murashige and Skoog medium (MS medium; Murashige and Skoog 1962) supplemented with 3.0% sucrose and 0.2% gellan gum. Established seedlings were maintained in vitro.
DNA extraction, amplification, cloning, and sequencing
Total genomic DNA was isolated from young leaves following modified cetyltrimethylammonium bromide method (Doyle and Doyle 1990). rbcL gene, trnK intron (including matK gene), ITS, and 5S-NTS (including 5S rDNA coding region) were amplified by polymerase chain reaction (PCR). The primers used are listed in Table 2. The cycle profile was an initial denaturation at 94°C (4 min); 35 cycles with 94°C (30 s), 57°C (30 s), and 72°C (60 s); and a final extension step at 72°C for 5 min. PCR products were column purified using QIAquick PCR Purification Kit (Qiagen, Tokyo, Japan) or gel purified using GeneClean III (BIO 101, Vista, CA, USA). The purified DNA was sequenced in both directions using ABI PRISM 310 DNA sequencer (Applied Biosystems, Foster City, CA, USA) with a BigDye Terminator Cycle Sequencing Kit (version 3.1, Applied Biosystems). When 3 or more arrays per somatic genomes were detected in rDNA-FISH analysis, the corresponding 5S-NTS and ITS were cloned into pGEM-T Easy vector (Promega Japan, Tokyo, Japan) and transformed into Escherichia coli DH5α competent cells. Recombinant clones were screened by colony direct PCR method, and were sequenced 3–10 clones per each sequence type using T7 (5′-TAATACGACTCACTATAGGG-3′) and SP6 (5′-TATTTAGGTGACACTATAG-3′) primers. To exclude artifactual nucleotide variations generated in the PCR reaction, the polymorphism observed in only 1 clone was removed from the analysis. The sequences reported in this paper were deposited in the GenBank database (Table 1).
Table 2 Primers used for PCR amplification, DNA sequencing and FISH probe preparation
DNA probes and labeling
To track the chromosomal locations of the 5S and 45S rDNA arrays, the entire region of the 5S rDNA unit and the 18S rDNA sequence were used as FISH probes, respectively. Primers used for PCR amplification are listed in Table 2. Sequence identities of the 5S and 18S rDNA fragments were confirmed by DNA sequencing (data not shown). To circumvent a contamination by dimeric or trimeric repeat sequences containing conservative 5S rDNA coding regions, 5S-NTS sequence type-specific probes were obtained by PCR amplification from clones. DNA fragments were biotin labeled through the random primed labeling technique (Feinberg and Vogelstein 1983) using Biotin-High Prime (Roche Applied Science, Indianapolis, IN, USA), or labeled in PCR with tetramethyl-rhodamine-5-dUTP (Roche Applied Science).
Chromosome preparation
Root tips that were 1–3 cm long were excised from in vitro plants and pretreated with 0.05% colchicine for 3–4 h at 18°C, fixed in 45% acetic acid, and then washed with distilled water. They were macerated in an enzymatic mixture containing 0.4% Cellulase Onozuka R-10 (Yakult Pharmaceutical Industry Co., Ltd., Tokyo, Japan) and 0.2% Pectolyase Y-23 (Kikkoman Co. Ltd., Tokyo, Japan) for 60 min at 37°C. After washing with distilled water, root tips were placed onto a glass slide and spread with ethanol–acetic acid (3:1). The slides were air dried for 40 h.
Fluorescence in situ hybridization
Chromosome preparation was treated with 100 μg ml−1 RNase A (NIPPON GENE Co., Ltd., Tokyo, Japan) for 120 min at 37°C in a humid chamber. After dehydration in a graded series of ethanol, a hybridization mixture containing 50% formamide, 10% dextran sulfate and DNA probes was dropped onto the slides. The preparation was sealed, denatured for 3 min at 78°C, and then incubated for 16 h at 37°C. Subsequently, the slides were rinsed in 2× SSC at 42°C for 10 min, 0.2× SSC at 42°C for 10 min, and 2× SSC/0.2% Tween-20 at room temperature for 10 min twice. The slides were blocked with 5% bovine serum albumin in 2× SSC/0.2% Tween-20 for 60 min at 37°C. Biotin-labelled probes were detected with streptavidin-Alexa Fluor 488 (Invitrogen, CA, USA) in 2× SSC for 2 h at 37°C in a humid chamber. The slides were washed in 2× SSC/0.2% Tween-20 for 10 min twice, and 2× SSC for 10 min twice at room temperature. The preparations were then mounted in Vectashield mounting medium containing 1.5 μg ml−1 4′,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories, Inc., CA, USA). Chromosome images were taken by a digital camera (CoolSNAP: Roper Scientific, Inc., Chiba, Japan) on a microscope (Olympus BX51; Olympus, Tokyo, Japan).
Phylogenetic analyses
Species of Antirrhinum, Buddleja, Torenia, and Jasminum were selected as outgroup taxa of plastid gene analysis based on previous broadscale phylogenetic analysis in Lamiales (Müller et al. 2004); their GenBank accession numbers are listed in Table 1. rbcL, trnK intron, ITS, and 5S-NTS nucleotide sequences were aligned using the Clustal X program (Thompson et al. 1994) implemented in MEGA4 (Tamura et al. 2007) and then refined by eye. Gaps and ambiguous sites were eliminated from datasets. Following phylogenetic analyses were separately performed for rbcL, trnK intron, ITS and 5S-NTS matrices. In addition, to enhance the reliability of plastid tree topology, the conjugate matrix of rbcL and trnK intron was analyzed. The congruence between rbcL and trnK matrices was confirmed using the incongruence length difference test (P = 0.22, Farris et al. 1994).
To independently test the reliability of the tree topology, phylogenetic analyses were performed under maximum parsimony (MP) analysis, maximum likelihood (ML) analysis, and Bayesian inference (BI). The MP tree was constructed using the close-neighbor-interchange search implemented in MEGA4, in which the search level was 3 and the initial trees were obtained with the random addition of sequences (10 replicates). The bootstrap percentages (BP; Felsenstein 1985) for the MP trees were calculated for 1,000 replications. For ML and BI searches, a best-fit model of nucleotide substitution was determined based on the evaluation of each dataset under AIC and hLRT criteria using MrModeltest (Nylander 2004). For rbcL, trnK intron, rbcL + trnK intron, ITS, and 5S-NTS datasets, HKY85+I+G, GTR+I+G, GTR+I+G, GTR+I+G, and K80+I+G models were optimized, respectively. ML trees were obtained using PhyML 3.0 (Guindon and Gascuel 2003). Topology search was carried out under the “Best of NNIs and SPRs” option with 10 random additions. Support for nodes was estimated by bootstrapping with 1,000 replications. BI was carried out by Metropolis-coupled Markov-chain Monte Carlo (MC3) analysis using MrBayes 3.1.2 (Ronquist and Huelsenbeck 2003). Topology search was performed by running four simultaneous chains initiated from random trees for 1,000,000 generations (heating parameter = 0.2), in which trees were sampled every 100 generation. At the end of the run, convergence was evaluated by visual inspection of a graph of likelihood as a function of generation. Because a conservative burn-in period was identified for 250,000 generations, the first 2,500 trees were discarded. A majority-rule consensus tree with posterior probabilities (PP) calculated for each clade was yielded from the 7,500 post-burn-in trees.
Results
Chromosome number
Table 3 summarizes the chromosomal characteristics of the B. liniflora complex at mitotic metaphase. Intraspecific variations of chromosome number were observed among accessions of B. filifolia: 4 accessions had a chromosome number of 2_n_ = 16 (diploid cytotype), whereas 1 accession exhibited 2_n_ = 32 (tetraploid cytotype). The other chromosome numbers are 2_n_ = 16 for B. aquatica and B. rorida, and 2_n_ = 32 for B. liniflora and B. guehoi.
Table 3 Karyotype characteristics in the B. liniflora complex
Chromosomal distribution of 5S and 45S rDNAs
Figure 1 shows the results of double-target FISH using the 5S and 45S rDNA probes to mitotic metaphase of the B. liniflora complex. All species were distinguishable in terms of FISH patterns of rDNAs. The number and distribution of 5S and 45S rDNAs are summarized in Table 3. Two signals of the 45S rDNA probe were observed in B. filifolia diploid cytotype, B. liniflora, and B. rorida, whereas four arrays were detected in B. aquatica, B. filifolia tetraploid cytotype, and B. guehoi. Two distal 45S rDNA signals of B. aquatica were strong as seen in those of other species, but the signals on the other two interstitial arrays were very weak. The 45S rDNA signals were located at secondary constrictions, except for 2 minor signals in B. aquatica. The 5S rDNA arrays were detected in 2 chromosomes of B. aquatica, 4 chromosomes of B. filifolia diploid cytotype, 8 chromosomes of B. filifolia tetraploid cytotype, 10 chromosomes of B. liniflora, and 12 chromosomes of B. guehoi. In B. rorida, 1 accession had 2 arrays whereas the other 2 accessions harbored 3 arrays. Hereafter, the former was designated as cytotype A, and the latter as cytotype B. In terms of proximo-distal (centromeric–telomeric) distribution of rDNA arrays, 5S and 45S rDNA arrays tended to occupy the proximal and distal positions, respectively.
Fig. 1
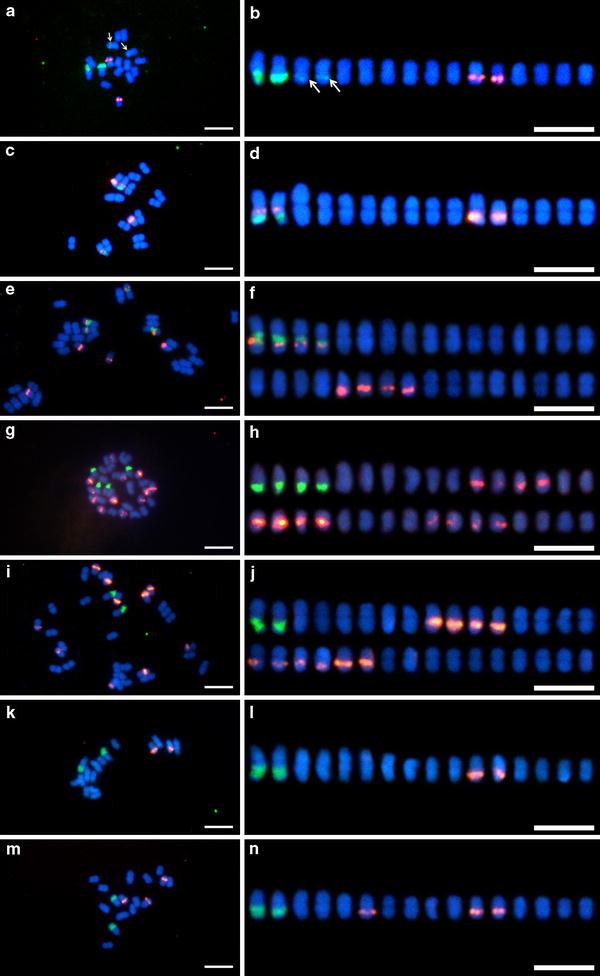
Fluorescence in situ hybridization of mitotic metaphase spreads (a, c, e, g, i, k, m) and chromosome alignments (b, d, f, h, j, l, n) of Byblis aquatica (a, b), B. filifolia diploid cytotype (c, d), B. filifolia tetraploid cytotype (e, f), B. guehoi (g, h), B. liniflora (i, j), B. rorida cytotype A (k, l), and B. rorida cytotype B (m, n). 5S rDNA (red fluorescence) and 45S rDNA (green fluorescence) probes were simultaneously detected. Chromosomes were counterstained with DAPI. Arrows minor 45S rDNA signals. Bars 5 μm
In B. filifolia diploid and tetraploid cytotypes, half of the 5S rDNA arrays were positioned at the chromosome arm carrying the 45S rDNA array. In the diploid cytotype, 5S and 45S rDNAs on chromosome arms occupied the proximal and distal sites, respectively. In the tetraploid cytotype, this positional relationship was inverted: interstitial 5S rDNA sites and proximal 45S rDNA sites.
Because an odd number of 5S rDNA arrays was observed in B. rorida cytotype B, assigning pairs of homologous chromosomes became difficult. Hence, the number of rDNA arrays per somatic chromosome complement but not at the haploid level is mentioned in the following sections.
Plastid dataset and tree topology
Cytogenetic screening revealed interspecies and intraspecies karyotype variations that guided the successive phylogenetic analyses. A representative 1 accession for each species and cytotype was analyzed in phylogenetic analyses. Unfortunately, we failed to obtain the PCR product of trnK intron from B. rorida cytotype B. Thus, this cytotype was excluded from the analyses of trnK intron and conjugated matrices. Figure 2 shows plastid gene phylogenies of Byblis and related genera. Although separate analyses of rbcL and trnK intron datasets did not show any topological incongruence in ingroup taxa, these datasets gave rise to less-resolved topology (Fig. 2a, b). The conjugated matrix of rbcL and trnK intron sequences resulted to a single most parsimonious tree with improved resolution (Fig. 2c). ML and BI trees resulting from the conjugated matrix exhibited essentially the same topology in comparison with the MP tree (data not shown).
Fig. 2
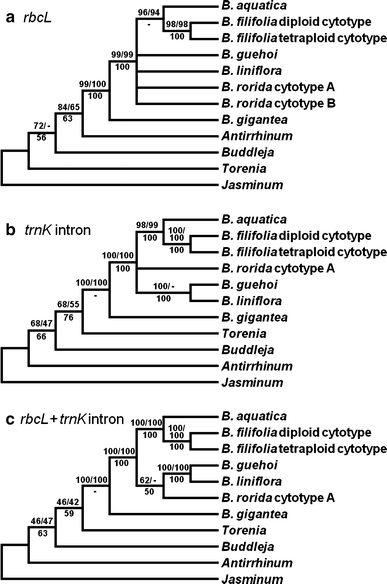
Plastid gene phylogenies of Byblis and its related taxa. a Strict consensus tree of 30 most parsimonious trees generated by analysis of the rbcL data matrix composed of 1,209 nucleotide positions (45 sites are parsimony informative). The consistency index (CI) is 0.750 and the retention index (RI) is 0.843. b Strict consensus tree of 2 most parsimonious trees yielded by the trnK intron dataset consisted of 2,266 positions (263 sites are parsimony informative). CI and RI are 0.797 and 0.863, respectively. c Single most parsimonious tree resulting from the rbcL + trnK intron conjugate dataset composed of 3,475 positions (308 sites are parsimony informative). CI and RI are 0.782 and 0.853, respectively. Numbers above branches are bootstrap percentages in MP/ML analyses. Bayesian posterior probabilities are indicated below the branches. A minus sign (−) denotes a clade not supported in the respective analysis
Plastid trees supported monophyly of not only Byblis but also the B. liniflora complex, with high confidence. In ingroup taxa, B. gigantea was the sister to five species of the B. liniflora complex. In the conjugated tree, the B. liniflora complex was divided into two clades, B. aquatica_–_B. filifolia and B. guehoi_–_B. liniflora_–_B. rorida, although the latter clade received low supports (BP in MP/BP in ML/PP in BI = 62/-/50). Byblis filifolia diploid and tetraploid cytotypes formed a well-supported clade (100/100/100), and this pair was sister to B. aquatica (100/100/100). Two tetraploid species, B. guehoi and B. liniflora, formed a clade with high support values (100/100/100). Byblis rorida cytotype A and B were indicated as sister lineage with a weak support in ML analysis of the rbcL dataset (BP = 51, data not shown), but MP and BI analyses did not resolve this clade.
Characterization of 5S rDNA sequences
By PCR amplification with universal primers (5SF and 5SR) following cloning, 1 sequence type of 5S-NTS was detected in B. aquatica (AQU1), B. gigantea (GIG1), B. rorida cytotype A (ROR-A1), and B. rorida cytotype B (ROR-B1); 2 sequence types in B. filiolia diploid cytotype (FIL-D1 and FIL-D2) and B. guehoi (GUE1 and GUE2); and 3 sequence types in B. filifolia tetraploid cytotype (FIL-T1, FIL-T2, and FIL-T3) and B. liniflora (LIN1, LIN2, LIN3) (Fig. 3b). Since considerable polymorphisms within and among species were observed, we carried out a detailed characterization of 5S-NTS. We designed sequence type-specific primer pairs, FIL-T2-1 and FIL-T2-2 for FIL-T2, FIL-T3-1 and FIL-T3-2 for FIL-T3, and LIN2-1 and LIN2-2 for LIN2 (Fig. 3a; Table 2). The presence of FIL-T2-like and FIL-T3-like sequences in B. filifolia diploid cytotype and LIN2-like sequence in B. guehoi were indicated by PCR screening (Fig. 3b). Using these sequence type-specific primers in combination with universal primers (5SF and 5SR), overlap fragments of these 5S-NTS sequence types were amplified, cloned, and then sequenced. The FIL-T2-like sequence in B. filifolia diploid cytotype was identical to FIL-D1/FIL-D2, as expected by sequence similarity in primer annealing sites (Fig. 3a), whereas the FIL-T3-like sequence was completely differentiated from FIL-D1/FIL-D2 (Fig. 3a). We designated FIL-T3-like 5S-NTS in B. filifolia diploid cytotype as FIL-D3. In addition, the LIN2-like sequence in B. guehoi was distinguishable from GUE1/GUE2 and therefore referred to as GUE3.
Fig. 3
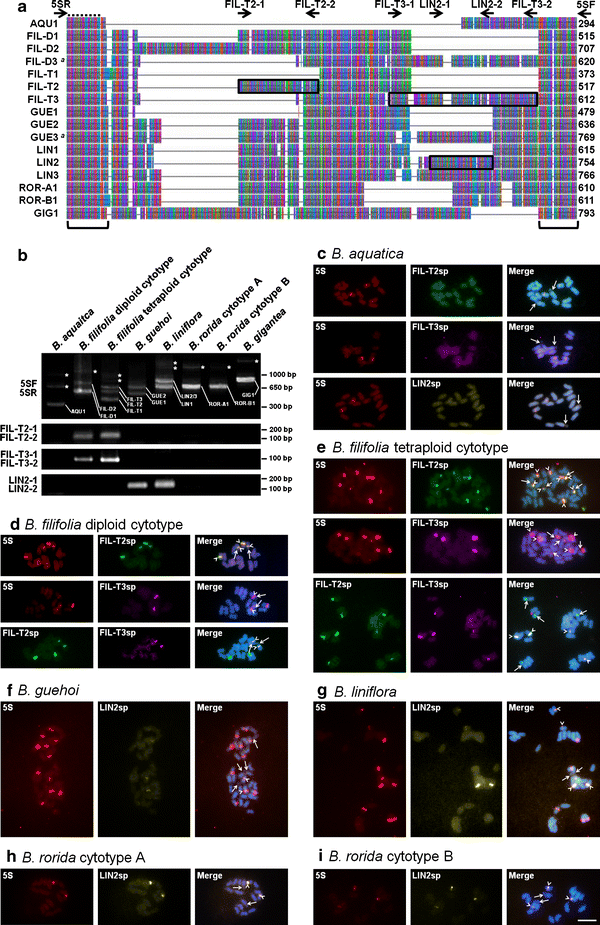
Molecular and cytogenetic characterization of 5S rDNA sequences in the Byblis liniflora complex. a A compressed representation of the 5S-NTS sequence alignment. A, T, G, and C nucleotides are indicated by green, red, purple, and blue, respectively. Bars missing position (insertion/deletion). Arrows positions of universal and sequence type-specific primers used for DNA sequencing and FISH probe preparation. Broken line 5S rDNA coding region. Brackets nucleotide positions used for phylogenetic analyses. DNA fragments used for sequence type-specific FISH are boxed. Sequence types, which are isolated by using sequence type-specific primers in combination with universal primers, are followed by a. The sequence length (excluding 5SF and 5SR primers) is noted at the end of each sequence. b PCR amplification patterns of 5S-NTS. PCR products are analyzed on a 2% agarose gel and visualized by ethidium bromide staining. Primer pairs used for PCR amplification are indicated on the left of each respective row. Sequence types corresponding to each band are indicated in the image. PCR bands of dimeric or trimeric 5S rDNA repeats are denoted by asterisk (*). Note that absence of bands corresponding to FIL-D3 and GUE3 sequences. c–i Simultaneous FISH detection of the 5S rDNA probe containing coding region and 5S-NTS sequence type-specific probe. FISH signal of the 5S rDNA probe containing coding region (5S), FIL-T2-specific probe (FIL-T2sp), FIL-T3-specific probe (FIL-T3sp), and LIN2-specific probe (LIN2sp) are pseudocolored red, green, purple, and yellow, respectively. Chromosomes were counterstained by DAPI. Arrows in c–i, secondary constrictions (the 45S rDNA sites). Arrowheads overlapping signals of 2 given probes. Bar 5 μm (c–i)
To localize specific 5S-NTS sequence types on chromosomes, we then carried out FISH analysis using FIL-T2-specific probe (FIL-T2sp), FIL-T3-specific probe (FIL-T3sp), and LIN2-specific probe (LIN2sp) (Fig. 3c–i). Based on sequence similarity, FIL-T2sp is predicted to recognize FIL-D1 and FIL-D2 in addition to FIL-T2 (Fig. 3a). Similarly, FIL-T3sp presumptively detects FIL-D3 in addition to FIL-T3, and LIN2sp seems to recognize GUE3, LIN2, LIN3, ROR-A1, and ROR-B1 (Fig. 3a). These sequence type-specific probes were hybridized to chromosome spreads of B. aquatica as a negative control, showing no apparent signal (Fig. 3c). FIL-T2sp hybridized to all arrays of B. filifolia diploid and tetraploid cytotypes (Fig. 3d), while FIL-T3sp specifically recognized 2 out of 4 and 4 out of 8 arrays, respectively (Fig. 3e). FIL-T3sp signals in the diploid cytotype were detected on 2 chromosomes carrying secondary constriction (the 45S rDNA sites) (Fig. 3d, arrows and arrowheads). Contrastingly, in the tetraploid cytotype, the signals were observed on 4 chromosomes without secondary constriction (Fig. 3e, arrows and arrowheads). These data suggested interlocus and intralocus heterogeneity of the 5S rDNA arrays in B. filifolia diploid and tetraploid cytotypes. LIN2sp was detected as two major sites along with some faint signals in B. guehoi and B. liniflora (Fig. 3f, g), suggesting interlocus heterogeneity. LIN2sp also recognized the 5S rDNA arrays of B. rorida cytotypes A and B (Fig. 3h, i).
5S-NTS dataset and tree topology
In this study, a total of 83 5S-NTS clones were sequenced and condensed into the 16 sequence types from 8 taxa. 5S-NTS length was quite variable from 294 bp in AQU1 to 793 bp in GIG1, owing to the presence of large indels that markedly reduced the amount of sequence available for comparison (Fig. 3a). A large fraction of NTS was inevitably discarded in phylogenetic analyses. The 5S-NTS dataset used for phylogenetic analyses contained partial 5S rRNA coding region (65 positions) and 5S-NTS (77 positions). Because of the difficulty in sequence alignment, outgroup taxa in plastid dataset could not be included to the 5S-NTS and ITS datasets. Alternatively, the trees were rooted with B. gigantea based on of plastid tree topology.
Maximum parsimony analysis generated a single most parsimonious tree (Fig. 4). MP, ML, and BI analyses yielded roughly congruent trees (data not shown). 5S-NTS sequence types in the B. liniflora complex were divided into two major clades as seen in plastid phylogenies: B. aquatica_–_B. filifolia (73/84/92) and B. guehoi_–_B. liniflora_–_B. rorida (97/51/99). In the MP tree, AQU1 was nested among 5S-NTS sequence types from B. filifolia with a low support (54/-/-); however, this clade did not appear in the ML and BI trees. Alternatively, in ML and BI analyses, 5S-NTS sequence types from B. filifolia diploid and tetraploid cytotypes formed a monophyletic clade (BP in ML/PP = 52/67; not shown). FIL-D3, which was isolated as a FIL-T3-like sequence, was predictably sister to FIL-T3 (91/87/93). On the other hand, ROR-A and ROR-B were nested in 5S-NTS sequence types from B. guehoi and B. liniflora with low confidences (61/40/52). Despite its original isolation as a LIN2-like sequence, GUE3 was indicated as sister to LIN3 (77/85/99).
Fig. 4
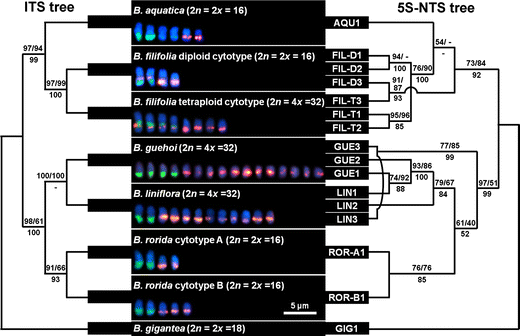
ITS (left) and 5S-NTS (right) phylogenies in the Byblis liniflora complex. Both ITS and 5S-NTS matrices yield a single most parsimonious tree. The ITS dataset is composed of 579 positions (53 sites are parsimony informative). The ITS tree yields CI and RI of 0.812 and 0.805, respectively. The 5S-NTS dataset consists of 142 positions (34 are parsimony informative). CI and RI of the 5S-NTS tree are 0.705 and 0.817, respectively. Numbers above branches are bootstrap percentages in MP/ML analyses. Bayesian posterior probabilities are indicated below the branches. A minus sign (−) denotes a clade not supported in the respective analysis. Somatic chromosomes mapped by the 5S (red) and 45S (green) rDNA probes are presented under the taxon name. Bar 5 μm for chromosomes
ITS dataset and tree topology
The length of ITS sequences in datasets ranged from 602 to 654 bp. Byblis aquatica, B. filifolia tetraploid cytotype, and B. guehoi, which harbored four 45S rDNA arrays, did not show any polymorphic sequence in an examination of clones (approximately 20 clones each were examined in colony PCR, and 5, 8, and 12 clones were sequenced, respectively). These data indicate an almost completion of interlocus homogenization of the 45S rDNA arrays in these taxa.
A single most parsimonious tree was obtained from this set of sequences (Fig. 4). MP, ML, and BI tree topologies were in full accordance with each other except for a collapsed clade of B. guehoi and B. liniflora in Bayesian analysis. Relationships in the B. liniflora complex indicated by the ITS tree were essentially identical to those of plastid and 5S-NTS phylogenies (Figs. 2, 4).
Discussion
Phylogenetic framework and morphological traits in the B. liniflora complex
In this study, we used maternally inherited plastid sequences (rbcL and trnK intron; Fig. 2) and biparentally inherited nuclear rDNA sequences (ITS and 5S-NTS; Fig. 4) for phylogenetic analyses. Comparing plastid and rDNA phylogenies, we found the topologies to be largely congruent. Byblis filifolia diploid and tetraploid cytotypes were suggested to be sister lineages. Morphological variations in B. filifolia are known to exist, for instance, flower size and petal color (Lowrie and Conran 1998; McPherson 2008). Among samples we analyzed, plants of the diploid cytotypes had navy-colored stigma and mauve-striped yellowish abaxial petal. In contrast, the tetraploid plants had white-colored stigma and whitish to yellowish abaxial petal. Extended sampling is needed to clarify the relationships between these morphological and cytogenetic characteristics in the B. filifolia lineage.
An affinity of B. filifolia and B. aquatica was suggested by both plastid and rDNA phylogenies. Their reproductive isolation was previously verified by a crossing experiment (Meyers-Rice 1993). Previous reports have suggested that seed morphology is a useful characteristic to define the different taxa in Byblis (Lowrie and Conran 1998, 2008; Conran et al. 2002b). These two species share relatively smooth anticlinal sculpturing of seeds presumably as a synapomorphy.
On the other hand, an association of B. guehoi and B. liniflora was observed. These two species share a putative synapomorphic structure—pedicels that are longer than leaves (Lowrie and Conran 2008). Byblis rorida was sister to the B. linflora_–_B. guehoi clade in plastid and rDNA trees. The three species share a morphological trait—denticulate sculpturing of seed coat.
Tetraploidizations in the B. liniflora complex
The chromosome counts in this study were essentially consistent with previous works (Peng and Kenton 1982; Conran et al. 2002a; Hoshi et al. 2007; Lowrie and Conran 2008; Fukushima et al. 2008). However, B. filifolia, which is thus far recognized as a diploid species, showed a tetraploidal chromosome number along with the diploid cytotype in the accessions we examined (Table 1). FISH analysis indicated the autopolyploidal karyotype for the tetraploid cytotype (Fig. 1), providing a further support for polyploidization within the lineage of B. filifolia. Inverted positional relationship between the 5S and 45S rDNA arrays on a chromosome arm of B. filifolia diploid and tetraploid cytotypes suggested the occurrence of a paracentric inversion at this chromosome arm (Fig. 1d, f). Although compositional discordance of 5S-NTS between the loci of diploid and tetraploid cytotypes observed in FISH with FIL-T3sp indicated unexplained complexity of the karyotype evolution in the taxa (Fig. 3d and e), the chromosome rearrangement would occur before tetraploidization because all four chromosomes carrying both 5S and 45S rDNA arrays in the tetraploid cytotype exhibited identical FISH pattern (Fig. 1f).
Peng and Kenton (1982) found the lack of multivalent formation during meiosis in B. liniflora, suggesting a functionally diploidal nature of this tetraploid species. Ribosomal DNA distribution in B. liniflora did not represent the exact doubling of any diploid species (Fig. 1j) and might help hamper multivalent formation. In phylogenetic analyses, B. guehoi and B. liniflora formed a clade, and the pair was sister to B. rorida (Figs. 2, 4). A candidate for at least 1 of the parental genome donor of these tetraploids is _B. rorida_-like species or its ancestor.
5S and 45S rDNA variabilities at the chromosomal distribution level
The copy number and chromosomal location of rDNA could be rapidly changed in plant genomes (Schubert and Wobus 1985; Raina and Mukai 1999). Schubert and Wobus (1985) postulated that the mobility of rDNAs in Allium is rendered by an adjacent transposable element or by recombination hot spots in the terminal heterochromatin blocks. In some members of Poaceae, _Enhancer/Supressor_-_mutator_-like (_En/Spm_-like) transposons and Ty3-gypsy retrotransposons localize inside rDNA cluster (Raskina et al. 2004; Belyayev et al. 2005; Altinkut et al. 2006). Transposase activity of _En/Spm_-like transposons increases during meiosis and seems to contribute to rDNA migration in Aegilops speltoides (Raskina et al. 2004; Altinkut et al. 2006). Similarly, in Nemesia and Oryza, the key mechanism facilitating diversification of rDNA distribution patterns is considered as transpositions rather than chromosome rearrangements (Datson and Murray 2006; Chung et al. 2008). FISH mapping revealed interspecies- and intraspecies-specific rDNA distribution in the B. liniflora complex (Fig. 1) without deviation of chromosome morphology (chromosome length and arm ratio; data not shown). Taking conservative karyotypes into account, a large contribution of transpositions to diversification of rDNA distribution patterns in the complex is conceivable. The copy number of migrated rDNA repeats would be amplified by unequal crossing over to the extent that these new sites can be detected by FISH. The array number of 5S rDNA (2–12 arrays) was more flexible compared to that of 45S rDNA (2–4 arrays) (Fig. 1 and Table 3). In the B. liniflora complex, generation of a novel locus via transposition may be more frequent in 5S rDNA than in 45S rDNA. Alternatively, amplification or maintenance of copy number within a novel array is more effective in the 5S rDNA.
Chromosomal rDNA distribution often varies among closely related taxa, even within species (Garrido et al. 1994). Byblis rorida exhibited intraspecies polymorphisms of the 5S rDNA distribution: cytotype A with 2 arrays and cytotype B with 3 arrays (Fig. 1l, n). We could not find any 5S-NTS polymorphism in B. rorida cytotype B (12 clones were examined in colony PCR and 8 clones were sequenced) (Fig. 4). Thus, most of the units in a surplus 5S rDNA array in cytotype B seem to be homogenized with the units of the other 2 arrays. Vaio et al_._ (2005) observed chromosomal polymorphisms in a population of Paspalum quadrifarium, and attributed the heterozygous karyotype with the odd number of 45S rDNA arrays to self-incompatibility in this species. The odd number of rDNA arrays was also found in Brachyscome lineariloba complex cytodeme E, which transmits specific chromosomes to progeny through microspores but not through megaspores (quasi-diploid; Carter et al. 1974; Adachi et al. 1997), similar to the permanent odd polyploid Rosa canina (Lim et al. 2005; Kovarik et al. 2008). Karyotype screening among populations and an investigation on reproductive strategy of B. rorida can provide an insight into the odd number of the 5S rDNA arrays.
In polyploid species, parental rDNA loci follow different evolutionary paths. The sum of the parental 45S rDNA arrays has been observed in various allopolyploid species, such as Oryza punctata (Chung et al. 2008). On the other hand, the decrease in the expected number of arrays has been observed in O. minuta and O. latrifolia (Chung et al. 2008). Likewise, a number of the 5S rDNA loci were conservative in some taxa (e.g., Sanguisorba, Mishima et al. 2002) but variable in others (e.g., Iris versicolor; Lim et al. 2007). At a glance, rDNA arrays seem to behave arbitrarily during the evolutionary history of the polyploid species. Differing from the estimation based on its tetraploidal chromosome number, two 45S rDNA arrays were detected in B. liniflora (Fig. 1j), suggesting complete or almost complete deletion of at least 2 arrays (Fukushima et al. 2008). On the other hand, B. filifolia tetraploid cytotype and B. guehoi retained 4 arrays (Fig. 1f, h). Although an apparent decrease of the 5S rDNA array number was not observed in 3 polyploids (Fig. 1f, h, j), the lack of information on parental genome obscured the evolutionary dynamics of rDNA site number in polyploid species.
5S and 45S rDNA variabilities at the nucleotide sequence level
A growing body of evidence indicates that there are three evolutionary patterns of different rDNA sequences separated by chromosomal location: (1) be retained and evolve independently without interaction, (2) be lost either through loss of entire array or through interlocus homogenization, and (3) be replaced by another rDNA type (Bao et al. 2010). We found one ITS sequence in Byblis species with two or four 45S rDNA arrays (Fig. 4). In contrast, multiple 5S-NTS sequence types were detected in species harboring 4 or more arrays (Fig. 4). Although orthology–paralogy relationships of 5S-NTS were largely obscured by extensive sequence modification in the B. liniflora complex (Fig. 3a), phylogenetic relationships suggested by the 5S-NTS tree were largely congruent with plastid and ITS phylogenies (Fig. 4). Based on our phylogenetic analysis, 5S-NTS polymorphisms within a genome would occur after the separation of B. aquatica_–_B. filifolia and B. guehoi_–_B. liniflora_–_B. rorida clades. Although network or phylogenetic analysis of entire sequences is not applicable due to a large amount of missing positions (Fig. 3a), 5S-NTS variants within a genome seem to be generated via recombination events between different sequence types. For example, the 5′ side of LIN2 is similar to that of LIN1, whereas the 3′ side of LIN2 resembles LIN3 (Fig. 3a). Likewise, the 5′ and 3′ sides of GUE2 had similarity with GUE3 and GUE1, respectively (Fig. 3a). These data indicated 5S-NTS sequence-type diversification in a genome via recombination events. FISH analysis suggested that sequence types recognized by FIL-T2sp and FIL-T3sp coexisted within an array (Fig. 3d, e). Colocalization of polymorphic sequences may facilitate the generation of novel sequence types in the B. liniflora complex.
PCR selection and variation in copy number among polymorphic sequences present in a genome lead to a biased representation of different paralogues (Wagner et al. 1994; Alvarez and Wendel 2003). Initially, 2 sequence types of 5S-NTS were found in B. filifolia diploid cytotype (15 clones were examined in colony PCR and 6 clones were sequenced), whereas 3 types were observed in the tetraploid cytotype (15 clones were examined in colony PCR and 13 clones were sequenced) (Fig. 3b). Using sequence type-specific primers, we found additional sequence type, FIL-D3, in the diploid cytotype (Fig. 3b). Sequence type-specific FISH analysis indicated an abundance of FIL-D3 in the 5S rDNA arrays of the diploid cytotype (Fig. 3d). GUE3, which was not represented in 5S-NTS pool of B. guehoi amplified with universal primers (36 clones were examined in colony PCR and 12 clones were sequenced) (Fig. 3b), was also detected by FISH using LIN2sp (Fig. 3f). Our results underline the risk in sequence analysis exclusively depending on universal primers.
Relationships between chromosomal distribution and sequence variation of 5S and 45S rDNAs
In cytogenetically well-characterized Gossypium and Nicotiana, 45S rDNA but not 5S rDNA is homogenized between loci in polyploid species (Wendel et al. 1995; Cronn et al. 1996; Fulnecek et al. 2002; Kovarik et al. 2004;). The 5S rDNA loci are located at the proximal region, whereas the 45S rDNA loci occupy the terminal locations in these two genera (Hanson et al. 1996; Fulnecek et al. 2002; Kovarik et al. 2004). The mechanism facilitating interlocus homogenization would be crossing over between rDNA repeats located on different chromosomes (Wendel et al. 1995). When crossing over occurs between proximally located arrays, inviable gametes could be generated more frequently. In contrast, telomeric or subtelomeric locations may allow interlocus crossing over without deleterious effect. After interlocus crossing over is established, intralocus homogenization would be completed via successive intralocus unequal crossing over. Under these assumptions, the chromosomal location has a substantial impact on the tempo of interlocus concerted evolution of tandemly repeated DNA elements (Zhang and Sang 1999). Our results in the B. liniflora complex appear to meet the above hypothesis. In all taxa we examined, other than B. filifolia tetraploid cytotype, the 45S rDNA arrays were more distal relative to the 5S rDNA arrays (Fig. 1; Table 3). ITS sequences seem to be well homogenized in the genome harboring four 45S rDNA arrays (Fig. 4). In B. guehoi and B. aquatica, ITS homogenization could have been prompted by relatively distal 45S rDNA positions. We believed that ITS uniformity in B. filifolia tetraploid cytotype may stem from a presumptive autotetraploidal origin. In B. liniflora, ITS can be homogenized through the entire locus loss of the 45S rDNA array. In contrast, the genomes harboring 4 or more 5S rDNA arrays surely had polymorphic 5S-NTS sequence types (Fig. 4). FISH with sequence type-specific probes indicated interlocus and intralocus heterogeneity in some 5S rDNA tandem repeats (Fig. 3d–g). Together, these data indicate that, in the B. liniflora complex, the homogenization of the 5S rDNA could not be completely accomplished within the genomes with multiple 5S rDNA arrays. Interlocus homogenization may have been hampered by their proximal location on chromosomes. Chromosomal position may have affected the contrasting evolutionary dynamics of rDNAs in the B. liniflora complex.
References
- Adachi J, Watanabe K, Fukui K, Ohmido N, Kosuge K (1997) Chromosomal location and reorganization of the 45S and 5S rDNA in the Brachyscome lineariloba complex (Asteraceae). J Plant Res 110:371–377
Article CAS Google Scholar - Albert VA, Williams SE, Chase MW (1992) Carnivorous plants: phylogeny and structural evolution. Science 257:1491–1495
Article CAS PubMed Google Scholar - Altinkut A, Raskina O, Nevo E, Belyayev A (2006) En/_Spm_–like transposons in Poaceae species: transposase sequence variability and chromosomal distribution. Cell Mol Biol Lett 11:214–230
Article CAS PubMed Google Scholar - Alvarez I, Wendel JF (2003) Ribosomal ITS sequences and plant phylogenetic inference. Mol Phylogenet Evol 29:417–434
Article CAS PubMed Google Scholar - Bao Y, Wendel JF, Ge S (2010) Multiple patterns of rDNA evolution following polyploidy in Oryza. Mol Phylogenet Evol 55:136–142
Article PubMed Google Scholar - Becerra JX (2003) Evolution of Mexican Bursera (Burseraceae) inferred from ITS, ETS, and 5S nuclear ribosomal DNA sequences. Mol Phylogenet Evol 26:300–309
Article CAS PubMed Google Scholar - Belyayev A, Raskina O, Nevo E (2005) Variability of the chromosomal distribution of Ty3-gypsy retrotransposons in the populations of two wild Triticeae species. Cytogenet Genome Res 109:43–49
Article CAS PubMed Google Scholar - Bruce AN (1905) On the activity of the glands of Byblis gigantea. Notes Roy Bot Gard Edinburgh 16:9–14
Google Scholar - Carter CR, Smith-White S, Kyhos DW (1974) The cytology of Brachycome lineariloba. 4. The ten chromosome quasidiploid. Chromosoma 44:439–456
Article Google Scholar - Chung MC, Lee YI, Cheng YY, Chou YJ, Lu CF (2008) Chromosomal polymorphism of ribosomal genes in the genus Oryza. Theor Appl Genet 116:745–753
Article CAS PubMed Google Scholar - Conran JG, Christophel DC (2004) A fossil Byblidaceae seed from Eocene South Australia. Int J Plant Sci 165:691–694
Article Google Scholar - Conran JG, Houben A, Lowrie A (2002a) Chromosome numbers in Byblidaceae. Aust J Bot 50:583–586
Article Google Scholar - Conran JG, Lowrie A, Moyle-Croft J (2002b) A revision of Byblis (Byblidaceae) in southwestern Australia. Nuytsia 15:11–20
Google Scholar - Cronn RC, Zhao X, Paterson AH, Wendel JF (1996) Polymorphism and concerted evolution in a tandemly repeated gene family: 5S ribosomal DNA in diploid and allopolyploid cottons. J Mol Evol 42:685–705
Article CAS PubMed Google Scholar - Datson PM, Murray BG (2006) Ribosomal DNA locus evolution in Nemesia: transposition rather than structural rearrangement as the key mechanism? Chromosome Res 14:845–857
Article CAS PubMed Google Scholar - Doyle JJ, Doyle JL (1990) Isolation of plant DNA from fresh tissue. Focus 12:13–15
Google Scholar - Farris JS, Källersjö M, Kluge AG, Bult C (1994) Testing the significance of incongruence. Cladistics 10:315–319
Article Google Scholar - Feinberg AP, Vogelstein B (1983) A technique for radiolabeling DNA restriction endonuclease fragments to high specific activity. Anal Biochem 132:6–13
Article CAS PubMed Google Scholar - Felsenstein J (1985) Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39:783–791
Article Google Scholar - Fukushima K, Nagano K, Hoshi Y (2008) Somatic chromosome differentiation in three species of the Byblis liniflora complex (Byblidaceae). Chromosome Bot 3:95–99
Article Google Scholar - Fulnecek J, Lim KY, Leitch AR, Kovarík A, Matyásek R (2002) Evolution and structure of 5S rDNA loci in allotetraploid Nicotiana tabacum and its putative parental species. Heredity 88:19–25
Article CAS PubMed Google Scholar - Garrido MA, Jamilena M, Lozano R, Ruiz Rejon C, Ruiz Rejon M, Parker JS (1994) rDNA site number polymorphism and NOR inactivation in natural populations of Allium schoenoprasum. Genetica 94:67–71
Article CAS PubMed Google Scholar - Guindon S, Gascuel O (2003) A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol 52:696–704
Article PubMed Google Scholar - Hanson RE, Islam-Faridi MN, Percival EA, Crane CF, Ji Y, McKnight TD, Stelly DM, Price HJ (1996) Distribution of 5S and l8S–28S rDNA loci in a tetraploid cotton (Gossypium hirsutum L.) and its putative diploid ancestors. Chromosoma 105:55–61
Article CAS PubMed Google Scholar - Hartmeyer I, Hartmeyer S (2005) Byblis filifolia als echte Karnivore rehabilitiert. Das Taublatt 3:4–5
Google Scholar - Hasebe M, Omori T, Nakazawa M, Sano T, Kato M, Iwatsuki K (1994) rbcL gene sequences provide evidence for the evolutionary lineages of leptosporangiate ferns. Proc Natl Acad Sci USA 91:5730–5734
Article CAS PubMed Google Scholar - Heslop-Harrison JS (2000) Comparative genome organization in plants: from sequence and markers to chromatin and chromosomes. Plant Cell 12:617–636
Article CAS PubMed Google Scholar - Higashiyama T, Inatsugi R, Sakamoto S, Sasaki N, Mori T, Kuroiwa H, Nakada T, Nozaki H, Kuroiwa T, Nakano A (2006) Species preferentiality of the pollen tube attractant derived from the synergid cell of Torenia fournieri. Plant Physiol 142:481–491
Article CAS PubMed Google Scholar - Hizume M (1993) Chromosomal localization of 5S rRNA genes in Vicia faba and Crepis capillaries. Cytologia 58:417–421
CAS Google Scholar - Hoshi Y, Fukushima K, Nagano K (2007) A comparative study of karyotypes in two species of Byblis (Byblidaceae). Chromosome Bot 2:39–43
Article Google Scholar - Kovarik A, Matyasek R, Lim KY, Skalická K, Koukalová B, Knapp S, Chase M, Leitch AR (2004) Concerted evolution of 18–5.8–26S rDNA repeats in Nicotiana allotetraploids. Biol J Linn Soc 82:615–625
Article Google Scholar - Kovarik A, Werlemark G, Leitch AR, Souckova-Skalicka K, Lim YK, Khaitová L, Koukalova B, Nybom H (2008) The asymmetric meiosis in pentaploid dogroses (Rosa sect. Caninae) is associated with a skewed distribution of rRNA gene families in the gametes. Heredity 101:359–367
Article CAS PubMed Google Scholar - Lee HL, Jansen RK, Chumley TW, Kim KJ (2007) Gene relocations within chloroplast genomes of Jasminum and Menodora (Oleaceae) are due to multiple, overlapping inversions. Mol Biol Evol 24:1161–1180
Article CAS PubMed Google Scholar - Liang H, Hilu KW (1996) Application of the matK gene sequences to grass systematic. Can J Bot 74:125–134
Article CAS Google Scholar - Lim KY, Werlemark G, Matyasek R, Bringloe JB, Sieber V, El Mokadem H, Meynet J, Hemming J, Leitch AR, Roberts AV (2005) Evolutionary implications of permanent odd polyploidy in the stable sexual, pentaploid of Rosa canina L. Heredity 94:501–506
Article CAS PubMed Google Scholar - Lim KY, Matyasek R, Kovarik A, Leitch A (2007) Parental origin and genome evolution in the allopolyploid Iris versicolor. Ann Bot 100:219–224
Article CAS PubMed Google Scholar - Lowrie A, Conran JG (1998) A taxonomic revision the genus Byblis (Byblidaceae) in northern Australia. Nuytsia 12:59–74
Google Scholar - Lowrie A, Conran JG (2008) Byblis guehoi (Byblidaceae), a new species from the Kimberley, Western Australia. Telopea 12:23–29
Google Scholar - McPherson S (2008) Byblis. In: McPherson S (ed) Glistening carnivores. The sticky-leaved insect-eating plants. Redfern Natural History Productions, Poole, pp 256–272
Google Scholar - Meyers-Rice B (1993) _Byblis_—notes on forms new to cultivation. Carnivorous Plant Newslett 22:39–40
Google Scholar - Mishima M, Ohmido N, Fukui K, Yahara T (2002) Trends in site-number change of rDNA loci during polyploid evolution in Sanguisorba (Rosaceae). Chromosoma 110:550–558
Article CAS PubMed Google Scholar - Müller KF, Borsch T, Legendre L, Porembski S, Theisen I, Barthlott W (2004) Evolution of carnivory in Lentibulariaceae and the Lamiales. Plant Biol 6:477–490
Article PubMed Google Scholar - Murashige T, Skoog F (1962) A revised medium for rapid growth and bioassays with tobacco tissue cultures. Physiol Plant 15:473–497
Article CAS Google Scholar - Nylander, JAA (2004) MrModeltest v2. Program distributed by the author. Evolutionary Biology Centre, Uppsala University
- Olmstead RG, Bremer B, Scott KM, Palmer JD (1993) A parsimony analysis of the Asteridae sensu lato based on _rbc_L sequences. Ann Mo Bot Gard 80:700–722
Article Google Scholar - Oxelman B, Backlund M, Bremer B (1999) Relationships of the Buddlejaceae s. l. investigated using parsimony jackknife and branch support analysis of chloroplast ndhF and rbcL sequence data. Syst Bot 24:164–182
Article Google Scholar - Peng CI, Kenton A (1982) Chromosome numbers of Byblis liniflora Salisb. (Byblidaceae). Ann Missouri Bot Gard 69:417–419
Article Google Scholar - Persson C (2000) Phylogeny of the neotropical Alibertia group (Rubiaceae), with emphasis on the genus Alibertia, inferred from ITS and 5S ribosomal DNA sequences. Am J Bot 87:1018–1028
Article CAS PubMed Google Scholar - Płachno BJ, Adamec L, Lichtscheidl IK, Peroutka M, Adlassnig W, Vrba J (2006) Fluorescence labelling of phosphatase activity in digestive glands of carnivorous plants. Plant Biol 8:813–820
Article PubMed Google Scholar - Raina SN, Mukai Y (1999) Detection of a variable number of 18S–5.8S-26S and 5S ribosomal DNA loci by fluorescent in situ hybridization in diploid and tetraploid Arachis species. Genome 42:52–59
Article CAS Google Scholar - Raskina O, Belyayev A, Nevo E (2004) Activity of the En/_Spm_-like transposons in meiosis as a base for chromosome repatterning in a small, isolated, peripheral population of Aegilops speltoides Tausch. Chromosome Res 12:153–161
Article CAS PubMed Google Scholar - Ronquist F, Huelsenbeck J (2003) Mrbayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19:1572–1574
Article CAS PubMed Google Scholar - Schubert I, Wobus U (1985) In situ hybridization confirms jumping nucleolus organizing regions in Allium. Chromosoma 92:143–148
Article Google Scholar - Sogin ML (1990) Amplification of ribosomal RNA genes for molecular evolution studies. In: Innis MA, Gelfand DH, Sninsky JJ, White TJ (eds) PCR protocols. A guide to methods and applications. Academic Press, New York, pp 307–322
Google Scholar - Steenis CGGJv (1971) Byblidaceae. Flor Males Bull Suppl Set. I 7:135–137
Google Scholar - Tamura K, Dudley J, Nei M, Kumar S (2007) MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol 24:1596–1599
Article CAS PubMed Google Scholar - Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
Article CAS PubMed Google Scholar - Vaio M, Speranza P, Valls JF, Guerra M, Mazzella C (2005) Localization of the 5S and 45S rDNA sites and cpDNA sequence analysis in species of the Quadrifaria group of Paspalum (Poaceae, Paniceae). Ann Bot 96:191–200
Article CAS PubMed Google Scholar - Wagner A, Blackstone M, Cartwright P, Dick M, Misof B, Snow P, Wagner GP, Bartels J, Murtha M, Pendleton J (1994) Surveys of gene families using polymerase chain reaction: PCR Selection and PCR Drift. Syst Biol 43:250–261
Google Scholar - Wendel JF, Schnabel A, Seelanan T (1995) Bidirectional interlocus concerted evolution following allopolyploid speciation in cotton (Gossypium). Proc Natl Acad Sci USA 92:280–284
Article CAS PubMed Google Scholar - White TM, Bruns T, Lee S, Taylo J (1990) Amplification and direct sequencing of fungal ribosomal RNA for phylogenetics. In: Innis MA, Gelfand DH, Sninsky JJ, White TJ (eds) PCR protocols: a guide to methods and applications. Academic Press, San Diego, pp 315–321
Google Scholar - Young ND, Steiner KE, de Pamphilis CW (1999) The evolution of parasitism in Scrophulariaceae/Orobanchaceae: plastid gene sequences refute an evolutionary transition series. Ann Mo Bot Gard 86:876–893
Article Google Scholar - Zhang D, Sang T (1999) Physical mapping of ribosomal RNA genes in peonies (Paeonia, Paeoniaceae) by fluorescent in situ hybridization: implications for phylogeny and concerted evolution. Am J Bot 86:735–740
Article CAS PubMed Google Scholar